

Abstract
During somatic hypermutation (SHM) of antibody variable (V) region genes, activation-induced cytidine deaminase (AID) converts dC to dU, and dUs can either be excised by uracil DNA glycosylase (UNG), by mismatch repair, or replicated over. If UNG excises the dU, the abasic site could be cleaved by AP-endonuclease (APE), introducing the single-strand DNA breaks (SSBs) required for generating mutations at A:T bp, which are known to depend upon mismatch repair and DNA Pol η. DNA Pol β or λ could instead repair the lesion correctly. To assess the involvement of Pols β and λ in SHM of antibody genes, we analyzed mutations in the VDJh4 3′ flanking region in Peyer’s patch germinal center (GC) B cells from polβ−/−polλ−/−, polλ−/−, and polβ−/− mice. We find that deficiency of either or both polymerases results in a modest but significant decrease in V region SHM, with Pol β having a greater effect, but there is no effect on mutation specificity, suggesting they have no direct role in SHM. Instead, the effect on SHM appears to be due to a role for these enzymes in GC B cell proliferation or viability. The results suggest that the BER pathway is not important during V region SHM for generating mutations at A:T bp. Furthermore, this implies that most of the SSBs required for Pol η to enter and create A:T mutations are likely generated during replication instead. These results contrast with the inhibitory effect of Pol β on mutations at the Ig Sμ locus, Sμ DSBs and class switch recombination (CSR) reported previously. We show here that B cells deficient in Pol λ or both Pol β and λ proliferate normally in culture and undergo slightly elevated CSR, as shown previously for Pol β-deficient B cells.
Keywords: Activation induced cytidine deaminase, Base excision repair, Mismatch repair, A:T mutations
1. Introduction
During the course of a humoral immune response, antibodies become increasingly effective for removal of the particular pathogen or immunogen that induced the response. This occurs in germinal centers (GCs) in lymphoid tissues and is due to somatic hypermutation (SHM) of the antibody variable (V) region genes, followed by selection of B cells producing antibody with increased affinity for the antigen. In addition, class switch recombination (CSR), which exchanges the Ig heavy chain constant region gene expressed, results in improved effector functions. SHM and CSR are both initiated by activation induced cytidine deaminase (AID), an enzyme highly expressed in activated B cells. AID deaminates cytosines in the expressed Ig V(D)J genes and IgH switch (S) regions, converting dC to dU [1–4]. AID-induced lesions can also lead to off-target mutations and DNA breaks that impact genomic stability [5–8].
The dU’s introduced by AID activity can be removed by two ubiquitous DNA repair pathways: base excision repair (BER) and mismatch repair (MMR). In the BER pathway, the dU base is excised by uracil DNA glycosylase (UNG) [9,10], leaving an abasic site with an intact sugar phosphate backbone, which is usually incised by AP endonuclease APE1, although during CSR in splenic B cells the homologous enzyme APE2 also appears to participate [11]. Both APEs leave a SSB with a 3′OH that can be extended by DNA polymerase [12,13]. DNA Polymerase β (Pol β) is the primary polymerase involved in replacing the excised nucleotide during BER, but Pol λ also contributes to repair of oxidative lesions [14,15]. Both enzymes have relatively error-free DNA polymerase activity and also have lyase activity, which is the activity that removes the 5′-deoxyribose phosphate group (dRP) that remains after cleavage of abasic sites by APE1 or APE2. The lyase activity is required in order to ligate the newly added nucleotide to the nucleotide 3′ to the SSB. Pol β and Pol λ perform short patch repair, i.e. they insert a single nucleotide at the abasic site, and the DNA ligase 3a-XRCC1 (X-ray cross-complementing protein 1) heterodimer then seals the nick to generate an intact DNA strand [12,16]. It is unknown whether B cells lacking both Pol β and Pol λ would have normal cell viability, SHM, or CSR, as they would be unable to perform short patch repair. However, it is known that in the absence of the lyase activity of Pol β or Pol λ, replicative high fidelity Pols δ or ε can perform displacement DNA synthesis at the lesion left by APE activity, inserting 2–13 nucleotides in what is termed long patch repair [12]. Subsequently, Fen1 excises the DNA flap that remains, followed by Ligase 1 activity [12,16].
The pathways used to remove dUs appear to differ between SHM and CSR. Ig S regions have a high density of AID target hotspot sequences (AGCT or WRC/GYW motifs), and it is essential for CSR that the AID-induced lesions are processed into DNA double-strand breaks (DSBs). Thus, the high density of AID hotspots likely results in SSBs sufficiently near on opposite strands to frequently form DSBs. UNG is required for CSR and for S region DSBs, demonstrating the importance of BER for generating DSBs required for CSR [10,17]. Also, in ape1+/−ape2−/− B cells, Sμ DSBs are greatly reduced (by ~80%). CSR is only reduced by ~50% in these cells [18], but they still have one allele of APE1, which is essential for viability [19]. DNA Pol β has been shown by chromatin immunoprecipitation to bind to S regions [20]. Theoretically, Pol β should reduce the numbers of S region DSBs and inhibit CSR. Indeed, that is what is observed [20]. It is possible that due to the high frequency of AID-induced lesions, DNA Pol β cannot repair all the SSBs before DSBs are generated.
