

Abstract
α-Synuclein is a small, natively unstructured protein with propensity to aggregate. α-Synuclein fibrils are major components of Lewy bodies that are hallmarks of many neurodegenerative diseases. The solution properties and aggregation behavior of α-synuclein has been well characterized, but despite numerous studies that address the role of α-synuclein in cells, a clear physiological function of this protein remains a mystery. Over a hundred review articles of α-synuclein have been written in the last decade, making it difficult to list all of the important studies that have added to our insight of α-synuclein physiology. Instead, we briefly review the status of α-synuclein research and propose a model based on the idea that α-synuclein may not have an intrinsic activity in cells but rather, it modifies the function of a group of protein partners that in turn affect cell processes. We propose that it is the loss of its cellular partners under oxidative conditions that promotes α-synuclein aggregation accelerating neuronal death.
Keywords: α-synuclein, protein aggregation, oxidative stress, phospholipase C
OVERVIEW
α-Synuclein is a small protein, comprising of 140 amino acids. α-Synuclein is conserved from fish to birds to mammals, and appears to be exclusive to vertebrates (for review see [1,2]). The protein is highly expressed in neuronal tissue and is found at high levels in hematopoietic cells. α-Synuclein has been the subject of intense study ever since its mutant forms were directly connected to familial Parkinson’s disease (see [3,4]). Strengthening the connection between α-synuclein and neurodegeneration is the observation that α-synuclein constitutes a major component in plaques called Lewy bodies that are found in neurodegenerative diseases such as Parkinson’s and Alzheimer’s (see for background [5,4,2,6]). These and subsequent studies launched a large body of work that focused on the structure and aggregation properties of α-synuclein (see [3]). While we now have a good understanding of the parameters that influence the transition of α-synuclein from an unstructured monomer, to a helical tetramer to a beta-stranded fibril (FIG 1), the cellular mechanisms responsible for initiating and promoting these aggregated states are unclear. Adding to our ignorance is the fact that the cellular function of α-synuclein remains unknown despite the many studies involving knock-out mice and cultured cells. Numerous reviews about different aspects of α-synuclein have been written and it is not our intention here to provide a comprehensive summary of the state of α-synuclein research. Rather, we will summarize the salient points about the biophysical properties and functional aspects of α-synuclein and present a hypothesis of the cellular role α-synuclein plays and a possible mechanism that may underlie α-synuclein -related diseases.
Figure 1. Cartoon of α-synuclein aggregates.

A neuron containing α-synuclein aggregates is on top. Below, from left to right, depicts a helical monomer that can associated to a tetramer or become unstructured monomer that can form fibrils.
Structural characterization of α-synuclein
α-Synuclein is is classified as a ‘natively unfolded’ protein [7]. When α-synuclein is bacterially expressed and purified using high heat ([7,8]), the resulting protein lacks secondary structure as seen by circular dichroism and nmr (see [9]). However, when purified from bacteria under non-denaturing conditions, α-synuclein appears as a helical tetramer [10], although some controversy remains as to the native state of the protein in cells [11,12]. When stored for several days at room temperature under non-denaturing conditions, purified α-synuclein aggregates to tetramers and higher order oligomers that are resistant to dissociation by SDS and other denaturants (see FIG 1). At high concentrations, α-synuclein will form β-stranded fibrils that resemble those found in neurodegenerative plaques [13]. Variations in environmental conditions change the propensity of the unstructured monomer to form helical tetramers and fibrils. While it is difficult to understand the behavior of α-synuclein point mutants associated with Parkinson’s disease from aggregation studies, they rationalize the early onset of neurodegeneration associated with multiple copies of the SNCA gene encoding α-synuclein (e.g. [14,15]) and the observation of aggregates in cultured cells when α-synuclein is over-produced. It is notable that studies of point mutants of α-synuclein that underlie familiar Parkinson’s disease suggest that neuropathicity results from α-synuclein oligomerization that precedes fibrillation (8).
α-Synuclein has three major domains. The amino terminal (residues 1–60) contains several imperfect ~11 amino acids repeats built around the hexamer KTKEGV although this motif is not strictly conserved. These repeats share similarity to a class of apolipoproteins that are associated with lipid binding which acquire helical conformations when lipid bound.
