

Synopsis
ER aminopeptidase 1 (ERAP1), ER aminopeptidase 2 (ERAP2) and Insulin Regulated aminopeptidase (IRAP) are three homologous enzymes that play critical roles in the generation of antigenic peptides. These aminopeptidases excise amino acids from N-terminally extended precursors of antigenic peptides in order to generate the correct length epitopes for binding onto MHC class I molecules. The specificity of these peptidases can affect antigenic peptide selection, but has not yet been investigated in detail. In the present study we utilized a collection of 82 fluorogenic substrates to define a detailed selectivity profile for each of the three enzymes and to probe structural and functional features of the primary specificity (S1) pocket. Molecular modeling of the three S1 pockets reveals substrate-enzyme interactions that are critical determinants for specificity. The substrate selectivity profiles suggest that IRAP largely combines the S1 specificity of ERAP1 and ERAP2, consistent with its proposed biological function. IRAP however, does not achieve this dual specificity by simply combining structural features of ERAP1 and 2, but rather by a unique amino acid change at position 541. Our results provide insights on antigenic peptide selection and may prove valuable in designing selective inhibitors or activity markers for this class of enzymes.
Keywords: Library, antigens, enzymes, aminopeptidases, peptides, kinetics, specificity
Introduction
Antigen presentation and processing
Cytotoxic T-lymphocytes identify infected or transformed cells by recognizing small antigenic peptides bound onto cell-surface receptors of the Major Histocompatibility Class I complex (MHC class I). These antigenic peptides are derived from the proteolysis of intra- or extra-cellular proteins and constitute an indicator of the health status of the cell. Aberrant generation of antigenic peptides can lead either to immune evasion or autoimmunity. Antigenic peptides are generated intracellular by complex proteolytic pathways [1]. A key component of these pathways is the proteasome, a large intracellular multi-subunit protease that generates fragments from intracellular or endocytosed proteins. Peptides generated by the proteasome are transported into the ER by a specialized ATP-dependent peptide transporter named Transporter associated with Antigen Processing (TAP) [2]. A similar but distinct pathway, called the cross-presentation pathway, operates in specialized intracellular vesicles that contain endocytosed extracellular proteins [3]. The proteasome-generated peptides usually have the correct C-terminus as the final antigenic peptides but also have N-terminal extensions that make them too large to bind onto MHC class I molecules that have stringent length requirements with a general preference for nonamers. Although these extensions vary from one to 6 amino acids long, the most common extension is one amino acid [4]. Inside the ER, resident aminopeptidases trim these antigenic peptide precursors to generate the mature antigenic peptides that can then bind onto nascent MHC class I molecules [5, 6].
ER aminopeptidase 1 (ERAP1) and ER aminopeptidase 2 (ERAP2) are two specialized aminopeptidases that reside in the ER and have been demonstrated to trim antigenic peptide precursors to generate mature antigenic peptides [7–10]. Recently a homologous aminopeptidase named IRAP (or PLAP) has been demonstrated to perform a similar function in cross-presentation vesicles [11]. These three aminopeptidases share about 50% sequence identity. As a result of their shared homology and function, it has been proposed recently that they constitute a distinct sub-family of aminopeptidases within the metalloprotease classification M1 [12]. ERAP1 is the best characterized of the three and has been shown to affect antigen presentation in vivo, shaping the pool of antigenic peptides and influencing immunodominance [13–17]. Inhibition of ERAP1 by the broad-spectrum metalloprotease inhibitor Leucinethiol was sufficient to replicate gene knockdown experiments in cells and to induce alterations in the repertoire of the antigenic peptides [14]. ERAP1 has unusual, for an aminopeptidase, enzymatic properties, preferring to trim longer peptides down to a length of 8–10 amino acids, the appropriate length for MHC class I binding [18]. It has relatively broad substrate specificity showing preferences for side chains throughout the peptide-substrate sequence [19]. IRAP shares some of the molecular properties of ERAP1 in generating mature antigenic epitopes, although recent findings suggest that it does so in distinct patterns suggesting differences in specificity [20].
The trimming specificity of the N-terminal amino acid from antigenic peptide precursors by aminopeptidases is a strong determinant for the generation of mature antigenic peptides and the determination of the antigenic peptide repertoire. A large number of antigenic peptide precursors carry only a single amino acid extension, whose trimming will be largely affected by the N-terminal specificity of the aminopeptidase [4]. The in vitro trimming preferences of ERAP1 have been recently demonstrated to largely determine antigenic peptide presentation in cultured cells [21]. Although highly homologous, ERAP1/2 and IRAP, do not have the same specificity. Using chromogenic substrates it has been reported that the preferred residue for ERAP1 is leucine, whereas for ERAP2 is arginine [22, 23]. IRAP can cleave both substrates [24]. The exact role of these specificity differences in the biological function of these enzymes is not clear, nor have they been investigated in any detail. In the present study we set forth to characterize in detail the shape, size and composition of the S1 specificity pocket of each enzyme, in an effort to better understand the molecular determinants that contribute to antigenic peptide repertoire generation. By a combination of substrate-library screening, molecular modeling and site-directed mutagenesis we unravel key features of the S1 pocket of these enzymes that are consistent with their distinct biological functions and may be valuable for the rational design of selective inhibitors or activity markers.
