

Abstract
Pancreatic β cell failure leads to diabetes development. During disease progression, β cells adapt their secretory capacity to compensate the elevated glycaemia and the peripheral insulin resistance. This compensatory mechanism involves a fine-tuned regulation to modulate the endoplasmic reticulum (ER) capacity and quality control to prevent unfolded proinsulin accumulation, a major protein synthetized within the β cell. These signalling pathways are collectively termed unfolded protein response (UPR). The UPR machinery is required to preserve ER homeostasis and β cell integrity. Moreover, UPR actors play a key role by regulating ER folding capacity, increasing the degradation of misfolded proteins, and limiting the mRNA translation rate. Recent genetic and biochemical studies on mouse models and human UPR sensor mutations demonstrate a clear requirement of the UPR machinery to prevent β cell failure and increase β cell mass and adaptation throughout the progression of diabetes. In this review we will highlight the specific role of UPR actors in β cell compensation and failure during diabetes.
1. Introduction
Type 2 diabetes (T2D) mellitus is a chronic metabolic disease with “epidemic” proportions. Its global prevalence was estimated to be 6.4% worldwide (285 million adults in 2010) and is predicted to rise to approximately 7.7% (439 million) by 2030 [1]. T2D is a multifactorial disorder resulting from an interaction between genetic and environmental conditions (sedentary lifestyle and Western diet) and characterized by a peripheral insulin resistance, hyperglycaemia, and pancreatic β cell dysfunctions. Two defects have been reported during diabetes development, a gradual deterioration of β cell functions and a reduction in pancreatic β cells mass. β cell failure is not limited to T2D but is rather a common feature of all forms of diabetes, including the autoimmune type 1 diabetes (T1D), autosomal dominant onset diabetes of young (MODY), Wolfram syndrome, and Wolcott-Rallison syndrome (WRS).
In the early stage of diabetes development, the response of pancreatic islets challenged by nutrients and/or insulin resistance is a hypersecretion of insulin to maintain normoglycaemia. To this end, an adaptative and compensatory response of β cells is required. The process of β cell compensation is a combination of β cell mass expansion and an increase of acute glucose-stimulated insulin secretion. Postmortem analyses of pancreas of nondiabetic obese patients show an increase of β cell volume, implying postnatal plasticity of β cell mass. Moreover the β cell compensation process is associated with an improved capacity of the secretory machinery to support increased insulin production. Subsequently, the production of large amounts of insulin by compensating islet β cells places a continuous demand on the ER for proper protein synthesis, folding, trafficking, and secretion. When the folding capacity of the ER is exceeded, misfolded or unfolded proteins accumulate in the ER lumen, resulting in ER stress.
The cytoprotective response to ER stress is the unfolded protein response (UPR). Paradoxically, UPR signalling activation leads to opposite cell fates, that is, adaptation/survival versus death. Increasing evidence links the endoplasmic reticulum (ER) stress to β cell deterioration and apoptosis [2, 3]. Recent experiments performed in db/db mice and ob/ob mice models at different times of disease progression revealed that the maintenance (or suppression) of adaptive UPR is associated with β cell compensation (or failure) in obese mice [4]. Moreover, Engin et al. recently showed a progressive loss of UPR mediator expression before the onset of diabetes in NOD mice [5]. The administration of the chemical chaperone tauroursodeoxycholic acid to rescue the deleterious ER stress response improved pathophysiological signs of diabetes with a recovery of β cell survival and adaptation to stress [5]. In addition, the authors showed a decline of the UPR mediator in both experimental models and T2D human islets, suggesting that decreased expression of β cell UPR actors can play a central role in β cell compensation and subsequently T2D occurrence [6].
2. The UPR Pathway
Three canonical ER resident molecules mediate UPR response, namely, protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6), which are maintained inactive by their association with the immunoglobulin heavy chain-binding protein (BiP, GRP78) in normal conditions (Figure 1(a)). The accumulation of unfolded proteins in the ER leads to the release of PERK, IRE, and ATF6 and their subsequent activation [7, 8]. The downstream signalling effectors from these pathways converge to the nucleus and activate UPR target genes, finally reducing the ER input (Figure 1(a)). Their action is bipartite, with an acute programme that attenuates the ER workload and a latent transcriptional one that builds ER capacity.
Figure 1.
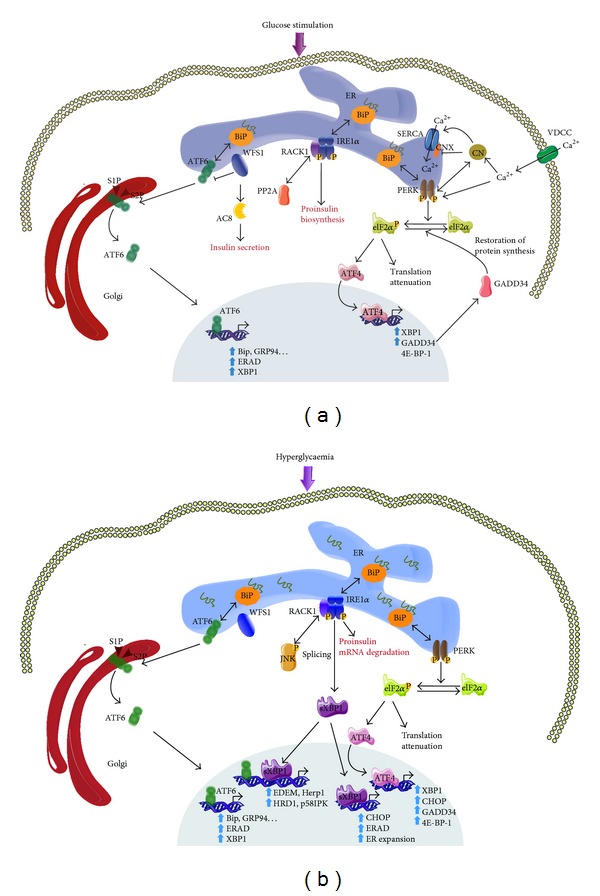
Physiological and physiopathological UPR activated pathways in β cells. (a) Under physiological conditions, increased proinsulin synthesis in response to postprandial glucose activates UPR to reduce ER stress and to promote β cell adaptation. The UPR triggers transcription of folding protein (BiP, GRP94,…), protein quality control (ERAD), UPR retrocontrol protein (GADD34), and attenuates protein translation (elF2α). Additionally, the UPR regulates calcium homeostasis via PERK, promotes proinsulin synthesis via IRE1α, and increases insulin secretion via a WFS1-AC8 pathway. (b) Under physiopathological conditions, the UPR is hyperactivated leading to IRE1α hyperphosphorylation, which in turn induces proinsulin mRNA degradation, JNK pathway activation, and XBP1 mRNA splicing. XBP1s alone or in synergy with ATF6 lead to expression of ER chaperon (Herp1, EDEM, HRD1, p58IPK, and ERAD) and subsequent ER expansion. Both ATF4 and sXBP1 increase CHOP mRNA expression. Under these conditions the UPR feedback is deregulated.