The middle region of the protein (residues 61–95) is more hydrophobic. This region, called the non-amyloid component (NAC) was the portion of α-synuclein first identified in Lewy bodies. Within this region is a 12 amino acid hydrophobic stretch that is thought to be responsible for α-synuclein aggregation. This hydrophobic stretch is not found in the other two members of synuclein family (β- and γ) that do not form fibrils and are not found in Lewy bodies even though they are also highly expressed in neuronal tissue. It is notable that γ-synuclein will form tetramers when purified from E. coli, but has a greatly reduced ability to aggregate into higher order oligomers as compared to α-synuclein (e.g. [14]).
Synuclein interacts with membranes
In one of the first biophysical studies of α-synuclein, Davidson and coworkers showed that when α-synuclein is incubated for several hours at 37°C with sonicated vesicles composed of at least 30% anionic lipids, it will interact with the lipids and form helical aggregates [16]. These helical aggregates were in sharp contrast to the β-sheet fibrils that typify neurodegenerative plaques, and led to the speculation that membrane bound synuclein has a high propensity to aggregate [17]. Subsequently, our lab quantified the binding of α-synuclein to membranes of varying composition in real time by measuring the changes in intrinsic α-synuclein fluorescence [6]. The addition of monomeric ‘random coil’ α-synuclein to membranes of varying compositions resulted in strong binding with a preference for lipids with negatively charged head groups and for the signaling lipid phosphatidyl inositol 4,5 bisphosphate (PIP2) [18]. In contrast to previous reports, these studies showed that membrane binding of α-synuclein under conditions of excess lipid, as would be expected in the cell, prevented α-synuclein oligomerization. Subsequent studies by other labs including the comprehensive work by Rhoades and coworkers have characterized the optimal membrane characteristics for α-synuclein [19]. It is also now believed that membrane interactions will compete with interactions between α-synuclein monomers and inhibit oligomerization. It is not clear if α-synuclein is membrane bound in cells since imaging studies tend to show the protein as being widely dispersed with a large perinuclear population (e.g. FIG 2.). However, it is notable that many of the proposed α-synuclein binding partners are membrane bound and/or participate in lipid signaling and other membrane events (see below).
Figure 2. Distribution of mCherry-α-synuclein in a HEK293 cells.
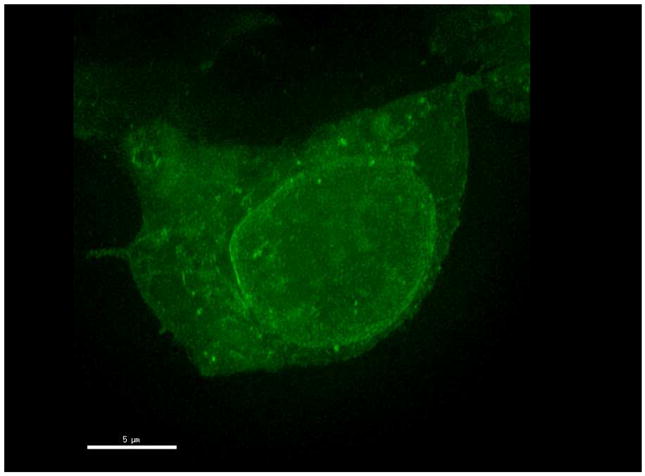
taken on a ZeissLSM780 in the facility at Cold Spring Harbor Laboratory.
Functional studies of α-synuclein
Despite its importance in disease, the cellular function of α-synuclein has not yet been clearly identified. It is known that its expression changes with development and is linked to song learning in songbirds, suggesting some role in synaptic plasticity [20]. Studies using cultured cells point towards a role of α-synuclein in neuronal development and dopamine function [21], however, the generation of knock-out mice did not offer other clues into its physiological roles on an organism level [22]. The mice were viable and almost fully functional with no major motor, behavioral or developmental defects. Even though α-synuclein aggregates are directly linked to cell toxicity (e.g. [23]), no major changes in the distribution of vesicular or presynaptic proteins were found in knock-out mice indicating that α-synuclein was not essential for neuronal differentiation and development. It is important to note that Murphy and coworkers did find minor changes in the lipid composition of endosomes and other subcelluar organelles [14,24].