Experimental
Protein expression and purification
Recombinant ERAP1 was produced by insect cell culture after infection with recombinant baculovirus carrying the ERAP1 coding sequence and isolated from the cell supernatant as previously described [19]. A recombinant and soluble form of IRAP was produced by 293F cells grown in suspension after transfection with a plasmid vector carrying the IRAP coding sequence as previously described [20].
For production of recombinant ERAP2, the sequence coding for full length human ERAP2 was inserted in the pFastBac1 vector between the BssHII and Notl restriction endonuclease sites. The final construct contained the 21-bp A-rich sequence derived from a lobster tropomyosin cDNA leader sequence adjacent to the initiation codon and a C-terminal x6 His tag for efficient expression and purification. The pFastBac1-ERAP2 vector was used to generate recombinant baculovirus according to the manufacturer’s instructions (Invitrogen, USA). The recombinant baculovirus was used to infect Hi5 cells grown in suspension in SF900II serum free medium. 3 days post-infection recombinant ERAP2 was found in the cell supernatant, harvested by centrifugation and isolated by Ni-NTA affinity chromatography as previously described for ERAP1 [19].
Mutagenesis
Site-directed mutagenesis for the construction of the E541R mutation in human IRAP, was performed using the Quickchange II XL kit according to the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA). The primers used for the mutagenesis were 5′-TCATCTGTTCAGTCTTCAGAACAAATTCGAGAAATGTTTGATTCTCTTTCC-3′ (sense) and 5′-GGAAAGAGAATCAAACATTTCTCGAATTTGTTCTGAAGACTGAACAGATGA-3′ (antisense). Successful mutagenesis was confirmed by DNA sequencing.
Library synthesis
Of the 82 fluorogenic substrates in the library, 61 have been described before [25]. All new compounds (D-amino acids-ACC, L-homoTyrosine-ACC, L-4-guanidino-phenylalanine-ACC and L-dehydrotryptophan-ACC) were synthesized using protocols described in [25]. HPLC purification and post-purification analysis of all new compounds were conducted on a Waters M600 solvent delivery module equipped with a Waters M2489 Detector system using preparative Waters Spherisorb S10ODS2 or analytical Waters Spherisorb S5ODS2 columns. Solvent composition: system A [water/0.1% TFA (trifluoroacetic acid)] and system B [acetonitrile/water 80%:20%(v/v) with 0.1% of TFA]. All substrates were at least 95% pure and were validated by ESI-MS at the mass spectrometry facility at Department of Chemistry of University of Wroclaw. The chemical structures for all 82 substrates can be found in the supplemental data section (Figure S1).
Fluorogenic assay
Trimming of the fluorogenic peptide substrates by ERAP1, ERAP2 and IRAP was followed using a TECAN infinite® M200 microplate fluorescence reader. The samples were excited at 380nm and fluorescence was recorded at 460nm. The reactions were followed for 5–10 min at 24°C. In all cases the rise in fluorescence was linear with time indicating steady-state kinetics. The slope of the time-course was used to calculate the reaction rate. L-AMC and R-AMC substrate controls were included in every plate to allow comparison between data collected from different plates.
Homology modeling
Multiple sequence alignment of human ERAP1 (isoform a, NP_057526.3), ERAP2 (NP_001123612.1) and IRAP (isoform a, NP_005566.2) was performed using ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2) with the default parameters (Figure S2). The good overall sequence identity of ERAP2 and IRAP with ERAP1 (49% and 44%, respectively), especially considering the higher degree of identity at the catalytic subsites of interest, provides a solid template for homology modeling. Based on the crystal structure of ERAP1 (PDB ID 2XDT,) ERAP2 and IRAP models were generated using Modeller 9v4 [26]. Residues Pro46 – Arg940 from 2XDT structure were used as template for the generation of ERAP2 (Arg61 – Thr960) and IRAP (Leu60 – Leu1025) models (excluded residues shown in Figure S2). The model with the lowest objective function value was selected for further optimization using AMBER 9 [27]. Hydrogen and missing heavy atoms, including Zn(II), disulfide and metal–ligand bonds were added using XLEaP. The AMBER-based parm99SB force field was applied to all protein atoms, while parameters for the Zn(II) coordination sphere were taken from [28]. Subsequently, the position of hydrogen atoms and the metal ion site was optimized with energy minimization in vacuum using a distance-dependent dielectric and a 20-Å cutoff for non-bonded interactions. The quality of ERAP2 and IRAP models was assessed using the Structural Analysis and Verification Server (http://nihserver.mbi.ucla.edu/SAVS), which exhibited a reasonable degree of quality by virtue of their sequence alignment.
Substrate docking
The substrate library was generated starting from the SMILES representation of each compound, then OMEGA [29] was used to calculate the initial 3D coordinates and QUACPAC (Openeye Inc.) to apply AM1-BCC atomic charges [30]. Docking of the substrates was performed using AutoDock 4.2 [31] with default parameters except for the number of docking rounds, which was increased to 100. Non-polar hydrogen atoms were merged and Kollman charges were applied to the protein atoms using AutoDockTools 1.4.5. Ligands were treated as fully flexible excluding amide bonds and guanidinium groups. The docked model chosen for analysis was amongst the highest binding energy conformations with proper orientation for substrate binding (i.e. the scissile bond C=O···Zn(II) distance < 2.5 Å). No further optimization of the predicted enzyme–substrate interactions was attempted. Electrostatic potential surfaces were generated using APBS and PME electrostatics packages [32]. VMD 1.8.6 and Pymol was used for visual inspection and rendering of the figures [33, 34].