PERK is a type 1 ER transmembrane kinase with a stress sensing luminal N-terminal domain. During ER stress PERK phosphorylates the α-subunit of eIF2-α on serine 51 leading to a delivery inhibition of the initiator methionyl-tRNAi to the ribosome and ultimately resulting in global protein translation attenuation [9] (Figure 1(a)). This phosphorylation event directly contributes to the reduction of ER stress and protects cells from ER stress-mediated apoptosis [10]. Intriguingly, the mRNA transcription of UPR target genes is selectively activated by eIF2α phosphorylation, as these polycistronic mRNAs have inhibitory upstream open reading frames (uPRFs) and are thus preferentially translated by the ribosome. These include the bZiP transcription factor 4 (ATF4) that acts as a regulator of UPR target genes such as C/EBP-homologous protein (CHOP) and growth arrest and DNA damage inducible gene 34 (GADD34), as well as genes involved in the redox balance and amino acid synthesis [11]. GADD34 interacts with the catalytic subunit of protein phosphatase (PP1c) and controls the level of eIF2-α phosphorylation by a negative feedback loop [12], allowing the restoration of an UPR basal state once ER stress is resolved.
IRE1 is a central regulator of UPR. Like PERK, IRE1 is also a type 1 transmembrane kinase with an N-terminal luminal domain that senses ER stress signalling. Two homologues of IRE1 have been described, IRE1α and IRE1β. IRE1α is expressed ubiquitously, showing high expression levels in the pancreas and placenta [13], whereas IRE1β is only expressed in the intestinal epithelium and lung [14]. IRE1 possesses kinase as well as endoribonuclease activities. Once the ER stress is triggered, IRE1 activates its RNase domain through its dimerization and transautophosphorylation and causes an unconventional splicing by the removal of 26-nucleotide intron from the X-box binding protein 1 (XBP1) mRNA (Figure 1(a)). The subsequent spliced XBP1 (XBP1s) mRNA encodes a leucine zipper transcription factor with a high transcriptional activity that upregulates genes encoding ER protein chaperones, ER associated protein degradation (ERAD), and lipid biosynthetic enzymes [15, 16]. IRE1 has also a nonspecific RNase activity that degrades mRNAs localized near the ER membrane, thereby reducing protein import into the ER lumen [17]. High levels of ER stress also activate the kinase activity of IRE1 and initiate a signalling cascade of apoptosis signal-regulating kinase 1 (ASK1)/cJun amino terminal kinase (JNK), which can participate in the apoptotic cell fate [18].
ATF6 is an ER located type 2 transmembrane protein with a basic leucine zipper DNA binding domain (Figure 1(a)). Two ubiquitously expressed isoforms of ATF6 have been described, ATF6α and ATF6β [19]. Under ER stress conditions, ATF6α translocates from the ER to the Golgi apparatus, where it is cleaved by Site-1 protease and Site-2 protease (S1P/S2P). The newly generated cytosolic fragment migrates to the nucleus and activates UPR gene transcription [20, 21]. The exclusive or the combined action of cleaved ATF6α and XBP1s is able to activate all three ER stress response elements: ERSE, UPRE, and ERSE2 [22]. ATF6β was first described as a repressor of ATF6α [23]. However, mouse embryonic fibroblasts generated from ATF6β null mice did not show altered UPR gene induction, suggesting a minor role for ATF6β in ER stress response [24]. In contrast, ATF6α null mice have a significant alteration in their UPR gene expression profile, suggesting a central role for ATF6α in ER protein quality control and protection against ER stress [24]. As the double ATF6α/β knockdown is lethal, the authors suggested that ATF6α and ATF6β provide a complementary function during early development [24]. Moreover ATF6 activity is regulated by the Wolfram syndrome 1 (WFS1) protein, which targets ATF6 to the E3 ubiquitin ligase HRD1, consequently resulting in its ubiquitination and proteasomal degradation [25]. A number of other ER stress transducers that share a high sequence homology with ATF6 have been identified, such as Luman, OASIS, BBF2H7, CREBH, and CREB4 (reviewed in [26]). However, despite their structural similarities, each ATF6 homolog seems to have specific functions in UPR regulated processes in specific organs and tissues [26].
2.1. β Cells Compensation and UPR Actors
β cell is a highly specialized secretory cell which responds to elevated postprandial glycaemia by increasing mRNA proinsulin translation and insulin secretion [27]. The periodic waves of proinsulin mRNA translation generate biosynthetic loads that induce UPR. To manage this burden imposed by proinsulin synthesis, β cells increase their ER size, as exemplified during diabetes development. In the prediabetic state, β cell must adapt its ER machinery to the new hyperglycaemic environment, promoting β cell compensation. The use of genetically modified mouse models and genetic studies from human diabetic patients demonstrated that UPR actors support this adaptation as well as β cell compensatory mechanism [4, 28].
2.2. Insulin Mutants and β Cells Compensation
Misfolding of proinsulin is associated with ER stress and severe dysfunctions leading to a massive destruction of β cells. In vivo evidences were observed in both Akita and Munich mice carrying cysteine residues mutations that interfere with disulphide bond formation [29, 30]. Interestingly, inactivation of the UPR induced proapoptotic Chop gene delayed the onset of diabetes in heterozygous Akita mice, suggesting a key role for CHOP in ER stress-mediated β cell apoptosis [30]. Diabetes of youth (MIDY) is a syndrome caused by a heterozygous mutation of the coding sequence of proinsulin leading to an autosomal-dominant and insulin-deficient diabetes [31]. This mutation has been shown to be the second most common cause of permanent neonatal diabetes related to ER stress [32, 33]. In line with these observations, inducible expression of the human analogue proinsulin C96Y mutation of Akita mice in rat insulinoma-1 (INS-1) caused ER stress and cell apoptosis. However, upregulation of UPR and ERAD seems to have a protective effect [34]. In vivo expression of the same proinsulin mutant driven by the weak Ins1 promoter induced both ER stress and pancreatic compensation [35]. Altogether these data demonstrate a clear link between misfolding of proinsulin and ER stress induction.