By contrast, SHM does not require DSBs. In fact, DSBs appear to be rarely induced in Ig V regions, since deletions or insertions are rare and most mutations are single-nucleotide substitutions. It is likely that AID activity is much more limited at V regions than at S regions for several reasons, including but probably not limited to, the lower density of AID target hotspots and lack of long R-loops [21–23]. Therefore, it seems unlikely that Pol β would be overwhelmed by lesions induced in V(D)J genes during SHM. Only limited analyses of the role of Pol β in SHM have been performed. Esposito et al. [24] observed a slightly reduced frequency of mutation within VDJ genes expressed after immunization with NP-chicken gammaglobulin, with a significant reduction in mutation to a specific amino acid in hypervariable region 1 that enhances NP-binding in polβ−/− relative to polβ+/+ germinal center (GC) cells. These results were interpreted to be due to reduced affinity selection caused by reduced B cell proliferation in the pol β−/− mice, consistent with their observation of reduced splenic B and T cell numbers in the Pol β-deficient mice [24]. By contrast, Poltoratsky et al. [25] reported that Pol β is down-regulated in the human BL2 GC B cell line, and that BL2 clones with the lowest levels of Pol β show increased V region SHM relative to other clones. These authors also observed by immunohistochemistry that Pol β levels are reduced in human tonsillar GC B cells relative to the surrounding stromal cells. They concluded that Pol β inhibits SHM. Pol λ-deficiency has been reported to have little or no effect on SHM in expressed VDJ genes in Peyer’s patch GC B cells [26]. There could be redundancy between Pols β and λ, and no one has examined mice with B cells that are deficient in both Pol β and Pol λ for effects on SHM or CSR.
In this study, we analyze the effect of combined deletion of the genes for DNA Pols β and λ on CSR in splenic B cells, and on SHM in the 3′ flanking region of recombined VDJh4 genes in Peyer’s patch GC B cells. We examined the 3′ flanking region to avoid the problem of confusing mutation itself with selection for high affinity antibodies during an immune response. Our experiments address the question of whether BER is involved in creating the SSBs that are used as entrance sites for the translesion DNA Pol η, which is required for producing A:T mutations during SHM [27–29]. Our data suggest that Pol β is not down-regulated in mouse GC B cells, as we detect a similar level to that in non-GC B cells. Although we find that SHM is reduced in mice deficient in Pols β and λ, a detailed analysis of mutation specificity indicates that this is likely due to an effect on cell proliferation and/or viability rather than a direct effect on SHM. Also, combined deficiency of Pol β and λ does not reduce SHM more than Pol β deficiency alone. We hypothesize that most of the SSBs required for AID-induced mutations at A:T bp are generated by DNA replication, rather than by BER. We discuss implications of these data for when AID might act during the cell cycle to initiate SHM.
2. Methods
2.1. Mice
All mouse strains were backcrossed to C57BL/6 for more than 8 generations. Polβ+/− mice [30] were bred to create polβ−/− embryos, or to polλ+/− mice [26] to create polβ+/−polλ+/− mice that were bred to create polβ+/−polλ−/− mice. These mice were bred and the embryos were harvested from the pregnant females just prior to birth, d 18 to 20 post coitus. Two x 106 fetal liver cells from polλ−/−, polβ−/−polλ−/−, or polβ−/− embryos were injected into the tail vein of sub-lethally irradiated (900R) CD45.1+ B6.SJL congenic mice (from Jackson Labs, then bred in our facility). Control (WT) animals received 2 × 106 bone marrow cells from WT C57BL/6 mice and all recipient mice were maintained on antibiotics. Spleens and Peyer’s patches were harvested from the recipients between 2 and 12 months after transfer. Reconstitution by CD45.2+ cells at the time of harvest was >90% in all mice, except for one that was 85%. Mice were housed in the Institutional Animal Care and Use Committee-approved specific pathogen-free facility at the University of Massachusetts Medical School. The mice were bred and used according to the guidelines from the University of Massachusetts Animal Care and Use Committee.
2.2. Purification of GC cells
Peyer’s patch cells were gently dispersed mechanically and filtered through 70 μm nylon mesh, and then incubated with anti-CD16/32 antibody (eBioscience) for 10 min on ice to block Fc receptors prior to the addition of B220-Alexafluor700, CD95-PE (eBioscience), GL7-FITC and CD45.2-APC (BD Pharmingen) for 30 min on ice. After washing, cells were resuspended in 7AAD and viable, GC (B220+CD95+GL7+) and non-GC (B220+CD95−GL7−) B cells were isolated on a FACSAriaII (>95% purity) and frozen for subsequent DNA or protein extraction.
2.3. Amplification, cloning and sequencing of J558VHFR3-JH4 3′intron segments
DNA extracted from GC B cells was amplified using Pfu Ultra II (Stratagene); primers were modified slightly from [27,31]. Primers for the first amplification were: forward 5′AGCCTGACAT-CTGAGGAC, and reverse 5′ GTGTTCCTTTGAAAGCTGGAC. Nested primers for the second amplification were: forward 5′ CCGGAATTC-CTGACATCTGAGGACTCTGC and reverse 5′ GATGCCTTTCTCC-CTTGACTC. The reaction conditions for the first primer set were 95 °C for 30 s, 57 °C for 30 s and 72 °C for 1 min for 30 cycles; and for the second primer set were the same but for 35 cycles. The PCR products were electrophoresed on 1% agarose gels; the 600 bp band was purified using QIAquick Gel Extraction Kit (Qiagen), dA tails were added with Taq polymerase, and cloned using TOPO TA cloning kit (Invitrogen), and sequenced by Macrogen USA.