Results
Screening strategy
We used a collection of 82 fluorogenic substrates to generate a selectivity profile for ERAP1, ERAP2 and IRAP. The experimental conditions for each screen were designed so that every substrate used would be assayed under sub-KM concentrations so that the rate of cleavage would linearly correlate to the kcat/KM value of each substrate and enzyme as previously described [25]. To ensure this, we first generated Michaelis-Menten plots for the best-known substrate for each enzyme. We used L-ACC for ERAP1 and IRAP and R-ACC for ERAP2. The KM for ERAP1 is larger than 1mM (estimated to be 1150±305μM) and the KM values for ERAP2 and IRAP are 90±3μM and 85±30μM respectively (data not shown). As a result we chose to do all screening assays at substrate concentrations below 10μM. After the preliminary screening, substrates for which no signal was measured were re-screened at 100μM concentration in an effort to quantify trimming rates for poor substrates. Using this strategy we estimate a minimum trimming rate difference between good and non-processed substrates of about 200-fold.
Selectivity profiles for ERAP1, ERAP2 and IRAP
The relative trimming rates for each of the 82 substrates with each of the three enzymes are shown in Figure 1. Rates are plotted as a fraction of the best substrate for each enzyme. The three enzymes share key preferences but also display marked differences. ERAP1 efficiently trimmed about 16 of the 82 substrates showing significant preference for hydrophobic and aromatic amino acids as well as for long aliphatic side chains. Accordingly, the best performing substrate was homo-tyrosine (hTyr). ERAP2 displayed a significantly different profile than ERAP1, with strong preferences for positively charged amino acids. Overall ERAP2 efficiently trimmed about 10 of the 82 substrates, with several key differences from ERAP1. The best two substrates, arginine and homo-arginine (hArg), had a guanidinium group, revealing a strong preference for extended chains with positively charged ends. Shorter hydrophobic side chains were processed to a smaller degree. Similarly to ERAP1, ERAP2 appeared to prefer extended carbon side chains but in contrast to ERAP1, ERAP2 displayed a very strong preference for a positive change at the end of those chains. The selectivity profile of IRAP was the most permissive of the three enzymes. IRAP was able to trim at least 25 of the 82 substrates in the library. Interestingly, in almost all cases IRAP was able to process the substrates that were trimmed by either ERAP1 or ERAP2. This finding suggests that IRAP has the combined specificity of ERAP1 and ERAP2. However some exceptions were evident since a few substrates were not processed by ERAP1/2 but trimmed by IRAP (cyclopentyl-Glycine, Abu, Bpa) and vice versa (3-NO2-tyrosine). These observations suggest that although IRAP can generally process the sum of substrates of ERAP1 and 2, it may use distinct molecular interactions to achieve this specificity.
Figure 1.

Selectivity profiles of ERAP1, ERAP2 and IRAP. Trimming rates were calculated for each substrate and then normalized for the best substrate for each enzyme. Error bars correspond to the standard deviation for 3–6 measurements. Substrates for which no bar is drawn failed to be hydrolyzed by the enzyme even when measured at 100μM substrate concentration. Panel A, Natural amino acid side chains in L- or D- configuration. Panel B, non-natural amino acid side chains. Panel C, reaction kinetics and specific rates for the best substrate for each enzyme (hTyr-ACC for ERAP1, Arg-ACC for ERAP2 and hArg-ACC for IRAP). Error bars indicate the standard deviation of 3 measurements.
Non-natural side chains probe the characteristics of the S1-pockets
We employed amino acids with unnatural side chains to gain insight on structural and functional features of the S1 pockets (Figure 1, panel B). Interestingly, we identified a much larger number of unnatural side chains as good substrates for all three enzymes. Accordingly, the best substrate for ERAP1 is hTyr, hArg is the second best substrate for ERAP2, and both of those substrates are optimal for IRAP. This observation suggests that the S1 pockets are not strictly optimized for natural amino acids but can easily accommodate more complex structures.
D-amino acid based substrates were poorly processed by all three enzymes, suggesting that the L configuration is a prerequisite for binding and/or catalysis. Under typical experimental conditions only ERAP2 was found to be able to trim D-arginine, albeit ~50-fold slower than L-arginine. Michaelis-Menten analysis of these two substrates suggested that the lower trimming rate was due to both changes on the KM (90±3 μM for L-Arg and 1053±304 μM for D-Arg) and kcat parameters (0.177±0.003 sec−1 for L-Arg and 0.038±0.018 sec−1 for D-Arg) (Figure S3). These findings suggest that the L-configuration is crucial for both binding and catalysis for this family of enzymes.
Similarly to human aminopeptidase N (CD13), the enzymes analyzed here had a very strong preference for amino acids with an amino group in the alpha position and were completely inactive toward substrates with a hydroxyl group in the alpha position such as Apns or amino acids with no amino group present in this position such as 6-Ahx or β-Ala [25]. This finding is consistent with the important role of the peptide N-terminus in substrate recognition [35].