2.3. PERK-elF2α-ATF4 Pathway in β Cells Compensation
Loss of function mutation in the EIF2AK3 gene-encoding PERK was associated with WRS, which has been confirmed by the functional characterisation of Perk knockout mice [36]. Embryonic development of these mice is normal but they exhibit a postnatal growth retardation, skeletal dysplasia, and progressive loss of β cells, associated with defects in ER secretory machinery and proinsulin folding [37, 38]. However, generation of β cell specific Perk knockout mice revealed that β cell death was not increased, but rather β cell proliferation and differentiation were repressed during the embryonic and postnatal state [39]. Other studies on these mice models demonstrated an impaired ER to Golgi anterograde trafficking, retrotranslocation out of the ER, and proteasomal degradation, showing requirement of PERK for ER and Golgi integrity and processing of ATF6 [40]. In contrast to the first rapport, specific β cell inducible Perk deletion in mice showed a rapid progression of insulin dependent diabetes regardless of mice age [41]. The authors showed, on the one hand, that this phenotype was due to an increased β cell proliferation after the induction of PERK deletion due to the increased activation cyclin D-dependent kinase activity. On the other hand, they showed a significant increase in β cell death, associated with an activation of other UPR actors and a disturbance in calcium homeostasis [41]. Moreover, a recent work demonstrated that PERK, in concert with calcineurin, regulates ER calcium reuptake through calnexin interaction and a negative regulation of the sarcoplasmic endoplasmic reticulum calcium ATPase pump (SERCA) [42] (Figure 1(a)). PERK thus appears to sense glucose by direct sensing of ER calcium levels, raising the possibility that the primary function of PERK in β cell is to modulate proinsulin quality control and trafficking.
Within the β cell, phosphorylation of elF2α is mostly PERK dependent in ER stress conditions (Figure 1(b)). Mice harbouring a homozygote knock-in mutation at the PERK-mediated phosphorylation site (Ser51Ala) of elF2α display a severe β cell deficiency detectable in late stage embryos and die within 18 h after birth as a consequence of hyperglycaemia associated with defective neoglucogenesis [43]. Heterozygote mice development is normal; however, when challenged with a high fat diet, they develop severe obesity, glucose intolerance, and impaired insulin release. In this genetic context reduced insulin content, fewer granules, ER distension, and a prolonged association of proinsulin with BiP have been observed. Thus, elF2α phosphorylation is required for UPR to prevent β cell failure when insulin demand is increased. These findings demonstrate a central role of elF2α phosphorylation in β cell adaptation during compensation [44]. The generation of conditional homozygote ser51Ala elF2α mutation in β cell confirmed this observation. These mice exhibit a high rate of β cell apoptosis, likely caused by hypoinsulinemia, severe glucose intolerance, and hyperglycaemia. Furthermore, the authors showed ER distension and mitochondrial damage associated with a lower basal expression of the majority of UPR genes and β cell antioxidant responsive genes. Altogether these data indicate that the correct UPR and antioxidant response controlled by PERK-elF2α signalling are required for β cell adaptation and survival [45].
Phosphorylation of elF2α leads to attenuate global translation of most mRNA although translation of ATF4 is selectively stimulated in this context [11] (Figure 1(b)). The role of ATF4 in insulin secretion and β cell survival is controversial. Pioneer studies showed that Atf4 knockout mice on a 129SV genetic background are lean, hypoglycaemic, and resistant to diet-induced obesity, probably as a result of increased energy expenditure. However, no effect was observed on plasma insulin level when mice were fed with normal diet, whereas insulin levels were shown to be lower in Atf4 null mice compared to wild-type mice when fed with a high fat diet [46, 47]. Paradoxically, other studies using Atf4 KO mice on C57BL6/J genetic background demonstrated that these mice are hypoglycaemic and hyperinsulinemic with an increased β cell mass and function. The phenotype of these mice could be secondary to an overexpression of osteocalcin in osteoblasts, a bone derived secreted molecule promoting insulin secretion and insulin sensitivity [48]. The genetic background may explain the differences observed in these Atf4 null mice models. The role of ATF4 in insulin synthesis and β cell adaptation to ER stress remains unclear, and further studies using a β cell specific Atf4 KO mouse model may be useful to answer this question.
ATF4 activation by elF2α also leads to the transcription of elF4-E binding protein (4E-BP-1), a well-documented gene involved in β cells adaptation to stress [49]. In fact, translation attenuation by elF2α phosphorylation is transient, subsequently leading to the feedback dephosphorylation by GADD34, whereas 4E-BP1 suppresses prolonged translation by the inhibition of cap-dependent translation [49, 50]. β cell specific 4e-bp1 KO mice are normal without any metabolic disorder when fed normal diet but are insulin resistant and show β cell defects under high fat diet due to induced ER stress [49, 51, 52]. Moreover, the inactivation of 4e-bp1 gene in Min6 cell line results in sensitization to ER stress and increased β cell loss and hyperglycaemia in diabetic mouse models [49]. These findings suggest a central role of 4E-BP1 in β cell adaptation to ER stress. In contrast, other groups indicated that suppression of 4E-BP1 expression is involved in beneficial effects of high-density lipoproteins on β cells survival [53], suggesting that the role of ATF4 in β cell compensation might depend on several cellular interactions.