2.4. Immunoblots
20 μg of whole cell extract in RIPA buffer was electrophoresed on gradient gels (4–20%, Pierce) and transferred to ImmobilonP membrane (Millipore). Blots were blocked with 5% powdered milk and probed with mouse anti-Pol β antibody (Neomarkers) and donkey anti-mouse-HRP (Santa Cruz) and developed with Pierce ECL western blot substrate.
2.5. B cell culture and CSR
Spleen B cells were prepared by T cell depletion with antibody and complement [32], and isolated on Lympholyte gradients. 100,000 B cells were cultured in 1 ml RPMI with 10% FBS and supplements as described [32]. Cultures contained 25 μg/ml LPS, 50 U/ml BLyS-FLAG, 10 ng/ml anti-IgD-dextran (FinaBio). To induce CSR to IgG2a, we also added 20 pg/ml IFN-γ (EBioscience). CSR and cell division were assayed by flow cytometry after 3 days of culture using F(ab)′2 anti-mouse-IgG2a-PE and -IgG3-PE antibodies (Southern Biotech), and dilution of the intracellular stain CFSE (Invitrogen Corp) as described [11].
3. Results
3.1. DNA Pol β is present in GC B cells
To determine whether SHM, which occurs in GC B cells, is affected by DNA Pol β, we first assessed whether DNA Pol β is present in these cells. GC B cells (B220+CD95+GL7+) were purified from Peyer’s patches of wild-type (WT) mice, as these are sites where B cells are continuously stimulated by gut microbes and therefore undergoing large amounts of SHM without immunization. We also sorted non-GC (B220+CD95−GL7−) B cells from Peyer’s patches for comparison. Immunoblot analysis indicated that Pol β is present at similar levels in both GC and non-GC B cells (Fig. 1). The amount is also comparable to that in splenic B cells induced to undergo CSR, which is similar to levels in unstimulated ex vivo splenic B cells [20]. We conclude that Pol β is not detectably down-regulated in Peyer’s patch GC B cells undergoing SHM.
Fig. 1.
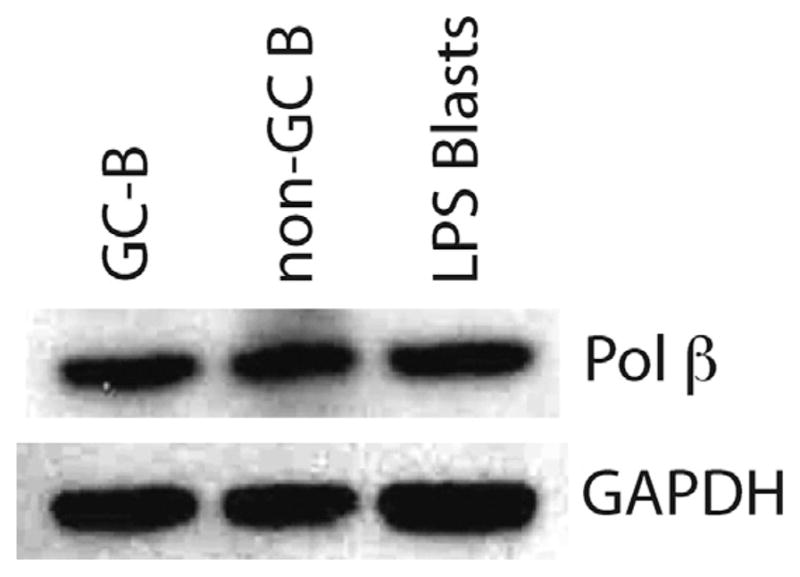
Pol βis present and not down-regulated in GC cells. Western blot of whole cell extracts from FACS-purified GC and non-GC WT B cells, or splenic B cells stimulated in vitro with LPS and anti-IgD-dextran for 48 h. The blot is representative of three independent experiments (three mice).
3.2. Reduced GC B cell numbers in Pol β- and Pol λ-deficient mice
Deletion of the DNA Pol β gene is a lethal mutation, and mice die at birth, although they survive to at least 18.5 days p.c., sufficient to obtain fetal liver cells that can be transferred into sub-lethally irradiated mice to reconstitute their hematopoietic system, as described previously [20,24]. The donor cells can then be identified by expression of CD45.2, as the congenic host mice express the CD45.1 allele. Although polλ−/− mice develop normally, in order to treat all 4 genotypes similarly, we created chimeric mice with hematopoietic systems lacking both DNA Pol β and λ genes (polβ−/−polλ−/−), Pol λ alone (polλ−/−), Pol β alone (polβ−/−), or WT (polβ+/+polλ+/+). Peyer’s patch cells were harvested from the chimeric mice between 2 and 12 months after fetal liver transfer, and CD45.2+B220+CD95+GL7+ cells were isolated by FACS. Although the average recovery of total Peyer’s patch cells did not differ between genotypes, the percent of Peyer’s patch B cells that were GC cells was reduced by an average of 50% in all of the Pol β-or Pol λ-deficient mice (Table 1). Consistent with the known importance of these polymerases for repair of oxidative damage in DNA [14,15], these results suggest that Pol β and Pol λ are required for normal levels of GC B cell proliferation and/or viability.
Table 1.