One approach to uncover α-synuclein function is to determine its cellular binding partners. Being ‘natively unfolded’, α-synuclein has been found to bind to many cellular proteins such as synphilin [25], the transcription factor Elk-1 [26], and Tat binding protein 1 [27] to name a few. Proteomics studies identified a large number of potential binding partners which generally supports the role of synuclein in dopamine signaling and mitochondrial damage, these studies expand rather than narrow the potential interaction partners of α-synuclein [28]. Studies in tissue slices and cultured cells point to a role of α-synuclein in vesicle transport, endocytosis and neural plasticity (e.g. [29]). For example, α-synuclein can bind and inhibit phospholipase D2 [30] which may influence vesicle release [31], and α-synuclein can bind to the dopamine transporters [32] correlating to a potential role in dopamine transport [33–35]. The ability of α-synuclein to bind to lipid membranes has led to the idea that it may form ion-selective channels although this does not appear to be the case. α-Synuclein has also been shown to bind to a Bcl-2 homolog that plays a role in mitochondrial disfunction, cytochrome C release and apoptosis correlating to the appearance of α-synuclein-cytochrome C complexes in Lewy bodies [36]. Important for the model presented here (see below) is the strong similarity of α-synuclein to fatty acid binding proteins 14-3-3 which act as chaperone regulating activities of enzymes, and as protein adaptors [37,38].
Because α-synuclein prefers membranes containing PIP2, our lab investigated the ability of α-synuclein to interact with the PIP2-hydrolyzing enzyme, phospholipase Cβ (PLCβ) (for review see [39,40]. Studies using purified proteins show that α-synuclein binds to PLCβ with a very strong affinity in solution [6]. Binding between the two proteins in cultured cells has also been noted [41]. α-Synuclein was found to bind to the same site on PLCβ as Gαq subunits, and α-synuclein and PLCβ may remain associated until Gαq becomes activated during signaling, its affinity for PLCβ increases and it can displace α-synuclein [41].
PLCβ is activated by Gαq which mediates signals from ligands such as angiotensin, acetylcholine, dopamine and bradykinin (see [42,43]) and activation of PLCβ leads to an increase in intracellular calcium. High calcium levels can result in activation of the calcium-sensitive protease calpain, which degrades PLCβ providing a negative feedback mechanism of signaling. We had noted that when the level of α-synuclein expression is increased by over-expression, the protein but not the transcript level of PLCβ similarly increases [44]. Alternate studies showed that α-synuclein protects PLCβ from calpain cleavage [41]. This latter finding correlates well with the observation that α-synuclein moves from the plasma membrane to the cytosol with Gαq activation (see [45]). It has recently been found that PLCβ can bind to and change the activity of a complex that promotes gene silencing by small RNAs [46]. This binding site overlaps with the α-synuclein binding site as well as Gαq and calpain. Thus, just the simple binding of α-synuclein to one of potentially many protein partners can impact cell function in a number of different ways.
Oxidation and environmental factors in α-synuclen diseases
Many environmental factors are thought to contribute to neurodegeneration due to synuclein pathology and of these, oxidative stress has been clearly established (for overview see [47]). The effect of oxidative stress on tissue and cells is profound. Oxidation results in modification of protein side chains and in α-synuclein, oxidation promotes nitrosylation tyrosine cross-linking (10, 11). Typically, oxidized proteins with modified side chains are degraded in proteosomes. However, this may not be the case for α-synuclein which appears to be degraded by a number of different pathways (see below and (12,13). Oxidative stress has been closely linked to Parkinson’s disease (see [48]), and oxidative stress in conjunction with elevated cellular calcium has been proposed to promote α-synuclein aggregation [49]. Interestingly, α-synuclein aggregation has been shown to disrupt mitochondria which may synergize the effects of oxidative stress [50].
α-Synuclein down-regulation
Adding to the confusion concerning α-synuclein function is its mechanism of down-regulation. Lewy bodies contain mono-ubiquitinated forms of α-synuclein and α-synuclein contains several active ubiquitination sites [51]. Although proteosomal degradation of α-synuclein has been reported, the most prevalent pathway appears to be through chaperone-assisted autophagy by lysozomes [52,53]. It is notable that denatured and aggregated proteins are often down-regulated by autophagy pathways (for overview see [54]). It is possible that different down-regulation pathways used for α-synuclein may result from the nature of the particular binding partner it is attached to, as well as its ability to self-associate which may differentially expose potential ubiquitination sites and modified side chains.
A general model of α-synuclein function and disfunction
This model is based on several key aspects of α-synuclein biology: 1 - α-synuclein lacks a clear cellular function and lacks a clear phenotype in knock-out animals, 2 – α-synuclein has the propensity to be an unstructured monomer, 3 – α-synuclein has the ability to bind to multiple proteins, 3 – the predominant degradation pathway of α-synuclein is chaperone-assisted autophagy, 4 - α-synuclein bears a strong resemblance to the 14-3-3 family of chaperone proteins, 5 – oxidation and mitochondrial disfunction play a key role in the progression of Parkinson’s disease.