Some of the substrates in the library have side chains of substantial size and would be expected to fit only in large S1 pockets. The S1 pocket of IRAP in particular appears to be able to accommodate the most bulky and hydrophobic substrates in the library (Bpa and Igl), while ERAP1 and ERAP2 processed them poorly. This result indicates that the S1 pocket of IRAP may be the largest of the three. Finally, lack of processing of conformationally restricted substrates like 1-Nal, 2-Nal or Bip by ERAP1, ERAP2 and IRAP suggests that although the pocket is large, it is well-defined and rigid so as to exclude side chain structures that are not flexible enough to adopt appropriate configurations. Overall, these observations suggest that it may be possible to optimize S1 recognition for each enzyme by incorporating bulky non-natural side chains in the substrate.
ERAP1&2 mixture behaves similarly to IRAP
Saveanu et al. [10] have previously suggested that ERAP1 and 2 operate in a concerted manner in the ER. In contrast, IRAP has been suggested to operate on a separate pathway of cross-presentation, distal from compartmentalized ERAP1 and 2 [11]. To investigate possible effects in S1 specificity when ERAP1 and 2 are mixed, we screened the L-substrate library in the presence of 2:1 molar ratio of ERAP1:ERAP2, according to the molar ratio of the two enzymes reported previously [10]. The resulting specificity profile was found to closely follow the sum of the individual selectivity profiles of each enzyme, revealing no strong synergism or allosteric effects under these experimental conditions (Figure 2). Again, the selectivity profile of the mixture of ERAP1 and 2 resembled closely the profile of IRAP, although some differences were obvious. We conclude that IRAP largely combines the specificity of ERAP1 and 2, but retains unique profile features that suggest differences in the molecular determinants of its S1 pocket.
Figure 2.

ERAP1 and ERAP2 were mixed at 2:1 molar concentration and the selectivity profile of the mix was compared to that of IRAP using the L-substrates.
The three enzymes present similarities primarily for the substrates they do not process efficiently
Despite their differences, the three enzymes presented some striking similarities in the substrates they were unable to process efficiently. None of the enzymes were able to process a proline side chain, presumably due to the absence of a free amino-terminal group to be recognized by the aminopeptidase GAMEN motif [12]. Very short hydrophilic side chains were not preferred presumably due to the hydrophobic nature of the S1 pockets. Beta-branched side chains such as valine, isoleucine and threonine were also poorly tolerated. Finally, negatively charged side chains were very poor substrates for all three enzymes. These observations suggest that the S1 pockets of the three enzymes share common structural features that exclude some categories of side chains from being effectively recognized.
Molecular modeling suggests critical features of the S1 pocket that control specificity
To understand the molecular basis for the specificity effects unraveled by the library screen, we utilized a recently released crystallographic structure of ERAP1 (PDB: 2XDT) to dock the best substrates and analyze the atomic level interactions in the S1 pocket. Since no crystallographic structures are available yet for ERAP2 and IRAP, we used the structure of ERAP1 to construct homology models of the other two aminopeptidases. The high homology shared between the three aminopeptidases (50% identity) and the relatively few amino acid differences in the vicinity of the S1 pocket, result in homology models of higher accuracy compared to our previous report [19].
Docking of model substrates, in combination with the positioning of key catalytic features of the enzyme (such as the residues of the HEXXH motif that binds the catalytic Zn(II) atom and the GAMEN motif that is responsible for the recognition of the N-terminus of the peptidic substrate) help define the spatial orientation of the S1 pockets (Figure 3). For all three enzymes the general shape and size of the pockets are similar, although IRAP has a larger exit channel towards the solvent. The S1 pocket is relatively large (being able to easily accommodate even the largest of the docked substrates) and elongated, originating from the catalytic site Zn(II) atom and forming a shallow channel towards the solvent. The channel is capped by residues from the C-terminal domain of the protein, forming a closed structure with minimal solvent access, suggesting that a conformational change may be necessary to allow substrate binding and product release. The overall electrostatic potential of the pocket is strongly negative, an observation that may explain the poor processing of negatively charged side chains. This potential is largely derived by the presence of two conserved glutamate residues that provide the terminal-NH2 docking site and by additional negatively charged side chains in the S1 pocket. ERAP2 has the most negatively charged residues of the three enzymes (E177, D198, D888) whereas IRAP has two (E426 and E541) and ERAP1 only one (E865). The only basic residue within the three S1 pockets belongs to ERAP1 (R430), leading to altered electrostatic potential near the top of the S1 pocket (Figure 3, blue colored region), an observation that supports the poorer ability of ERAP1 to trim positively charged P1 substrates.
Figure 3.

Surface representation of the S1 pocket for ERAP1, ERAP2 and IRAP colored by electrostatic potential. The best substrates for each enzyme are shown as stick models in the predicted conformations.