Reduced GC B cell recovery in polβ−/− and polλ−/− mice.
| Months since transfer: | % Germinal center B cells
|
||
|---|---|---|---|
| 4 | 6 | 12d | |
| WT | 5.1a (1)b | 4.3 (1) | 7.6 (2) |
| polβ−/− | 2.8 (1) | 1.8 (1) | n.d. |
| polλ−/− | n.d.c | n.d. | 3.5 (2) |
| polβ−/−polλ−/− | n.d. | n.d. | 3.6 (2) |
Average GC cells as a percent of B220+ PP cells.
Number of individual mice.
Not determined.
Reconstitution by transferred cells was greater than 97% 12 months after transfer.
3.3. Mutations in the 3′ Jh4 flank are reduced in Peyer’s patch GC cells from Pol β- and Pol λ-deficient mice
We analyzed Ig V region SHM by examining the 493 bp region 3′ to the Jh4 gene, a non-coding region that does not undergo antigen-specific selection, in order to analyze the mutation pattern in the absence of selection for antigen binding. Also, because the sequences are from Peyer’s patches from unimmunized mice, they are likely to be from B cells responding to many different antigens, and thus not skewed to a limited number of clones. To obtain the sequences, we amplified this region from genomic DNA using high-fidelity Pfu polymerase and nested primers complementary to the framework 3 region of the J558L family and 3′ to Jh4, as others have previously reported [27,31]. We counted all mutations in non-identical sequences in the dataset from each mouse, and only included sequences that had mutations in the calculation of mutation frequency. The numbers of unmutated sequences did not correlate with genotype and varied between mice, suggesting that they might be from a few contaminating non-GC cells. To determine the mutation spectra, i.e., their nucleotide specificity, we include identical mutations only when they are present in sequences with different CDR3’s, as this indicates they were independently derived.
Results shown in Table 2 demonstrate that mutation frequency is reduced by an average of 24% in polλ−/− cells, and by an average of 35% in polβ−/− and polβ−/−polλ−/− GC B cells relative to WT cells, and the reduction in all three is highly significant. However, there is no difference in the nucleotide specificity of the substitution mutations, i.e., no difference in the proportion of the mutations that are at G:C bp, nor the proportion of transitions at G:C bp, nor the proportion of mutations at AID target hotspots relative to other nucleotide positions in any of the polymerase-deficient mice. Although we examined SHM in chronically-stimulated Peyer’s patch B cells, it has also been shown that under acute antigen stimulation, Pol β is not important for SHM [24]. The SHM results for polλ−/− cells agree qualitatively with a previous smaller scale analysis of SHM in Peyer’s patch germinal center B cells from polλ−/− mice, in which it was found that mutation frequency was slightly decreased, and that there was no effect on base specificity [26]. However, the proportion of mutations that are deletions and insertions was significantly lower in polλ−/− mice than in WT mice (Table 2), and Pol λ is known to generate small deletions due to a propensity for strand slippage [33,34]. Pol λ is also known to contribute to repair of DNA ends to make them good substrates for NHEJ during V(D)J recombination [35]. As the proportion of insertions/deletions was not significantly reduced in the polβ−/−polλ−/− mice, this result is difficult to interpret, especially given the small numbers of insertion/deletion mutations. Due to the lack of effect of these polymerases on the specificity of mutations, these results suggest that neither DNA Pol β nor Pol λ has a direct effect on SHM, and further suggest that the reduction in mutation frequency is most likely due to reduced cell proliferation and/or viability in the polymerase-deficient cells. These polymerases are important for repair of oxidative lesions [14,15], which should be numerous in these highly proliferating GC B cells.
Table 2.
SHM mutation frequency and base specificity in Pol β- and Pol λ-deficient B cells.a
| Genotype | Total mutationsa | Total nts present in mutated sequences | Mutation frequency | % of Mutations at G:C bpc,d (%) | % of G:C mutations that are transitions (%) | Frequency of mutations at hotspotsd (% at hotspots) | Frequency of insertions/deletionsd |
|---|---|---|---|---|---|---|---|
| WT (4 mice) | 2468 | 92,046 | 26.8 × 10−3 | 39.7 | 56.6 | 3.5 × 10−3 (21%) | 0.29 × 10−3 (29)e |
| Pol β KO (5 mice) | 1194 | 58,617 | 20.3 × 10−3 p < 0.00012 |
41.5 | 61.3 | 3.4 × 10−3 (24%) | 0.05 × 10−3 (3) p = 0.005b |
| Pol β KO (5 mice) | 2301 | 133,820 | 17.2 × 10−3 p < 0.0001 |
43.4 | 58.5 | 2.8 × 10−3 (26%) | 0.14 × 10−3 (19) N.S.f |
| DBL Pol β+λ (2 mice) | 578 | 34,017 | 17.0 × 10−3 p < 0.0001 |
41.9 | 54.6 | 3.2 × 10−3 (22%) | 0.12 × 10−3 (4) N.S. |
These data are from: 2 WT mice (9, 10 months), 3 Pol λ KO 5 months old, and 5 Pol β KO 9–10 months old, and 2 WT, 2 Pol λ, 2 DKO mice each 14 months old. All cells were from Peyer’s patches of fetal liver transfer recipients, sorted for CD45.2, B220, CD95 and GL7. All calculations include only the 493 bp JH4 3′ intron segments that contain mutations. Nucleotide totals only include sequences with mutations. Total mutations include indels.
All p values indicate difference from WT mutation frequencies (chi-square test).
