

Abstract
MATR3 is an RNA/DNA binding protein that interacts with TDP-43, a major disease protein linked to amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia. Using exome sequencing, we identified mutations in MATR3 in ALS kindreds. We also observed MATR3 pathology in the spinal cords of ALS cases with and without MATR3 mutations. Our data provide additional evidence supporting the role of aberrant RNA processing in motor neuron degeneration.
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by progressive paralysis and respiratory failure leading to death, typically within two to three years of symptom onset. Much attention has focused on the discovery of causal genes on the basis that understanding the pathophysiology underlying motor neuron degeneration would provide rational targets for therapeutic development. These efforts have been successful to the point that the genetic etiology of two thirds of the familial form of ALS and 11% of the more common sporadic form of the disease is now known1. Nevertheless, the discovery of additional genes would allow complete mapping of the cellular pathways underlying this fatal neurological condition.
Here, we applied exome sequencing to a Caucasian family in which several individuals had been diagnosed with ALS and dementia (Fig. 1a) with the aim of identifying the causative mutation.
Figure 1. Pedigrees of patients with MATR3 mutations.

Mutant alleles are indicated by mt, whereas wild-type alleles are indicated by wt3. Genotypes of presumed obligate carriers are in brackets. Red asterisks indicate individuals who underwent clinical examination.
We found two novel, heterozygous, missense variants that segregated with disease within this kindred, namely p.Ala436Val (chr5:126156748, C>T) in LMNB1 and p.Phe115Cys (chr5:138643448, T>G) in MATR3. Neither variant was present in population polymorphism databases (including the Exome Sequencing Project (n = 13,000 control chromosomes), the 1000 Genomes Project (n = 2,184 chromosomes) and dbSNP), or in the Human Gene Diversity Panel (HGDP, n = 2,102 chromosomes screened in our laboratory). The MATR3 variant was also not present in an additional 5,190 neurologically normal subjects genotyped in our laboratory, bringing the total number of control chromosomes that did not carry this transversion to 27,666.
A p.Ser85Cys (chr5:138643358, C>G) mutation in MATR3 was previously reported as the cause of autosomal dominant, distal, asymmetrical myopathy with vocal cord paralysis in a large multi-generational family (Fig. 1b)2,3. Neurophysiological studies and muscle biopsies of affected members were variably reported to be consistent with either a neurogenic or a myopathic pattern.
In light of our genetic findings, the senior author (BJT) and the neurologist who initially reported this family (HF) re-evaluated the p.Ser85Cys MATR3 family. Affected individuals developed progressive respiratory failure resulting in death, typically after fifteen years of illness. Pathologically brisk knee reflexes, indicative of upper motor neuron lesions, were present in four of six examined patients. One patient also had brisk upper limb reflexes, as well as tongue fasciculations and a brisk jaw jerk. All of the examined cases displayed a “split-hand” pattern of weakness suggestive of a lesion in the anterior horn of the cervical spinal cord, a sign commonly observed in ALS patients4. These clinical findings supported reclassification of this condition as slowly progressive ALS, and the presence of upper motor neuron signs in the form of brisk reflexes ruled out myopathy as the only cause of disease in this family.
To determine the frequency of MATR3 mutations as a cause of ALS, we examined exome sequence data from 108 additional familial ALS cases. We identified a p.Thr622Ala (chr5:138658372, A>G) missense change in MATR3 in a 66-year-old Sardinian diagnosed with familial ALS. This variant was present in a first-degree cousin, who had also presented with typical, rapidly progressive ALS at the age of 64 (Fig. 1c). In addition, custom re-sequencing of genes linked to neurodegeneration in 96 British ALS cases identified a p.Pro154Ser (chr5:138643564, C>T) missense variant in MATR3 in an individual diagnosed with sporadic disease (Fig. 1d and Supplementary Fig. 1). Again, neither mutation was present in population polymorphism databases or in HGDP (n = 17,286 control chromosomes). Though these data are supportive, additional studies are required to confirm the pathogenicity of these variants, especially p.Pro154Ser, which was found in a single sporadic case and consequently lacks segregation data. We did not find any additional mutations in the LMNB1 gene.