2.4. IRE1α-XBP1 Pathways in β Cells Compensation
IRE1α is the major isoform expressed in the pancreas and plays a central role in β cell adaptation to ER stress (Figure 1(b)). IRE1α is required for embryonic development as demonstrated by the embryonic lethality of global IRE1α KO mice. IRE1α plays a crucial role in insulin biosynthesis. The generation of IRE1α conditional KO mice revealed that IRE1α deletion caused mild hyperinsulinemia and hyperglycaemia and a lower body mass under normal diet [54]. Physiological activation of UPR by glucose results in IRE1α phosphorylation, without increasing XBP1 mRNA splicing. Moreover, knockdown of IRE1α in INS-1 insulinoma cell line resulted in decreased proinsulin biosynthesis or insulin content without impacting global protein synthesis or insulin secretion, suggesting a beneficial effect of IRE1α activation by transit exposure to glucose in β cells [55]. However, chronic exposure to high glucose leads to hyperphosphorylation of IRE1α, which in turn results in selective degradation of proinsulin mRNA [56]. This may be part of the β cell protective mechanism from apoptosis under chronic hyperglycaemia induced ER stress. This adaptative mechanism combined to UPR activation may explain the reduced insulin secretion in type 2 diabetic patients in the absence of β cell death. IRE1α dephosphorylation is mediated by proteins phosphatase A2 (PP2A) through ternary complex containing the scaffold protein RACK1 (receptor for activated C kinase 1). Under glucose stimulation or ER stress, RACK1 mediates IRE1α, RACK1, and PP2A complex formation and promotes IRE1α dephosphorylation by PP2A, thereby inhibiting IRE1α activation and attenuating IRE1α-dependent increase in insulin production. Moreover, IRE1α activation is increased and RACK1 abundance is decreased in db/db mice [57]. The endoplasmic activity of IRE1α is also involved in the activation of a key metabolic enzyme, AMP-activated kinase (AMPK), in response to nitric oxide (NO) and ER stress in β cells [58]. AMPK is a holoenzyme activated by changes in AMP/ATP ratio, shifting from glucose to the use of lipids as an energy source in order to respond to cellular demand [59]. Activated AMPK by GTPase dynamin related protein 1 (DRP1) phosphorylation prevents ER and mitochondrial alteration in stressed β cells [60]. In addition IRE1α modulates nuclear factor κ light chain enhancer (NF-κB) target gene expression and IL-1β activation under mild ER stress, which could contribute to chemokine-induced β cell death [61].
Upon UPR mediating IRE1α activation, XBP1 splicing is the major event. Several reports indicated that XPB1s target genes and its downstream effect are cell specific and might be dependent on the activating pathways. Like IRE1α, XBP1 deficient mice died between 10.5 and 14.5 day after birth because of cardiac myocyte defects [62]. Heterozygous Xbp1 mice exhibited significant increase in body mass associated with a progressive hyperinsulinemia and glucose intolerance when fed with a high fat diet [63]. These mice showed increased ER stress and decreased insulin receptor expression in the liver. The β cell-specific deletion of XBP1 in mice resulted in a modest hyperglycaemia and glucose intolerance caused by decreased insulin secretion [64]. The loss of XBP1 markedly decreased the number of insulin granules and impaired proinsulin processing. Further analysis revealed that XBP1 deficiency not only participated in the ER stress in β cells but also caused constitutive hyperactivation of its upstream activator, IRE1α, which could degrade a subset of mRNAs encoding proinsulin-processing enzymes [64]. In summary, β cell defects in XBP1 mutant mice result from a combined effect of XBP1 suppression on canonical UPR and its negative feedback activation of IRE1α. Altogether these findings suggest a dual and opposite role for IRE1α in β cells. A precise regulated feedback circuit involving IRE1α and its product XBP1s is required to achieve optimal insulin secretion and glucose control. In contrast, sustain production of XBP1s leads to inhibition of PDX1 and MAFA expressions, promoting β cells dysfunction and apoptosis [54].
2.5. ATF6 Pathways in β Cell Compensation
Both ATF6 isoforms are required for positive regulation of UPR. However, the transcriptional activity of ATF6β is lower than that of ATF6α. The double knockdown of the two isoforms caused an embryonic lethality demonstrating overlapping functions of ATF6α and ATF6β, which are essential for embryonic development [24]. ATF6α KO mice demonstrated a severe hypoglycaemia suggesting that suppression of ATF6α increased insulin sensitivity [65]. Treatment of these mice with a pharmacological ER stress inducer leads to liver dysfunction and steatosis [66]. Furthermore, when fed with a high fat diet, ATF6α null mice developed insulin resistance associated with impaired insulin secretion and lower insulin content, reinforcing the idea of a key role of ATF6α in β cells adaptation and insulin resistance [65]. Recently, a basal expression of active ATF6α was demonstrated to be essential for β cell survival even under unstressed conditions. Interestingly, specific functions of ATF6α have been revealed depending on its interaction with XBP1. When ATF6α is acting alone, it induces the expression of a cluster of genes involved in protein folding such as BiP and GRP94. When it heterodimerizes with XBP-1 they are modulating the expression of specific class of target genes, such as genes involved in protein degradation (EDEM, Herpude1, HRD1, and p58IPK) [24]. In contrast, a deleterious effect of active ATF6α overexpression on β cell function and expression of insulin, PDX1, and MAFA in INS-1 cells was shown [67]. Interestingly, some reports demonstrated that some ATF6 variants are associated with type 2 diabetes and new onset diabetes after transplantation (NODAT), suggesting potential links between ATF6α and human diabetes pathophysiology [68, 69]. It is important to note that, from the myriad of ATF6 homolog described until now, only old astrocyte specifically induced substance (OASIS) was identified in β cell [70]. However, microarray analysis of INS-1 β cell line overexpressing the active form of OASIS showed its implication in extracellular matrix production and protein transport but not in the classical ER stress response [70].
A great interest has been focused on the ATF6α negative regulator WFS1 because of its association with the Wolfram syndrome, a rare genetic disorder [71, 72]. Loss of function mutation in the wfs1 gene encoding wolframin protein caused neurodegenerative disorders characterised by juvenile onset diabetes mellitus, optic atrophy, and hearing impairment [73, 74]. WFS1 KO mice exhibit an activated ER stress especially in β cells, leading to β cell loss through impaired cell cycle progression and increased apoptosis [75]. Conditional WFS1 knockdown in β cell induced diabetes as a result of enhanced ER stress and apoptosis [76]. Moreover WFS1 is essential for glucose and glucagon-like peptide 1 (GLP1) stimulated AMP production and regulation of insulin biosynthesis and secretion [77]. Under glucose stimulation, WFS1 translocates from the ER to plasma membrane, where it stimulates cyclic adenosine monophosphate [73] synthesis through an interaction with adenylyl cyclase 8 (AC8), which subsequently promoted insulin secretion [77] (Figure 1(a)). A recent report using induced pluripotent stem (iPS) cells to create β cells from individuals with Wolfram syndrome confirmed these observations. In this study, WFS1 deficient β cells showed increased levels of UPR genes and decreased insulin content, leading to β cell dysfunctions as previously described in mouse models [78].