The results have been corrected for the base composition of the J4 3′ DNA segment analyzed.
The calculations for base specificity and insertions/deletions exclude identical mutations found in sequences with the same CDR3 from the same mouse.
Numbers of insertions/deletions.
N.S. not significant.
3.4. Pol λ and Pol β have a small inhibitory effect on CSR
Previously we showed that Pol β deficiency increases CSR to IgG2a by 1.6-fold, and to additional isotypes if CSR is induced under sub-optimal conditions in which SSBs might be limiting [20]. We asked whether Pol λ-deficiency also increases CSR. Splenic B cells from polλ−/− and polλ−/−polβ−/− chimeric mice were induced to switch in optimal culture conditions to IgG3 or IgG2a. As shown in Fig. 2, both polλ−/− and polβ−/−polλ−/− B cells showed a consistent, slight increase in the level of CSR to both IgG2a and IgG3, with cells from the doubly-deficient mice showing a greater increase (up to 1.5-fold). As shown previously for Pol β-deficient splenic B cells [20], we observed no effect on cell recovery, viability or proliferation of cultured B cells.
Fig. 2.

CSR is slightly increased in the absence of Pol β and Pol λ. (A) Representative profile and (B) quantification of CSR in CFSE-stained spleen B cells stimulated for 3 days and assayed by flow cytometry (n = 3 mice of each chimera).
In conclusion, there is a difference in the role for DNA Pol β in V region SHM in comparison with CSR induced in culture. Pol β is likely to be important for proliferation or survival of GC B cells from Peyer’s patches but does not appear to have a direct effect on SHM. By contrast, Pol β– deficient B cells proliferate normally when induced to switch in culture, have a slightly increased frequency of CSR, a two-fold increase in S region DSBs, and a two-fold increase in mutations at the Sμ region, mostly due to an increase of mutations at A:T bp [20].
4. Discussion
DNA Pol β and Pol λ accurately repair SSBs after APE1 acts, and likely also after APE2, by adding a single nucleotide complementary to the template DNA (short patch repair), thus inserting a dC at AID-induced lesions. In addition, the lyase activity of these enzymes creates a substrate for DNA ligase3a, leading to error-free repair of the DNA strand. These activities might predict that SHM would be increased in the absence of Pol β and Pol λ, similar to the increase in mutations found in the unrearranged (germline, GL) 5′ Sμ region in B cells undergoing CSR in culture [20]. But instead, we observed a decrease in SHM, which theoretically could be explained by the fact that in the absence of both Pol β and Pol λ, long patch repair occurs, involving recruitment of high fidelity replicative polymerase (Pol δ or Pol ε) [12,36]. These enzymes perform accurate displacement DNA synthesis. It is possible that recruitment of replicative polymerase would decrease recruitment of error-prone translesion DNA polymerases, which could decrease mutations at A:T bp or alter the ratios of mutations at AID target hotspots (WRC/GYW motifs) relative to non-hotspot sites. However, we do not observe any effect on the base specificity of mutations in these mice. The fact that there is no consistent difference in the specificity of mutations or their location relative to the AID target hotspots in the Pol β and Pol λ-deficient mice suggests that neither DNA Pol β or λ acts directly during SHM at V(D)J regions.
Instead, our data suggest that the reduction in SHM observed in Peyer’s patch GC B cells in mice lacking either or both Pol β and Pol λmight be explained by a reduction in proliferation and/or survival, consistent with the fewer GC B cells recovered from these mice. As Pol β, and Pol λ to a lesser extent, are important for repairing SSBs arising after oxidative base damage [14,15], it is not surprising that GC B cells, which are rapidly dividing, would survive less well when they lack these enzymes. The importance of Pol β for cell survival is demonstrated by the fact that polβ−/− mice die before birth, and the lower numbers of B and T cells found in mice reconstituted with polβ−/− fetal liver cells compared with mice reconstituted with WT fetal liver cells [24]. Also, as Pol λ contributes to NHEJ [37,38], polλ−/− cells could have reduced efficiency of DSB repair. This effect on proliferation and/or survival could result in sub-optimal selection of high-affinity GC B cells, and this could explain the reduced frequency of a specific mutation that is important for NP-binding in polβ−/− mice [24].
It is clear that the effect of Pol β on V region SHM differs from its inhibitory effect on mutations introduced into the 5′ region of the GL Sμ region in splenic B cells stimulated to undergo CSR, but that have not yet switched. It is thought that Sμ mutations occur due to unsuccessful attempts to initiate CSR. Polβ−/− splenic B cells have ~2-fold increased frequency of S region mutations, with mutations at A:T bp increased more than G:C mutations [20]. This suggests that in the absence of Pol β, SSBs in S regions are more numerous, providing increased entrance sites for error-prone DNA Pol η [39,40]. Consistent with an inhibitory effect on SSBs, DSBs are increased in polβ−/− splenic B cells undergoing CSR [20]. It has been proposed that the mutation mechanisms for V regions and GL Sμ are the same, but our data indicate that Pol β deficiency has different effects on the mutations at these two DNA regions. Pol β repairs SSBs and this is inhibitory for CSR. A priori Pol β activity could also be inhibitory for SHM at A:T bp, but we find it is not. These differences suggest that AID-induced lesions are treated differently in the V and S regions, consistent with the different requirements for SSBs vs DSBs for SHM and CSR, respectively, and perhaps in part due to the many fewer AID target hotspots in V regions than in S regions.