Key residues that control specificity
By analyzing the interactions between docked substrates and protein side chains we were able to define the residues that line the S1 pocket for the three enzymes. These residues are listed in Table I and indicated in the alignment in Figure S2. Half of these residues are conserved between the three enzymes (positions 184, 314, 319, 433, 864 and 868 in ERAP1 numbering) and presumably contribute to the common general characteristics of the pocket. Five of these conserved residues are non-polar (Pro 184, Phe 314, Met 319, Phe 433 and Phe 864 in ERAP1) and may support the preference of all three enzymes for non-polar P1 substrates. Interestingly, Phe433/450/544 comes in close proximity to the beta-carbon of the substrate backbone leading to unfavorable steric hindrance with any substrates with beta-branched side chains such as valine, threonine or isoleucine (Figure 4A). Instead, Phe433/450/544 residue is predicted to provide favorable aromatic–π interactions with the guanidinium groups of Arg and hArg P1-bearing substrates as well as favorable CH–π interactions with linear aliphatic chains (Figure 4C). Accordingly, the longer hTyr, hLeu and Nle are even or better substrates in comparison to Tyr, Leu and Ile. On the opposite side of the pocket, Met319/336/430 is predicted to make contacts with the Cβ atom of the substrates leading to steric hindrance for substrates with D-configuration and decreased binding affinity (Figure 4B). D-substrates are predicted to bind with a relatively different configuration of their scissile peptide bond compared to L-substrates (compare Figure 4A with 4B), consistent with the reduced catalytic efficiency we observed (lower kcat).
Table I.
Residues of ERAP1, ERAP2 and IRAP that are predicted to provide key interactions with substrates and help form the S1 specificity pocket. Conserved residues between the three enzymes are grayed.
| ERAP1 | ERAP2 | IRAP |
|---|---|---|
| His160 | Glu177 | Tyr272 |
| Gln181 | Asp198 | Gln293 |
| Pro184 | Pro201 | Pro296 |
| Phe314 | Phe331 | Phe425 |
| Gln315 | Ala332 | Glu426 |
| Ser316 | Pro333 | Ala427 |
| Met319 | Met336 | Met430 |
| Arg430 | Gln447 | Glu541 |
| Phe433 | Phe450 | Phe544 |
| Phe864 | Phe887 | Phe956 |
| Glu865 | Asp888 | Pro957 |
| Ser868 | Ser891 | Ser960 |
Figure 4.
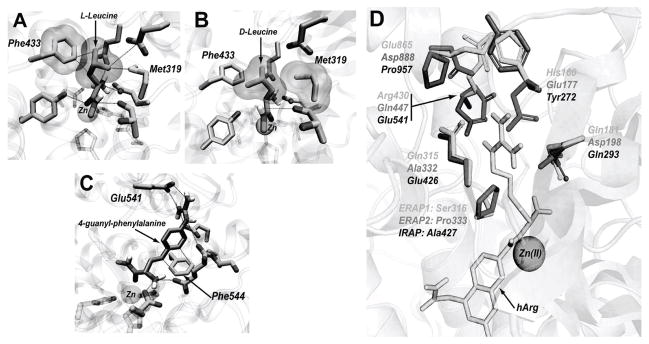
Key residues that define the S1 pocket. Panel A, Phe433 in ERAP1 is stacked closely with a leucine side chain of the substrate. Panel B, Met319 makes unfavorable steric interactions with the Cβ of D-leucine, leading to an altered binding conformation of the scissile peptide bond. Panel C, Simulated interactions between a 4-guanyl-phenylalanine side chain with Phe544 and Glu541 in IRAP. Panel D, The six non-conserved amino acids that define the S1 pocket of each enzyme; the predicted conformation of homo-arginine is depicted in yellow.
Six of the 12 residues that define the S1 pockets vary between the enzymes and contribute to differences between the three S1 pockets that underlie changes in specificity (Figure 4D). Of these residues, two were found to be of particular importance for interactions that appear critical for the differences in specificity between the three enzymes. The polar residue at position 181/198/293 (ERAP1/ERAP2/IRAP numbering) is a glutamine in ERAP1 and IRAP but is an aspartate in ERAP2. Its positioning adjacent to the GAMEN motif makes it appropriate for interactions with positively charged side chains and has been shown to be important for the selectivity of ERAP2 by site-directed mutagenesis [36]. Interestingly, although IRAP is, similarly to ERAP2, able to process substrates with positively changed side chains, it does not contain an aspartate at position 181/198/293 but resembles ERAP1 by having a glutamine. This observation raises the question on how IRAP is able to recognize positive charges in the S1 pocket. Docking of positively charged substrates in IRAP suggests that at least a non conserved, negatively charged amino acid in IRAP, Glu541, may be a candidate residue for providing salt-bridge interactions to stabilize positively charged substrates in IRAP’s S1 cavity (Figure 4C). ERAP1 has an arginine residue at the equivalent position (R430) and ERAP2 has a glutamine (Q447).
Mutagenesis confirms that Glu 541 in IRAP is important for positively charged substrate recognition
To test the prediction that Glu541 in IRAP is important for the enzyme’s preference for positively charged side chains, we used site-directed mutagenesis to change the Glu541 in IRAP to an arginine, the equivalent residue in ERAP1. The IRAP E541R variant was expressed in recombinant form and purified to homogeneity (Figure S4). We probed the substrate selectivity of the mutant IRAP using the L-substrate library and compared it to the wild-type protein (Figure 5). As predicted, the mutant IRAP had an altered selectivity profile and was much less potent in trimming positively charged residues (noted by arrows in figure 5). In this context the selectivity profile of E541R IRAP was similar to the profile of ERAP1. Michaelis-Menten analysis using the fluorigenic substrate R-AMC (L-arginine-7-amido-4-methyl coumarin) indicated that the difference in specificity for IRAP E541R was primarily due to loss in affinity (KM) (Figure 6). We concluded that Glu541 in IRAP is largely responsible for allowing IRAP to mimic the substrate preferences of ERAP2, without losing the preferences of ERAP1.