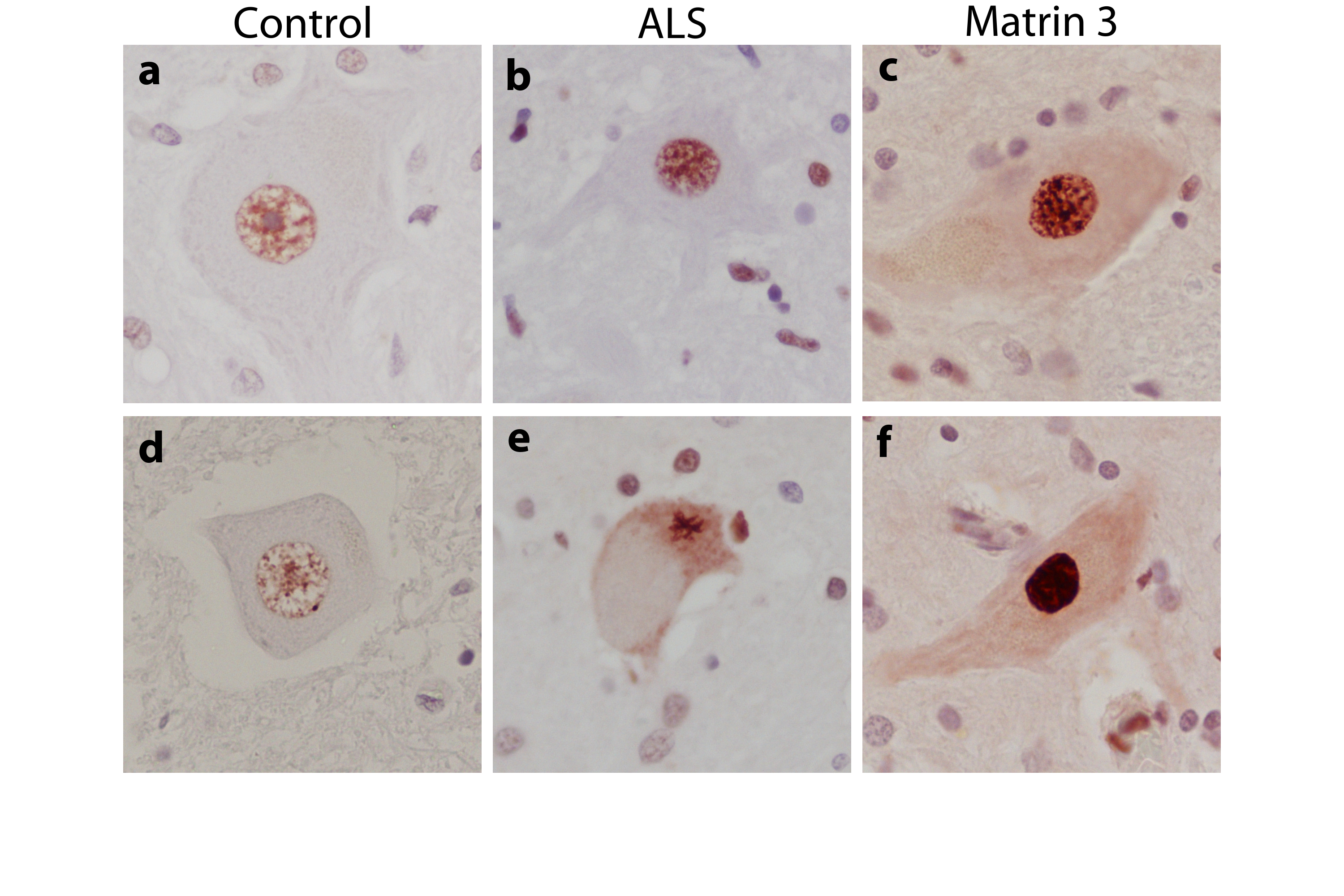
We examined subcellular distribution of MATR3 using immunohistochemistry. In control subjects, MATR3 was detected in a granular staining pattern within the nuclei of motor neurons and surrounding glial cells (Fig. 2a). In ALS patients, MATR3 was observed in the nuclei of remaining motor neurons and occasionally within the cytoplasm (Fig. 2b). In a patient harboring the p.Phe115Cys MATR3 mutation, MATR3 immunoreactivity was intense within the nucleus of all motor neurons and diffuse cytoplasmic staining was evident in many neurons (Fig. 2c). Cytoplasmic inclusions were absent in this individual. However, we detected rare MATR3-positive cytoplasmic inclusions in an ALS patient known to carry the C9ORF72 repeat expansion (Supplementary Fig. 2).