The DSBs required for CSR are introduced into the Sμ region during the G1 phase of the cell cycle, and they are also repaired/recombined during G1 phase [41,42], consistent with the fact that NHEJ is the primary repair pathway for performing S-S recombination [43,44]. Furthermore, UNG has been shown to function only during G1 phase to repair AID-induced dU lesions in cultured splenic B cells during CSR, and also in vivo during SHM [45]. UNG is essential for CSR, due to the requirement for DSBs. Chromatin immunoprecipitation assays have shown that AID recruits UNG to S regions during CSR; interestingly, this recruitment depends upon the C terminus of AID [46], which is required for CSR but not for SHM [47,48]. The interaction between AID and UNG does not appear to be direct [46], and we hypothesize that it might be mediated by a protein(s) that specifically binds S regions, of which a few have been described [49–53].
The phenotypes of UNG-deficiency on CSR and SHM clearly differ. Deletion of UNG actually increases the frequency of V(D)J region SHM, indicating that the BER pathway sometimes accurately repairs AID-induced deaminations [10,45,54,55]. Ung−/− mice have a large increase in the proportion of transition mutations at G:C bp (increasing from 65% to become 95% of G:C mutations), but only a small decrease in mutations at A:T bp (15–25%) [10,56–60]. The increase in transition mutations relative to transversions is expected if U:G mismatches are not processed by UNG, because dU can be read as dT by most replicative DNA polymerases, resulting in a transition mutation [61]. Without UNG, SSBs would not be generated at AID induced lesions, thus reducing sites for entrance of Pol η and subsequent mutations at A:T bp. Thus, it is surprising that the effect on A:T mutations is small. That neither UNG nor Pol β have a marked effect on mutations at A:T bp suggests that most of these mutations do not depend on generation of SSBs through the BER pathway, and also suggests that another mechanism is used for generating SSBs during SHM. In other words, these data predict that APE is not important for creating SSBs required for mutations at A:T bp.
By contrast, V region mutations at A:T bp are reduced by ~80% in mice deficient for the MMR proteins, Msh2, Msh6, or Exo1 [29,58,62]. Deletions of both the Msh2 and DNA Pol η genes eliminates 99% of A:T mutations [29]. As 50–60% of the V region mutations in WT mice are at A:T bp, it is clear that SSBs, MMR, and Pol η activities are very important for SHM. The role of MMR in general is to repair errors due to misincorporation during S phase DNA replication. Msh2-Msh6 recognizes and binds to mismatches generated during replication, and recruits Mlh1-Pms2 and Exo1, which together cause excision from a SSB located 5′ to the mismatch and removal of the base misincorporated during DNA replication [63]. Also, Mlh1-Pms2 creates additional SSBs on the DNA strand with a pre-existing SSB to create additional entrance sites for excision and repair synthesis [64]. MMR uses SSBs near the mismatch to identify the newly synthesized strand in order to determine which strand to excise. The 5′ ends of newly synthesized Okazaki fragments and the 3′ end of the leading strand have been proposed to serve this function [63]. Recently, it has also been shown that SSBs created by removal by RNaseH2 of rNTPs misincorporated into the leading strand also serve this function [65].
It is clear that AID can act during G1 phase [41,42,66], and thus AID would introduce dU into a DNA strand that serves as template during the subsequent S phase. If UNG does not excise the dU prior to replication, the dU can be read as dT, creating transition mutations [61], but this is not a substrate for MMR. If UNG does excise the dU during G1 phase and APE creates a SSB, A:T mutations can be produced using this SSB. However, this cannot be the predominant pathway as A:T mutations are only reduced by an average of 20% in UNG-deficient mice.
By contrast, if AID acts at V(D)J genes during S phase at the replication fork, it could deaminate either the template or daughter strands, thus producing U:G mismatches that when repaired by MMR would use SSBs on either the dU or dG strands for recruitment of Exo1. Since AID requires a single-strand DNA substrate, it might more frequently deaminate the template strand. The use by MMR of single strand DNA ends or other SSBs generated during DNA replication would eliminate the requirement for the BER pathway to generate the SSBs required for mutations at A:T bps. Supporting the hypothesis that A:T mutations require S phase SSBs is a recent report that B lymphoma cells cultured under conditions promoting fast replication have a greater proportion of V region mutations at A:T bp than those cultured under conditions that cause slow replication [67].
The hypothesis that DNA breaks generated during DNA synthesis are involved in creating most of the A:T mutations might explain why A’s are more frequently mutated than T’s [68], since DNA replication across the V(D)J segment might originate from the same direction in most cells, and Pol η has a higher mutation rate using template T than template A [27,28,69–71]. Our dataset also showed this preference, with the ratio of A to T mutations ranging from 1.7 to 2.0 (corrected for sequence specificity). One strand might more often serve as the leading strand and the other as the lagging strand during DNA replication across V(D)J regions, and this would affect the frequency of SSBs on the two strands and their positions relative to the U:G mismatch. Alternatively, there might be differential activities on the transcribed and non-transcribed DNA strands [70,72].