Figure 5.

Selectivity profile of IRAP E541R mutation compared to wild-type protein. Data have been normalized for Leucine. Arrows highlight the most significant changes brought about by the mutation.
Figure 6.
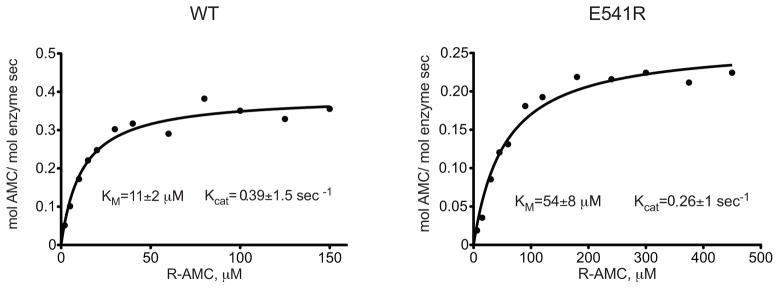
Michaelis-Menten kinetics for hydrolysis of the substrate R-AMC by E541R IRAP as well as the WT enzyme. The enzymatic parameters KM and Kcat are depicted for each enzyme.
Discussion
The importance of antigenic peptide precursor trimming by aminopeptidases has emerged the last few years as both a necessary step for antigenic peptide generation but also as a novel paradigm of regulation of the adaptive immune response [17, 37]. However the discovery that three distinct aminopeptidases participate in antigen processing has raised important questions regarding the regulation of antigenic peptide generation that are far from answered. We hypothesized that the necessity for multiple aminopeptidases performing what is seemingly an identical role, lies in key differences between the specificity of these enzymes. Towards testing this hypothesis, we systematically characterized the S1 specificity of ERAP1, ERAP2 and IRAP. We discovered that these three enzymes share many features between their S1 binding pockets but at the same time, have key differences that may help explain their distinct biological functions.
Our analysis suggests that to a large extent, IRAP combines the N-terminal specificity of ERAP1 and ERAP2. This is consistent with the recently proposed function of IRAP in a distinct processing compartment inside the cell [11]. ERAP1 and ERAP2 have been proposed to function in tandem inside the ER, with ERAP2 behaving as an accessory protease, assisting ERAP1 in trimming sequences that would otherwise be poorly processed. IRAP however appears to act alone inside cross-presentation compartments and as a result it needs to be able to process both ERAP2 and ERAP1 substrates. ERAP2 gains the ability to process positively charged amino acids by a key change at position 181/198/293 [36]. However, this particular change reduces its affinity for hydrophobic chains, specializing it for positively charged amino acids. IRAP however, cannot afford this option; it needs to be able to trim both ERAP1 and ERAP2 substrates. IRAP achieves this not by altering position 181/198/293 but by altering position 430/447/541 instead, allowing it to combine both specificities. This elegant solution to this apparent specificity problem is indicative on how key amino acid changes inside a specificity pocket can guide selectivity in this family of aminopeptidases. Generally it has proven difficult to alter the primary specificity of proteases by single amino acid replacements because the S1 pocket is influenced by a large number of interactions and may even be intrinsically disordered, as seen for example in the trypsin/chymotrypsin family [38]. However, certain scaffolds tolerate specificity switching by single residue substitutions, for example the chymase/granzyme family [39].
Regardless of the differences between their specificity, the three enzymes share some striking similarities for the side chains they fail to recognize. Neither enzyme can process negatively charged side chains presumably due to the strong negative electrostatic potential of the general region of the S1 pocket. Furthermore, all three enzymes fail to process substrates with side chains that carry beta-carbon or oxygen branching (such as valine, isoleucine or threonine) due to the limited space in the S1 pocket and the stringent stereochemical requirements for the recognition of the N-terminus of the substrate by the conserved GAMEN motif. Phenylalanine 544 plays a key role in this phenomenon and is critical for enzyme activity [40]. These findings however, raise a crucial question. If all the aminopeptidases that perform antigenic peptide processing before MHC class I loading cannot process efficiently common aminoacids such as valine, isoleucine, threonine, glutamate or aspartate, how do such antigenic peptide precursors get processed? Inspection of the SYFPEITHI antigen database reveals that many antigenic peptides may be derived from precursors that would require the excision of such amino acids (http://www.svfpeithi.de/). One notable example is the antigenic peptide from human ovalbumin, SIINFEKL, that can be processed by ERAP1 efficiently although a common precursor sequence contains a glutamate (ESIINFEKL) [9]. A possible answer to this question lies on the specificity of ERAP1 for amino acids distal to the N-terminus of the substrate [19] a property that may be shared by ERAP2 and IRAP. An alternative explanation would include the participation of a currently unidentified accessory aminopeptidase.