2.6. The UPR/ER Stress Induction of β Cell Apoptosis
As discussed above, the UPR regulates both survival and death effectors. It is now clear that the three unfolded protein sensors—IRE1a, PERK, and ATF6—influence the life-death decision. The inability of UPR outputs to restore homeostasis may generate continuous signalling from these sensors, tipping the balance in favour of apoptosis. The ER might actually serve as a site where apoptotic signals are generated and integrated to elicit the death response. ER stress leads to apoptosis by activating both mitochondrial dependent and independent pathways. Several stimuli have been linked to ER stress-induced apoptosis including hyperglycaemia, exposure to long-chain free fatty acids (e.g., palmitate) [79–81], hyperinsulinemia occurring in the prediabetic stage [82], glucose deprivation [83], islet amyloid polypeptide (IAPP) expression [84], and exposure to inflammatory cytokine [85]. Players involved in the cell death response include PERK/elF2α-dependent transcriptional induction of proapoptotic transcription factor CHOP which represses Bcl-2 [86], IRE1-mediated activation of ASK1/JNK [18], and cleavage and activation of procaspase 12 (caspase 4 in humans) [87, 88].
CHOP has retained a special attention as a central mediator of apoptosis. Its expression is low under physiological condition but is strongly induced upon ER stress [89]. The induction of CHOP is regulated by ATF4 [11, 90] and ATF6 [91–93] and its role in ER stress-induced apoptosis was demonstrated both in vitro and in vivo [94]. Mice lacking CHOP are protected from renal toxicity of the ER stressor tunicamycin, an inhibitor of glycosylation [94]. CHOP deletion promotes β cells survival in both genetic and diet-induced insulin resistant mice models [30, 92]. Pancreatic β cells are also sensitive to oxidative stress, but β cells from CHOP knockout mice are protected and maintain insulin secretion under oxidative stress [92, 95]. Moreover, islets from these mice showed resistance to NO, a chemical agent implicated in β cells disruption in type 1 diabetes [96]. In contrast, CHOP deficiency in a genetic background of nonobese diabetic mice (NOD-Chop−/−) did not affect the development of insulitis, diabetes, and β cells apoptosis [97]. Interestingly, CHOP knockout mice on a C57BL/6 background showed a different phenotype, with abdominal obesity and hepatic steatosis, while preserving normal glucose tolerance and insulin sensitivity [98].
Under ER stress CHOP positively regulates the expression of genes involved in apoptosis including GADD34 [50, 99], the ER oxidoreductin 1 α (ERO1α) [100], death receptor 5 (DR5) [101], and the pseudokinase tribbles related 3 (TRB3) [102]. As shown for CHOP deletion, genetic inactivation of these genes protected against β cell ER stress-induced apoptosis [100, 103–105]. Additionally, CHOP represses the expression of the antiapoptotic gene Bcl2 and enhances oxidant injury [106]. Finally, deletion of CHOP was reported to prevent the cytokine-mediated cleavage of caspase 9 and caspase 3 and subsequent β cells apoptosis by reducing cytokine-induced NF-κB activity and the expression of key NF-κB target genes involved in apoptosis and inflammation [107].
ER stress-mediated apoptosis can also be signalled through IRE1α dependent activation of JNK pathway [18]. IRE1α interacts with TBF receptor associated factor 2 (TRAF2) and ASK1 mediating JNK phosphorylation [18, 108]. The analysis of ASK1 deficient mice showed that ASK1 loss of function attenuated insulin resistance, cardiac inflammation and fibrosis, vascular endothelial dysfunction, and remodelling induced by diet-induced obesity [109]. Moreover, deletion of ASK1 in homozygous Akita mice protected β cells from ER induced apoptosis and delayed the onset of diabetes [110]. The IRE1α/TRAF2 complexes also contributes to ER stress-induced apoptosis by promoting the clustering of procaspase-12 and its activation by cleavage in response to ER stress [111]. In addition, the IRE1α/TRAF2 complex interacts with IKK, an inhibitor of NF-κB, mediating its activation and promoting cell apoptosis in response to ER stress [112, 113]. Finally, members of Bcl2 family including BAX, BAK, BIM, and PUMA have been reported to directly interact with IRE1α demonstrating a physical link between members of the core apoptotic pathway and the UPR [114, 115]. In contrast, IRE1α forms a stable protein complex with Bax inhibitor-1 (BI-1) protein, suppressing cell death [116]. The IRE1α/BI-1 association decreased the ribonuclease activity of IRE1α and seemed to be required for early adaptive responses against ER stress-induced apoptosis [116]. The control of IRE1α activity appears to be central in the mechanism protecting β cells from ER stress-induced apoptosis. Further studies are needed to understand the various aspect of IRE1α regulation and the contribution of the others actors of UPR in the ER stress-induced apoptosis.
2.7. The UPR in Human Diabetes
Clear evidence of the existence of ER stress in human β cells has been reported in the last decade [2, 3, 6, 117]. First analysis of islets from human T2D patients showed an ER extension but modest signs of ER stress marker in human pancreatic samples and isolated islets. However, glucose stimulation induced increased UPR in T2D islets cells [3]. Some markers of ER stress are increased in T1D human islets with partial ER stress [117, 118]. A recent report shows an alteration in the expression of specific branches of UPR mediators in T2D β cells [6]. These findings support the hypothesis of a decline in β cell adaptation/compensation during the progression of diabetes in human.