Figure 2. Lumbar spinal cord tissue immunostained for MATR3 and counterstained with hemotoxylin.

(a) Control spinal cord exhibits MATR3 nuclear immunoreactivity in some motor neurons, with weak glial cell immunostaining. (b) ALS cases exhibit strong nuclear immunoreactivity with cytoplasmic immunoreactivity present in some motor neurons either diffusely or in cytoplasmic puncta. Strong glial immunostaining is also noted in ALS patients. (c) Patient with the p.Phe115Cys MATR3 mutation exhibits strong nuclear staining, as well as cytoplasmic staining in many cells. Images are taken at 20× magnification and insets are at 40× magnification. Scale bars represent 50um.
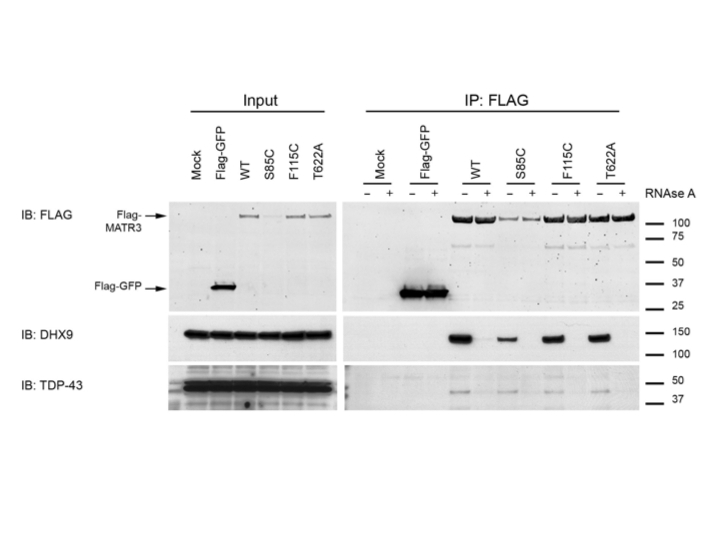
MATR3 is a 125kDa nuclear matrix protein that is known to bind DNA and RNA. Previous unbiased screens found that MATR3 is a protein interactor of TDP-43, an RNA binding protein that is known to cause ALS5,6. To confirm this interaction, we performed co-immunoprecipitation of FLAG-tagged MATR3 variants with endogenous TDP-43 in HEK293FT cells. As the genetic data were strongest for the p.Phe115Cys and the p.Ser85Cys, we selected these variants, as well as the p.Thr622Ala variant for which proof of pathogenicity was less clear, for further scrutiny. We found a reliable interaction that, interestingly, was increased by the p.Ser85Cys mutation, but not with the p.Phe115Cys or p.Thr622Ala variants (Fig. 3). We also noted that p.Ser85Cys MATR3 was expressed at lower steady state levels than other variants, suggesting a structural effect of the mutation (Fig. 3). There was no alteration in interaction between MATR3 and a second protein interactor, DHX9, demonstrating that the effect of p.Ser85Cys is specific for TDP-43 and not generalized to all interactions of MATR3. Both interactions were abolished by RNase (Supplementary Fig. 3), demonstrating that they are RNA-dependent. Co-immunoprecipitation of endogenous protein confirmed that MATR3 and TDP-43 interact at the endogenous level (Supplementary Fig. 4). Also consistent with an interaction, MATR3 and TDP-43 co-aggregated in skeletal muscle tissue of a patient carrying the p.Ser85Cys mutation (Supplementary Fig. 5).