The important role of these aminopeptidases in antigen presentation in combination with their distinct selectivity profiles suggests that selective inhibition of a single one, may lead to subtle alterations of the antigenic peptide repertoire that can be used to modulate the immune response. Recently, polymorphic variation in ERAP1/2 has been linked with predisposition to autoimmunity, viral infection and cancer suggesting that manipulation of the activity of these aminopeptidases may have an important therapeutic potential [41]. Indeed, use of the non-selective general metalloproteinase inhibitor leucine-thiol in cultured cells, has been recently demonstrated to alter antigen presentation [17]. Furthermore we recently demonstrated that polymorphic variation in ERAP1 can affect antigen processing in vitro [42]. Therefore, the development of highly potent and selective inhibitors for this class of enzymes may constitute a useful approach towards the modulation of the adaptive immune response. In addition, the development of highly selective substrates can be useful for investigating established pathogenic links and developing diagnostic and prognostic markers. Our results suggest that although these three enzymes are highly homologous, they still carry key differences in their S1 pockets that can be exploited for the design of selective inhibitors or specific activity markers that can be used to follow antigen processing in vivo or ex vivo.
In summary, we have performed a detailed analysis of the S1 specificity of ERAP1, ERAP2 and IRAP, three enzymes that process antigenic peptide precursors and are crucial to the functioning of the adaptive immune system. By combining small-substrate library screening, molecular modeling and mutagenesis we revealed key differences and similarities between the three enzymes that underlie their biological function. Furthermore, our analysis can facilitate efforts towards the rational design of small molecular weight selective inhibitors and activity markers that can be used to manipulate and characterize the adaptive immune response.
Acknowledgments
We thank Dr. Florentia Fostira for help with confirming mutagenesis reactions by DNA sequencing and Paulina Piatek for help with synthesis of fluorogenic substrates.
Funding
This work was funded in part by a NCSR Demokritos internal grant (E.S.) and by the Foundation for Polish Science and the State for Scientific Research Grant N N401 042838 (M.D.). I.E. and D.G. acknowledge financial support by the National Centre for Scientific Research “Demokritos” graduate fellowship program.
Abbreviations used in this paper
- ACC
7-amino-4-carbamoylmethylcoumarin
- AMC
7-amino-4-methylcoumarin
- ERAP
ER aminopeptidase
- IRAP
Insulin-regulated aminopeptidase
Footnotes
Author contribution
EZ performed and analyzed the enzymatic experiments. AP and DV performed and helped interpret the molecular modeling. DG, IE, AH and LS designed the ERAP2 and IRAP expression systems, purified the recombinant enzymes and helped interpret the specificity data. AG, MP and MD designed and synthesized the substrate library and helped interpret the specificity data. MD, AP and GS helped design the study, interpret the data and write the paper. ES conceived and supervised the project, helped analyze the data and wrote the paper. All authors read and approved the final manuscript.
References
- 1.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC classI-presented peptides. Annu Rev Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 2.Lauvau G, Kakimi K, Niedermann G, Ostankovitch M, Yotnda P, Firat H, Chisari FV, van Endert PM. Human transporters associated with antigen processing (TAPs) select epitope precursor peptides for processing in the endoplasmic reticulum and presentation to T cells. J Exp Med. 1999;190:1227–1240. doi: 10.1084/jem.190.9.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 4.Cascio P, Hilton C, Kisselev AF, Rock KL, Goldberg AL. 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. Embo J. 2001;20:2357–2366. doi: 10.1093/emboj/20.10.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roelse J, Gromme M, Momburg F, Hammerling G, Neefjes J. Trimming of TAP-translocated peptides in the endoplasmic reticulum and in the cytosol during recycling. J Exp Med. 1994;180:1591–1597. doi: 10.1084/jem.180.5.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serwold T, Gaw S, Shastri N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol. 2001;2:644–651. doi: 10.1038/89800. [DOI] [PubMed] [Google Scholar]
- 7.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 8.Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M, Goldberg AL. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–1176. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 9.York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 10.Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, Schomburg L, Fruci D, Niedermann G, van Endert PM. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- 11.Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR, Niedermann G, van Endert P. IRAP Identifies an Endosomal Compartment Required for MHC Class I Cross-Presentation. Science. 2009;325:213–217. doi: 10.1126/science.1172845. [DOI] [PubMed] [Google Scholar]