5. Concluding remarks
Because we find no change in the base specificity or location of mutations in the Jh4 3′ flanking region in GC B cells from polβ−/−, pol λ−/− mice, or pol β−/−pol λ−/− mice, our data suggest that Pol β does not have an direct role in SHM and support a previous report concluding the same for Pol λ [26]. Our data indicate that Pol β is expressed in GC cells, presumably because it is required for repair of oxidative damage expected in these highly replicating cells. Furthermore, these data, along with the finding of others that UNG is not important for mutations at A:T bp, suggest that most of the SSBs required for generation of A:T mutations during SHM are not due to the BER pathway, and most likely are created instead by DNA replication. Thus, these results imply that during SHM of V(D)J regions, AID acts during DNA replication, perhaps at the fork immediately after DNA synthesis, in order to generate mutations at A:T bp, which comprise ~60% of the mutations in WT cells.
Acknowledgments
We thank the University of Massachusetts Flow Cytometry Core Facility for their help with cell sorting. The research was supported by grants from the NIH: RO1-AI23283 (J.S.) and R03-AI092528 (C.E.S.).
Abbreviations
- AID
activation induced cytidine deaminase
- BER
base excision repair
- CSR
class switch recombination
- DSB
double-strand DNA break
- GC
germinal center
- MMR
mismatch repair
- Pol
polymerase
- SHM
somatic hypermutation
- SSB
single-strand DNA break
- UNG
uracil-N-DNA glycosylase
Footnotes
Conflict of interest statement
The authors have no commercial/financial conflict of interest.
References
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 2.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 3.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–104. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 4.Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM, 3rd, Gearhart PJ. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 6.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 7.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–350. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- 9.Lindahl T. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat Res. 2000;462:129–135. doi: 10.1016/s1383-5742(00)00024-7. [DOI] [PubMed] [Google Scholar]
- 10.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 11.Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hadi MZ, Ginalski K, Nguyen LH, Wilson DM., 3rd Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J Mol Biol. 2002;316:853–866. doi: 10.1006/jmbi.2001.5382. [DOI] [PubMed] [Google Scholar]
- 14.Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. DNA polymerase λ mediates a back-up base excision repair activity in extracts of mouse embryonic fibroblasts. J Biol Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- 15.Braithwaite EK, Kedar PS, Stumpo DJ, Bertocci B, Freedman JH, Samson LD, Wilson SH. DNA polymerases β and λ mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE. 2010;5:e12229. doi: 10.1371/journal.pone.0012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent upon AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrader CE, Guikema JE, Wu X, Stavnezer J. The roles of APE1, APE2, DNA polymerase β and mismatch repair in creating S region DNA breaks during antibody class switch. Philos Trans R Soc Lond B Biol Sci. 2009;364:645–652. doi: 10.1098/rstb.2008.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Stavnezer J. DNA polymerase β is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med. 2007;204:1677–1689. doi: 10.1084/jem.20070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang FT, Yu K, Balter BB, Selsing E, Oruc Z, Khamlichi AA, Hsieh CL, Lieber MR. Sequence-dependence of chromosomal R-loops at the immunoglobulin heavy chain Sμ class switch region. Mol Cell Biol. 2007;27:5921–5932. doi: 10.1128/MCB.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kenter AL. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol. 2012;24:281–286. doi: 10.1016/j.smim.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esposito G, Texido G, Betz UA, Gu H, Muller W, Klein U, Rajewsky K. Mice reconstituted with DNA polymerase β-deficient fetal liver cells are able to mount a T cell-dependent immune response and mutate their Ig genes normally. Proc Natl Acad Sci USA. 2000;97:1166–1171. doi: 10.1073/pnas.97.3.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poltoratsky V, Prasad R, Horton JK, Wilson SH. Down-regulation of DNA polymerase β accompanies somatic hypermutation in human BL2 cell lines. DNA Repair (Amst) 2007;6:244–253. doi: 10.1016/j.dnarep.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertocci B, De Smet A, Flatter E, Dahan A, Bories JC, Landreau C, Weill JC, Reynaud CA. Cutting edge: DNA polymerases μ and λ are dispensable for Ig gene hypermutation. J Immunol. 2002;168:3702–3706. doi: 10.4049/jimmunol.168.8.3702. [DOI] [PubMed] [Google Scholar]
- 27.Martomo SA, Yang WW, Wersto RP, Ohkumo T, Kondo Y, Yokoi M, Masutani C, Hanaoka F, Gearhart PJ. Different mutation signatures in DNA polymerase η- and MSH6-deficient mice suggest separate roles in antibody diversification. Proc Natl Acad Sci USA. 2005;102:8656–8661. doi: 10.1073/pnas.0501852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA. Contribution of DNA polymerase η to immunoglobulin gene hypermutation in the mouse. J Exp Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delbos F, Aoufouchi S, Faili A, Weill JC, Reynaud CA. DNA polymerase η is the sole contributor of A/T modifications during immunoglobulin gene hyper-mutation in the mouse. J Exp Med. 2007;204:17–23. doi: 10.1084/jem.20062131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 31.McDonald JP, Frank EG, Plosky BS, Rogozin IB, Masutani C, Hanaoka F, Woodgate R, Gearhart PJ. 129-derived strains of mice are deficient in DNA polymerase ι and have normal immunoglobulin hypermutation. J Exp Med. 2003;198:635–643. doi: 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrader CE, Edelmann W, Kucherlapati R, Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J Exp Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 34.Bebenek K, Garcia-Diaz M, Blanco L, Kunkel TA. The frameshift infidelity of human DNA polymerase λ. Implications for function. J Biol Chem. 2003;278:34685–34690. doi: 10.1074/jbc.M305705200. [DOI] [PubMed] [Google Scholar]