Figure 3. Immunoprecipitation of MATR3 with TDP-43.

(a) FLAG-MATR3 was expressed in HEK293FT cells, immunoprecipitated using anti-FLAG antibody, and probed with TDP-43 and DHX9 antibodies. (b) Graphs show mean +/− SEM based on 10 replicate immunoprecipitation experiments. Differences in interaction between MATR3 and TDP-43 were tested with Wilcoxon signed rank test (**p<0.01).
Differential effects of different mutations within the same gene have been reported for other neurodegenerative diseases, such as LRRK2 (a cause of familial Parkinson’s disease)7 and FUS (a cause of familial ALS)8. Therefore, the lack of effect of p.Phe115Cys and p.Thr622Ale on MATR3/TDP-43 interaction does not necessarily preclude their pathogenicity, as it is possible that these mutations disrupt other cellular processes in a manner that would not be detected by our assays. The variants found in MATR3 were distributed across the length of protein, perhaps disrupting different nearby domains. Indeed, multiple functions have been associated with MATR3, including RNA processing6, retention of hyper-edited RNA9, gene silencing through interaction with Ago-containing complexes10, chromatin organization11, and mediating neuronal cell death in response to NMDA glutamate receptor activation12. We also note that the p.Ser85Cys variant only alters interaction with TDP-43 and not another MATR3 partner, DHX9. However, and reminiscent of mutations in VAPB13, p.Ser85Cys is notably less stable than other MATR3 variants. We infer that, of the genetic variants tested, p.Ser85Cys has the strongest effect on protein structure and this is correlated with a change in affinity for TDP-43. The structural basis of this interaction will need to be resolved in future studies.
In this regard, it is interesting to note that the p.Ser85Cys mutation in MATR3 was associated with slowly progressive form of ALS, whereas individuals carrying the p.Phe115Cys mutation typically died from respiratory failure within five years of symptom onset. Similar phenotype variability has been observed for other ALS genes. For example, the A4V mutation of SOD1 is associated with an aggressive form of the disease with an average survival of only nine months after symptom onset14. In contrast, the homozygous D90A mutation in the same gene is associated with an indolent course with patients developing respiratory failure after ten years of illness14. Furthermore, the phenotype observed in some patients carrying MATR3 mutations combined features of ALS and myopathy. This clinical pattern is markedly similar to that observed in patients with mutations in VCP, HNRNPA1 and HNRNPA2B1, and the term “multisystem proteinopathy” has been used to reflect this broad pleiotropy 15,16.
Exome sequencing data from the original USALS#3 family, as well as the 108 familial ALS cases, have been made available on dbGaP (accession number phs000101). The public release of such data allows other ALS researchers around the world to access, reanalyze and combine it with their own sequence data, thereby accelerating the pace of gene discovery.