- 12.Tsujimoto M, Hattori A. The oxytocinase subfamily of M1 aminopeptidases. Biochim Biophys Acta. 2005;1751:9–18. doi: 10.1016/j.bbapap.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 13.York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci US A. 2006;103:9202–9207. doi: 10.1073/pnas.0603095103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol. 2006;7:103–112. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- 15.Yan J, Parekh VV, Mendez-Fernandez Y, Olivares-Villagomez D, Dragovic S, Hill T, Roopenian DC, Joyce S, Van Kaer L. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med. 2006;203:647–659. doi: 10.1084/jem.20052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Firat E, Saveanu L, Aichele P, Staeheli P, Huai J, Gaedicke S, Nil A, Besin G, Kanzler B, van Endert P, Niedermann G. The role of endoplasmic reticulum-associated aminopeptidase 1 in immunity to infection and in cross-presentation. J Immunol. 2007;178:2241–2248. doi: 10.4049/jimmunol.178.4.2241. [DOI] [PubMed] [Google Scholar]
- 17.Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8:101–108. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 18.Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci USA. 2005;102:17107–17112. doi: 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evnouchidou I, Momburg F, Papakyriakou A, Chroni A, Leondiadis L, Chang SC, Goldberg AL, Stratikos E. The internal sequence of the peptide-substrate determines its N-terminus trimming by ERAP1. PLoS ONE. 2008;3:e3658. doi: 10.1371/journal.pone.0003658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Georgiadou D, Hearn A, Evnouchidou I, Chroni A, Leondiadis L, York IA, Rock KL, Stratikos E. Placental leucine aminopeptidase efficiently generates mature antigenic peptides in vitro but in patterns distinct from endoplasmic reticulum aminopeptidase 1. J Immunol. 2010;185:1584–1592. doi: 10.4049/jimmunol.0902502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hearn A, York IA, Rock KL. The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J Immunol. 2009;183:5526–5536. doi: 10.4049/jimmunol.0803663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hattori A, Kitatani K, Matsumoto H, Miyazawa S, Rogi T, Tsuruoka N, Mizutani S, Natori Y, Tsujimoto M. Characterization of recombinant human adipocyte-derived leucine aminopeptidase expressed in Chinese hamster ovary cells. J Biochem (Tokyo) 2000;128:755–762. doi: 10.1093/oxfordjournals.jbchem.a022812. [DOI] [PubMed] [Google Scholar]
- 23.Tanioka T, Hattori A, Masuda S, Nomura Y, Nakayama H, Mizutani S, Tsujimoto M. Human leukocyte-derived arginine aminopeptidase. The third member of the oxytocinase subfamily of aminopeptidases. J Biol Chem. 2003;278:32275–32283. doi: 10.1074/jbc.M305076200. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto H, Rogi T, Yamashiro K, Kodama S, Tsuruoka N, Hattori A, Takio K, Mizutani S, Tsujimoto M. Characterization of a recombinant soluble form of human placental leucine aminopeptidase/oxytocinase expressed in Chinese hamster ovary cells. Eur J Biochem. 2000;267:46–52. doi: 10.1046/j.1432-1327.2000.00949.x. [DOI] [PubMed] [Google Scholar]
- 25.Drag M, Bogyo M, Ellman JA, Salvesen GS. Aminopeptidase fingerprints, an integrated approach for identification of good substrates and optimal inhibitors. J Biol Chem. 2010;285:3310–3318. doi: 10.1074/jbc.M109.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 27.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papakyriakou A, Spyroulias GA, Sturrock ED, Manessi-Zoupa E, Cordopatis P. Simulated interactions between angiotensin-converting enzyme and substrate gonadotropin-releasing hormone: novel insights into domain selectivity. Biochemistry. 2007;46:8753–8765. doi: 10.1021/bi700253q. [DOI] [PubMed] [Google Scholar]
- 29.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 31.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLano WL. The Pymol Molecular Graphics System. DeLano Scientific; San Carlos, CA: http://www.pymol.org. [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Tsujimoto M, Goto Y, Maruyama M, Hattori A. Biochemical and enzymatic properties of the M1 family of aminopeptidases involved in the regulation of blood pressure. Heart Fail Rev. 2008;13:285–291. doi: 10.1007/s10741-007-9064-8. [DOI] [PubMed] [Google Scholar]
- 36.Goto Y, Tanji H, Hattori A, Tsujimoto M. Glutamine-181 is crucial in the enzymatic activity and substrate specificity of human endoplasmic-reticulum aminopeptidase-1. Biochem J. 2008;416:109–116. doi: 10.1042/BJ20080965. [DOI] [PubMed] [Google Scholar]
- 37.Georgiadou D, Stratikos E. Cellular mechanisms that edit the immunopeptidome. Current Proteomics. 2009;6:13–24. [Google Scholar]
- 38.Hung SH, Hedstrom L. Converting trypsin to elastase: substitution of the S1 site and adjacent loops reconstitutes esterase specificity but not amidase activity. Protein Eng. 1998;11:669–673. doi: 10.1093/protein/11.8.669. [DOI] [PubMed] [Google Scholar]
- 39.Caputo A, James MN, Powers JC, Hudig D, Bleackley RC. Conversion of the substrate specificity of mouse proteinase granzyme B. Nat Struct Biol. 1994;1:364–367. doi: 10.1038/nsb0694-364. [DOI] [PubMed] [Google Scholar]
- 40.Albiston AL, Pham V, Ye S, Ng L, Lew RA, Thompson PE, Holien JK, Morton CJ, Parker MW, Chai SY. Phenylalanine-544 plays a key role in substrate and inhibitor binding by providing a hydrophobic packing point at the active site of insulin-regulated aminopeptidase. Mol Pharmacol. 2010;78:600–607. doi: 10.1124/mol.110.065458. [DOI] [PubMed] [Google Scholar]
- 41.Haroon N, Inman RD. Endoplasmic reticulum aminopeptidases: Biology and pathogenic potential. Nat Rev Rheumatol. 2010;6:461–467. doi: 10.1038/nrrheum.2010.85. [DOI] [PubMed] [Google Scholar]
- 42.Evnouchidou I, Kamal RP, Seregin SS, Goto Y, Tsujimoto M, Hattori A, Voulgari PV, Drosos AA, Amalfitano A, York IA, Stratikos E. Cutting Edge: Coding Single Nucleotide Polymorphisms of Endoplasmic Reticulum Aminopeptidase 1 Can Affect Antigenic Peptide Generation in vitro by Influencing Basic Enzymatic Properties of the Enzyme. J Immunol. 2011 Jan;17:2011. doi: 10.4049/jimmunol.1003337. [DOI] [PMC free article] [PubMed] [Google Scholar]