3. Conclusion
The β cell has a marked capacity to adapt to environment changes by increasing its mass and function. Diabetic signs occur when this adaptative mechanism fails to compensate for the increasing insulin demand. Activation of UPR actors is triggered in the early stage of the compensatory mechanism and may play a central role in β cell adaptation and subsequent functions. Further studies are required to understand the physiological significance and the direct implication of ER stress and UPR in the early stage of diabetes physiopathology. Moreover, the relationships among UPR actors, their activation, and β cell fate (adaptation/survival versus β cell dysfunction/apoptosis) remain to be fully clarified. Theoretically, the size of β cell mass is controlled by a balance between proliferation and apoptosis. Either increase of β cell apoptosis or decrease in β cell adaptation and compensation could, therefore, reduce the β cell mass in T2D patients. Studies carried out during diabetes development are required to better understand the mechanism of compensatory capacity and subsequent β cell loss in humans. This is of particular interest, since it could have beneficial impact for the treatment of metabolic diseases such as diabetes.
It is important to note that most UPR molecules have an adaptive function in β cells. Their role in the switch from survival to apoptosis is clearly demonstrated in vitro and in animal models but it remains unclear whether the same mechanisms occur in human β cell. Isolating and culturing primary β cells may be very stressful and do not perfectly reflect the in vivo context. Therefore the use of alternative method such as immunohistochemistry is powerful to determine the role of each branch of UPR in diabetes.
Acknowledgments
The authors apologize to all their colleagues whose work could not be cited due to space limitations. We thank members of the Annicotte's lab and Froguel's lab for discussions. The author's research is supported by the French Agence Nationale de la Recherche (ANR-10-LABX-46), Association pour la Recherche sur le Diabète, Lille2 University, Conseil Régional Nord-Pas de Calais and Lille Métropole Communauté Urbaine, Société Francophone du Diabète-Laboratoires SERVIER.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Research and Clinical Practice. 2010;87(1):4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Fonseca SG, Burcin M, Gromada J, Urano F. Endoplasmic reticulum stress in β-cells and development of diabetes. Current Opinion in Pharmacology. 2009;9(6):763–770. doi: 10.1016/j.coph.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marchetti P, Bugliani M, Lupi R, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50(12):2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- 4.Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in beta-cell gene expression and progression to diabetes. Diabetes. 2013;62(5):1557–1568. doi: 10.2337/db12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engin F, Yermalovich A, Ngyuen T, et al. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Science Translational Medicine. 2013;5(211) doi: 10.1126/scitranslmed.3006534.211ra156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engin F, Nguyen T, Yermalovich A, Hotamisligil GS. Aberrant islet unfolded protein response in type 2 diabetes. Scientific Reports. 2014;4, article 4054 doi: 10.1038/srep04054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 8.Nadanaka S, Yoshida H, Mori K. Reduction of disulfide bridges in the lumenal domain of ATF6 in response to glucose starvation. Cell Structure and Function. 2006;31(2):127–134. doi: 10.1247/csf.06024. [DOI] [PubMed] [Google Scholar]
- 9.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 10.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 11.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y-Y, Cevallos RC, Jan E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2α phosphorylation. The Journal of Biological Chemistry. 2009;284(11):6661–6673. doi: 10.1074/jbc.M806735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & Development. 1998;12(12):1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertolotti A, Wang X, Novoa I, et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. The Journal of Clinical Investigation. 2001;107(5):585–593. doi: 10.1172/JCI11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee A-H, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 17.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 18.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 19.Haze K, Okada T, Yoshida H, et al. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochemical Journal. 2001;355, part 1:19–28. doi: 10.1042/0264-6021:3550019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong M, Luo S, Baumeister P, et al. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. The Journal of Biological Chemistry. 2004;279(12):11354–11363. doi: 10.1074/jbc.M309804200. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. Journal of Biochemistry. 2004;136(3):343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 23.Thuerauf DJ, Morrison L, Glembotski CC. Opposing roles for ATF6α and ATF6β in endoplasmic reticulum stress response gene induction. The Journal of Biological Chemistry. 2004;279(20):21078–21084. doi: 10.1074/jbc.M400713200. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Developmental Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 25.Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. The Journal of Clinical Investigation. 2010;120(3):744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asada R, Kanemoto S, Kondo S, Saito A, Imaizumi K. The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. Journal of Biochemistry. 2011;149(5):507–518. doi: 10.1093/jb/mvr041. [DOI] [PubMed] [Google Scholar]
- 27.Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature. 1980;283(5742):100–102. doi: 10.1038/283100a0. [DOI] [PubMed] [Google Scholar]
- 28.Omikorede O, Qi C, Gorman T, et al. ER stress in rodent islets of Langerhans is concomitant with obesity and beta-cell compensation but not with beta-cell dysfunction and diabetes. Nutrition & Diabetes. 2013;3, article e93 doi: 10.1038/nutd.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herbach N, Rathkolb B, Kemter E, et al. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56(5):1268–1276. doi: 10.2337/db06-0658. [DOI] [PubMed] [Google Scholar]
- 30.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. The Journal of Clinical Investigation. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Støy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(38):15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edghill EL, Flanagan SE, Patch A-M, et al. Insulin mutation screening in 1,044 patients with diabetes mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57(4):1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M, Hodish I, Haataja L, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends in Endocrinology and Metabolism. 2010;21(11):652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartley T, Siva M, Lai E, Teodoro T, Zhang L, Volchuk A. Endoplasmic reticulum stress response in an INS-1 pancreatic β-cell line with inducible expression of a folding-deficient proinsulin. BMC Cell Biology. 2010;11, article 59 doi: 10.1186/1471-2121-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodish I, Absood A, Liu L, et al. In vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes. 2011;60(8):2092–2101. doi: 10.2337/db10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delépine M, Nicolino M, Barrett T, Golamaully M, Mark Lathrop G, Julier C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nature Genetics. 2000;25(4):406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 37.Zhang P, McGrath B, Li S, et al. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Molecular and Cellular Biology. 2002;22(11):3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in Perk−/− mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metabolism. 2006;4(6):491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao Y, Sartori DJ, Li C, et al. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Molecular and Cellular Biology. 2012;32(24):5129–5139. doi: 10.1128/MCB.01009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang R, McGrath BC, Kopp RF, et al. Insulin secretion and Ca2+ dynamics in beta-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. The Journal of Biological Chemistry. 2013;288(47):33824–33836. doi: 10.1074/jbc.M113.503664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular Cell. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 44.Scheuner D, Vander Mierde D, Song B, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nature Medicine. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 45.Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metabolism. 2009;10(1):13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seo J, Fortuno ES, III, Suh JM, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes. 2009;58(11):2565–2573. doi: 10.2337/db09-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Research. 2010;20(2):174–184. doi: 10.1038/cr.2010.4. [DOI] [PubMed] [Google Scholar]
- 48.Yoshizawa T, Hinoi E, Dae YJ, et al. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. The Journal of Clinical Investigation. 2009;119(9):2807–2817. doi: 10.1172/JCI39366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamaguchi S, Ishihara H, Yamada T, et al. ATF4-mediated induction of 4E-BP1 contributes to pancreatic β cell survival under endoplasmic reticulum stress. Cell Metabolism. 2008;7(3):269–276. doi: 10.1016/j.cmet.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 50.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α . Journal of Cell Biology. 2001;153(5):1011–1021. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.le Bacquer O, Petroulakis E, Paglialunga S, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. The Journal of Clinical Investigation. 2007;117(2):387–396. doi: 10.1172/JCI29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsukiyama-Kohara K, Poulin F, Kohara M, et al. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nature Medicine. 2001;7(10):1128–1132. doi: 10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- 53.Pétremand J, Bulat N, Butty A-C, et al. Involvement of 4E-BP1 in the protection induced by HDLs on pancreatic β-cells. Molecular Endocrinology. 2009;23(10):1572–1586. doi: 10.1210/me.2008-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwawaki T, Akai R, Kohno K. IRE1α disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0013052.e13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Lipson KL, Ghosh R, Urano F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE. 2008;3(2) doi: 10.1371/journal.pone.0001648.e1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qiu Y, Mao T, Zhang Y, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1α activation in pancreatic β cells. Science Signaling. 2010;3(106, article ra7) doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meares GP, Hughes KJ, Naatz A, et al. IRE1-dependent activation of AMPK in response to nitric oxide. Molecular and Cellular Biology. 2011;31(21):4286–4297. doi: 10.1128/MCB.05668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. The Journal of Clinical Investigation. 2013;123(7):2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wikstrom JD, Israeli T, Bachar-Wikstrom E, et al. AMPK regulates ER morphology and function in stressed pancreatic beta-cells via phosphorylation of DRP1. Molecular Endocrinology. 2013;27(10):1706–1723. doi: 10.1210/me.2013-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miani M, Colli ML, Ladriere L, Cnop M, Eizirik DL. Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1beta in pancreatic rat beta-cells via the IRE1alpha/XBP1s pathway. Endocrinology. 2012;153(7):3017–3028. doi: 10.1210/en.2011-2090. [DOI] [PubMed] [Google Scholar]
- 62.Masaki T, Yoshida M, Noguchi S. Targeted disruption of CRE-Binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochemical and Biophysical Research Communications. 1999;261(2):350–356. doi: 10.1006/bbrc.1999.0972. [DOI] [PubMed] [Google Scholar]
- 63.Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 64.Lee A-H, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(21):8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Usui M, Yamaguchi S, Tanji Y, et al. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism. 2012;61(8):1118–1128. doi: 10.1016/j.metabol.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 66.Yamamoto K, Takahara K, Oyadomari S, et al. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Molecular Biology of the Cell. 2010;21(17):2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seo H-Y, Yong DK, Lee K-M, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases insulin gene expression via up-regulation of orphan nuclear receptor small heterodimer partner. Endocrinology. 2008;149(8):3832–3841. doi: 10.1210/en.2008-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thameem F, Farook VS, Bogardus C, Prochazka M. Association of amino acid variants in the activating transcription factor 6 gene (ATF6) on 1q21-q23 with type 2 diabetes in Pima Indians. Diabetes. 2006;55(3):839–842. doi: 10.2337/diabetes.55.03.06.db05-1002. [DOI] [PubMed] [Google Scholar]
- 69.Fougeray S, Loriot M-A, Nicaud V, Legendre C, Thervet E, Pallet N. Increased body mass index after kidney transplantation in activating transcription factor 6 single polymorphism gene carriers. Transplantation Proceedings. 2011;43(9):3418–3422. doi: 10.1016/j.transproceed.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 70.Vellanki RN, Zhang L, Guney MA, Rocheleau JV, Gannon M, Volchuk A. OASIS/CREB3L1 induces expression of genes involved in extracellular matrix production but not classical endoplasmic reticulum stress response genes in pancreatic β-cells. Endocrinology. 2010;151(9):4146–4157. doi: 10.1210/en.2010-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boutzios G, Livadas S, Marinakis E, Opie N, Economou F, Diamanti-Kandarakis E. Endocrine and metabolic aspects of the Wolfram syndrome. Endocrine. 2011;40(1):10–13. doi: 10.1007/s12020-011-9505-y. [DOI] [PubMed] [Google Scholar]
- 72.Barrett TG, Bundey SE, Fielder AR, Good PA. Optic atrophy in Wolfram (DIDMOAD) syndrome. Eye. 1997;11(6):882–888. doi: 10.1038/eye.1997.226. [DOI] [PubMed] [Google Scholar]
- 73.Cryns K, Sivakumaran TA, van den Ouweland JMW, et al. Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Human Mutation. 2003;22(4):275–287. doi: 10.1002/humu.10258. [DOI] [PubMed] [Google Scholar]
- 74.Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframin mutations, wolfram syndrome, and associated diseases. Human Mutation. 2001;17(5):357–367. doi: 10.1002/humu.1110. [DOI] [PubMed] [Google Scholar]
- 75.Yamada T, Ishihara H, Tamura A, et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Human Molecular Genetics. 2006;15(10):1600–1609. doi: 10.1093/hmg/ddl081. [DOI] [PubMed] [Google Scholar]
- 76.Riggs AC, Bernal-Mizrachi E, Ohsugi M, et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia. 2005;48(11):2313–2321. doi: 10.1007/s00125-005-1947-4. [DOI] [PubMed] [Google Scholar]