In summary, our genetic data identified mutations of the MATR3 gene as a rare cause of familial ALS and broadens the phenotype associated with this gene beyond the previously reported distal myopathy. This provides further insight into the importance of RNA metabolism in this fatal neurodegenerative disease. Future efforts to unravel the precise mechanism by which defects in RNA processing lead to motor neurodegeneration may provide novel targets for the design of rational therapies
METHODS
Methods and any associated references are available in the online version of the paper.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported in part by the Intramural Research Programs of the NIH, National Institute on Aging (Z01-AG000949-02), and the National Institute of Neurological Disorders and Stroke (NINDS). The work was also supported by the Packard Center for ALS Research at Hopkins (B.J.T.), the ALS Association (B.J.T., A.C.), Ontario Research Fund (E.R.), the UK MND Association (J.H., R.W.O. Grant 11/6075), the Medical Research Council UK (J.H.), the Wellcome Trust/MRC Joint call in Neurodegeneration Award (J.H. grant WT089698), the MRC Neuromuscular Centre (JH), the National Institute for Health Research Biomedical Research Unit (JH), Biomedical Research Centre (AP), MRC/MNDA Lady Edith Wolfson fellowship (P.F.), AriSLA (A.C., B.J.T.), the Italian Health Ministry (Ricerca Sanitaria Finalizzata 2007, A.C.), Fondazione Vialli e Mauro ONLUS (A.C.), Federazione Italiana Giuoco Calcio (A.C., M.S., B.J.T.) and Compagnia di San Paolo (A.C., G.R.), the Adelis Foundation (V.E.D.), the European Community’s Health Seventh Framework Programme (FP7/2007-2013) under grant agreements 259867 (A.C., M.S., C.D.), EuroMOTOR, BMBF, German Network for Motoneuron Disease (M.S. - Grant TP4) and from the NIH grant NS061867 (R.B.). DNA samples for this study were obtained in part from the NINDS repository at the Coriell Cell Repositories (http://www.coriell.org/). We thank the patients and research subjects who contributed samples for this study.
Footnotes
Author contributions
J.O.J., A.B., R.C., A.E.R, H.A.P., Y.A., G. Marangi, B.J.W., S.M., M. Shaoi, A. Pittman, P.F., M.B.H., R.H.B., A. Pestronk and C.C.W performed laboratory-based experiments and data analysis, and revised the report; E.P.P., H.F., R.W.O., A.M., K.C.S., E.R., L.Z., V.E.D., G.B., G. Mora, A. Calvo, J.D.R., ITALSGEN, C.D., M. Sendtner, G.R., M. Sabatelli and A. Chiò collected clinical information and DNA samples from patients, and revised the report; J.R.G. and M.A.N. performed data analysis and revised the report; J.H., A.B.S., J.P.T., M.R.C. and R.B. supervised laboratory-based experiments and data analysis, and revised the report; B.J.T. supervised the project and wrote the manuscript.
Declaration of competing financial interests
B.J.T., A.B.S. and J.H. have a patent pending on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9ORF72. J.D.R. has patents pending on antisense therapy for C9ORF72 and associated biomarkers, and is Director of the Packard Center for ALS Research at Hopkins.
References
- 1.Renton AE, Chiò A, Traynor BJ. Nat. Neurosci. 2013 doi: 10.1038/nn.3584. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feit H, et al. Am. J. Hum. Genet. 1998;63:1732–1742. doi: 10.1086/302166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senderek J, et al. Am. J. Hum. Genet. 2009;84:511–518. doi: 10.1016/j.ajhg.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisen A, Kuwabara S. J. Neurol. Neurosurg. Psychiatry. 2012;83:399–403. doi: 10.1136/jnnp-2011-301456. [DOI] [PubMed] [Google Scholar]
- 5.Ling SC, et al. Proc. Natl. Acad. Sci. USA. 2010;107:13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salton M, et al. PLoS One. 2011;6:e23882. doi: 10.1371/journal.pone.0023882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cookson MR. Biochem. Soc. Trans. 2012;40:1070–1073. doi: 10.1042/BST20120165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Blitterswijk M, et al. PLoS One. 2013;8:e60788. doi: 10.1371/journal.pone.0060788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Carmichael GG. Cell. 2001;106:465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 10.Hock J, et al. EMBO Rep. 2007;8:1052–1060. doi: 10.1038/sj.embor.7401088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma H, Siegel AJ, Berezney R. J. Cell Biol. 1999;146:531–542. doi: 10.1083/jcb.146.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giordano G, et al. J. Neurochem. 2005;94:808–818. doi: 10.1111/j.1471-4159.2005.03235.x. [DOI] [PubMed] [Google Scholar]
- 13.Aliaga L, et al. Hum. Mol. Genet. 2013;22:4293–4305. doi: 10.1093/hmg/ddt279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiò A, et al. Amyotroph. Lateral Scler. 2009;10:310–323. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson JO, et al. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HJ, et al. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.