- 35.Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases μ, λ, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006;25:31–41. doi: 10.1016/j.immuni.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 37.Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, Koiwai O, Hsieh CL, Schwarz K, Lieber MR. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol Cell. 2004;16:701–713. doi: 10.1016/j.molcel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Lee JW, Blanco L, Zhou T, Garcia-Diaz M, Bebenek K, Kunkel TA, Wang Z, Povirk LF. Implication of DNA polymerase λ in alignment-based gap filling for nonhomologous DNA end joining in human nuclear extracts. J Biol Chem. 2004;279:805–811. doi: 10.1074/jbc.M307913200. [DOI] [PubMed] [Google Scholar]
- 39.Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ. MSH2-MSH6 stimulates DNA polymerase η, suggesting a role for A:T mutations in antibody genes. J Exp Med. 2005;201:637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arakawa H, Moldovan GL, Saribasak H, Saribasak NN, Jentsch S, Buerstedde JM. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, Redon C, Ried T, Bonner WM, Honjo T, Nussenzweig MC, Nussenzweig A. AID is required to initiate Nbs1/γ-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179:6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- 43.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–131. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 44.Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Ann Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharbeen G, Yee CW, Smith AL, Jolly CJ. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med. 2012;209:965–974. doi: 10.1084/jem.20112379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, Stavnezer J. AID binds cooperatively with UNG and Msh2-Msh6 to Ig switch regions dependent upon the AID C terminus. J Immunol. 2011;187:2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, Nonoyama S, Tashiro J, Ikegawa M, Ito S, Kinoshita K, Muramatsu M, Honjo T. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. 2003;4:843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- 48.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. 2003;12:501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 49.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates J, Herron B, 3rd, Otto M, Zan H, Fu H, Casali P. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zan H, White CA, Thomas LM, Mai T, Li G, Xu Z, Zhang J, Casali P. Rev1 recruits Ung to switch regions and enhances dU glycosylation for immunoglobulin class switch DNA recombination. Cell reports. 2012;2:1220–1232. doi: 10.1016/j.celrep.2012.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, Alt FW, Losson R, Reina-San-Martin B. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208:1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zahn A, Daugan M, Safavi S, Godin D, Cheong C, Lamarre A, Di Noia JM. Separation of function between isotype switching and affinity maturation in vivo during acute immune responses and circulating autoantibodies in UNG-deficient mice. J Immunol. 2013;190:5949–5960. doi: 10.4049/jimmunol.1202711. [DOI] [PubMed] [Google Scholar]
- 55.Storb U, Shen HM, Nicolae D. Somatic hypermutation: processivity of the cytosine deaminase AID and error-free repair of the resulting uracils. Cell cycle. 2009;8:3097–3101. doi: 10.4161/cc.8.19.9658. [DOI] [PubMed] [Google Scholar]
- 56.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 57.Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. EMBO J. 2006;25:585–595. doi: 10.1038/sj.emboj.7600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6−/−Ung−/− double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- 59.Perez-Duran P, Belver L, de Yebenes VG, Delgado P, Pisano DG, Ramiro AR. UNG shapes the specificity of AID-induced somatic hypermutation. J Exp Med. 2012;209:1379–1389. doi: 10.1084/jem.20112253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Zhao Y, Wang JY. Analysis of Ig gene hypermutation in Ung−/−Polh−/− mice suggests that UNG and A:T mutagenesis pathway target different U:G lesions. Mol Immunol. 2013;53:214–217. doi: 10.1016/j.molimm.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 61.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 62.Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, Scharff MD. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 63.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLα in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 65.Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol Cell. 2013;50:437–443. doi: 10.1016/j.molcel.2013.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Faili A, Aoufouchi S, Gueranger Q, Zober C, Leon A, Bertocci B, Weill JC, Reynaud CA. AID-dependent somatic hypermutation occurs as a DNA single-strand event in the BL2 cell line. Nat Immunol. 2002;3:815–821. doi: 10.1038/ni826. [DOI] [PubMed] [Google Scholar]
- 67.Kano C, Ouchida R, Kokubo T, Wang JY. Rapid cell division contributes to efficient induction of A/T mutations during Ig gene hypermutation. Mol Immunol. 2011;48:1993–1999. doi: 10.1016/j.molimm.2011.06.218. [DOI] [PubMed] [Google Scholar]
- 68.Milstein C, Neuberger MS, Staden R. Both DNA strands of antibody genes are hypermutation targets. Proc Natl Acad Sci USA. 1998;95:8791–8794. doi: 10.1073/pnas.95.15.8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase η error spectrum. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 70.Pavlov YI, Rogozin IB, Galkin AP, Aksenova AY, Hanaoka F, Rada C, Kunkel TA. Correlation of somatic hypermutation specificity and A–T base pair substitution errors by DNA polymerase η during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci USA. 2002;99:9954–9959. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frieder D, Larijani M, Collins C, Shulman M, Martin A. The concerted action of Msh2 and UNG stimulates somatic hypermutation at A. T base pairs. Mol Cell Biol. 2009;29:5148–5157. doi: 10.1128/MCB.00647-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Unniraman S, Schatz DG. Strand-biased spreading of mutations during somatic hypermutation. Science. 2007;317:1227–1230. doi: 10.1126/science.1145065. [DOI] [PubMed] [Google Scholar]