- 77.Fonseca SG, Urano F, Weir GC, Gromada J, Burcin M. Wolfram syndrome 1 and adenylyl cyclase 8 interact at the plasma membrane to regulate insulin production and secretion. Nature Cell Biology. 2012;14(10):1105–1112. doi: 10.1038/ncb2578. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 78.Shang L, Hua H, Foo K, et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes. 2014;63(3):923–933. doi: 10.2337/db13-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology. 2006;147(7):3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 80.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. Journal of Cell Science. 2008;121(14):2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cnop M, Abdulkarim B, Bottu G, et al. RNA-sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes. 2013 doi: 10.2337/db13-1383. [DOI] [PubMed] [Google Scholar]
- 82.Wang H, Kouri G, Wollheim CB. ER stress and SREBP-1 activation are implicated in B-cell glucolipotoxicity. Journal of Cell Science. 2005;118(17):3905–3915. doi: 10.1242/jcs.02513. [DOI] [PubMed] [Google Scholar]
- 83.Carlson SG, Fawcett TW, Bartlett JD, Bernier M, Holbrook NJ. Regulation of the C/EBP-related gene gadd153 by glucose deprivation. Molecular and Cellular Biology. 1993;13(8):4736–4744. doi: 10.1128/mcb.13.8.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang C-J, Lin C-Y, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress-mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56(8):2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- 85.Cardozo AK, Ortis F, Storling J, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54(2):452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 86.Anding AL, Chapman JS, Barnett DW, Curley RW, Jr., Clagett-Dame M. The unhydrolyzable fenretinide analogue 4-hydroxybenzylretinone induces the proapoptotic genes GADD153 (CHOP) and Bcl-2-binding component 3 (PUMA) and apoptosis that is caspase-dependent and independent of the retinoic acid receptor. Cancer Research. 2007;67(13):6270–6277. doi: 10.1158/0008-5472.CAN-07-0727. [DOI] [PubMed] [Google Scholar]
- 87.Rao RV, Hermel E, Castro-Obregon S, et al. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. The Journal of Biological Chemistry. 2001;276(36):33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 88.Hitomi J, Katayama T, Eguchi Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. Journal of Cell Biology. 2004;165(3):347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X-Z, Lawson B, Brewer JW, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Molecular and Cellular Biology. 1996;16(8):4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. Journal of Molecular Biology. 2002;318(5):1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 91.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Structure and Function. 2008;33(1):75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 92.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. The Journal of Clinical Investigation. 2008;118(10):3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochemical Journal. 2002;366, part 2:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & Development. 1998;12(7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ariyama Y, Tanaka Y, Shimizu H, et al. The role of CHOP messenger RNA expression in the link between oxidative stress and apoptosis. Metabolism. 2008;57(12):1625–1635. doi: 10.1016/j.metabol.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 96.Oyadomari S, Takeda K, Takiguchi M, et al. Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Satoh T, Abiru N, Kobayashi M, et al. CHOP deletion does not impact the development of diabetes but suppresses the early production of insulin autoantibody in the NOD mouse. Apoptosis. 2011;16(4):438–448. doi: 10.1007/s10495-011-0576-2. [DOI] [PubMed] [Google Scholar]
- 98.Maris M, Overbergh L, Gysemans C, et al. Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia. 2012;55(4):1167–1178. doi: 10.1007/s00125-011-2427-7. [DOI] [PubMed] [Google Scholar]
- 99.Kojima E, Takeuchi A, Haneda M, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. The FASEB Journal. 2003;17(11):1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 100.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & Development. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamaguchi H, Wang H-G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. The Journal of Biological Chemistry. 2004;279(44):45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 102.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. The EMBO Journal. 2005;24(6):1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cunha DA, Igoillo-Esteve M, Gurzov EN, et al. Death protein 5 and p53-upregulated modulator of apoptosis mediate the endoplasmic reticulum stress-mitochondrial dialog triggering lipotoxic rodent and human beta-cell apoptosis. Diabetes. 2012;61(11):2763–2775. doi: 10.2337/db12-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jang YY, Kim NK, Kim MK, et al. The effect of tribbles-related protein 3 on ER stress- suppressed insulin gene expression in INS-1 cells. Korean Diabetes Journal. 2010;34(5):312–319. doi: 10.4093/kdj.2010.34.5.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liew CW, Bochenski J, Kawamori D, et al. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse β cells. The Journal of Clinical Investigation. 2010;120(8):2876–2888. doi: 10.1172/JCI36849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCullough KD, Martindale JL, Klotz L-O, Aw T-Y, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Molecular and Cellular Biology. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Allagnat F, Fukaya M, Nogueira TC, et al. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death & Differentiation. 2012;19(11):1836–1846. doi: 10.1038/cdd.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes & Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yamamoto E, Dong Y-F, Kataoka K, et al. Olmesartan prevents cardiovascular injury and hepatic steatosis in obesity and diabetes, accompanied by apoptosis signal regulating Kinase-1 inhibition. Hypertension. 2008;52(3):573–580. doi: 10.1161/HYPERTENSIONAHA.108.112292. [DOI] [PubMed] [Google Scholar]
- 110.Yamaguchi K, Takeda K, Kadowaki H, et al. Involvement of ASK1-p38 pathway in the pathogenesis of diabetes triggered by pancreatic ss cell exhaustion. Biochimica et Biophysica Acta. 2013;1830(6):3656–3663. doi: 10.1016/j.bbagen.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 111.Yoneda T, Imaizumi K, Oono K, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. The Journal of Biological Chemistry. 2001;276(17):13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 112.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Molecular and Cellular Biology. 2006;26(8):3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tonnesen MF, Grunnet LG, Friberg J, et al. Inhibition of nuclear factor-κB or Bax prevents endoplasmic reticulum stress- but not nitric oxide-mediated apoptosis in INS-1E cells. Endocrinology. 2009;150(9):4094–4103. doi: 10.1210/en.2009-0029. [DOI] [PubMed] [Google Scholar]
- 114.Hetz C, Bernasconi P, Fisher J, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α . Science. 2006;312(5773):572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 115.Rodriguez DA, Zamorano S, Lisbona F, et al. BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1α . The EMBO Journal. 2012;31(10):2322–2335. doi: 10.1038/emboj.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lisbona F, Rojas-Rivera D, Thielen P, et al. BAX Inhibitor-1 is a negative regulator of the ER stress sensor IRE1α . Molecular Cell. 2009;33(6):679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marhfour I, Lopez XM, Lefkaditis D, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia. 2012;55(9):2417–2420. doi: 10.1007/s00125-012-2604-3. [DOI] [PubMed] [Google Scholar]
- 118.Hartman MG, Lu D, Kim M-L, et al. Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Molecular and Cellular Biology. 2004;24(13):5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]