In general, biophysical studies suggest that α-synuclein is a plastic protein that can bind as a helical monomer or adapt other conformations to interact with a variety of cellular proteins and membranes, and these many partners prevent α-synuclein self-association and aggregation. Additionally, we speculate that α-synuclein does not have an intrinsic function but serves to alter / modify function of interacting partners or possibly membrane properties. The large number of potential interacting partners as well as the multiple effects that any individual interaction may have, as exemplified by PLCβ, can result in a multitude of direct and indirect cellular effects. This wide range of interactions explains the ability of cell to produce high levels of α-synuclein without greatly affecting cell properties as seen when knock-out mice are compared to control groups.
Over-expression of α-synuclein has been shown to cause inclusions and neuronal death (6) but under native conditions, if α-synuclein binds many partners, what triggers its self-association and subsequent large scale aggregation? We propose that aggregation is initiated and promoted by the loss of α-synuclein binding partners through events such as oxidation (FIG 3). This hypothesis is based on our finding that over-expression of α-synuclein results in the formation of oligomers which can be reversed by over-expression of a binding partner (i.e.PLCβ), and conversely, down-regulation of a binding partner (i.e. PLCβ) promotes α-synuclein aggregation [44]. Importantly, we have found that oxidative stress results in a pronounced reduction in the level of many cellular proteins including PLCβ that are down-regulated by ubiquitin-mediated pathways, while the level of α-synuclein, which is primarily down-regulated by chaperone-assisted autophagy (10), appears unchanged [44]. While our previous studies focused on PLCβ, many other cellular proteins and potential α-synuclein binding partners are lost when cells are subject to oxidative stress. The loss in protein partners would give rise to higher levels of unbound α-synuclein which may result in homo-oligomerization. In turn, aggregation will promote degradation of mitochondria. A cartoon describing this model is given in FIG 3. Support for this model comes from studies α-synuclein aggregation can be ablated using a peptide that blocks α-synuclein / PLCβ association thus suggesting a target for drug development [44]. It is also important to note that oxidative stress would be expected to modify α-synuclein side chains which may promote aggregation.
Figure 3. Model of α-synuclein aggregation in cells.

The top left depicts α-synuclein monomers in different conformations which depend on the nature of the protein or membrane it is bound (green and blue circles). When these proteins are degraded by oxidation or other stress conditions (orange, spiky circle), α-synuclein is released as unstructured monomer which is poised to aggregate.
This model for the mechanism underlying the aggregation of α-synuclein in cells is very general and more details are needed in regards to the specific effects of oxidation on α-synuclein and its partners, mitochondrial damage and down-regulation mechanisms. It is also expected that complementary and novel mechanisms of aggregation will come to light as our understanding of α-synuclein’s role in the cell progresses.
Acknowledgments
We would like to thank Vijaya Narayanan and Yuanjian Guo for their contribution to the studies from our lab highlighted in this paper. This work was supported by NIH GM05312.
References
- 1.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21(6):249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 2.George JM. The synucleins. Genome Biol. 2002;3:3002.3001–3002.3006. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsian A, Racette B, Zhang ZH, Chakraverty S, Rundle M, Goate A, et al. Mutation, sequence analysis, and association studies of alpha-synuclein in Parkinson’s disease. Neurology. 1998;51(6):1757–1759. doi: 10.1212/wnl.51.6.1757. [DOI] [PubMed] [Google Scholar]
- 4.Lundvig D, Lindersson E, Jensen PH. Pathogenic effects of α-synuclein aggregation. Molecular Brain Research. 2005;134(1):3–17. doi: 10.1016/j.molbrainres.2004.09.001. http://dx.doi.org/10.1016/j.molbrainres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61(2):121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 6.Narayanan V, Scarlata S. Membrane binding and self-association of alpha-synuclein. Biochem. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 7.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 8.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345(1):27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 9.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of α-synuclein in its free and lipid-associated states. Journal of Molecular Biology. 2001;307(4):1061–1073. doi: 10.1006/jmbi.2001.4538. http://dx.doi.org/10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 10.Bartels T, Choi JG, Selkoe DJ. [agr]-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–110. doi: 10.1038/nature10324. http://www.nature.com/nature/journal/v477/n7362/abs/nature10324.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LTT, Liao J, et al. A soluble α-synuclein construct forms a dynamic tetramer. Proceedings of the National Academy of Sciences. 2011;108(43):17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coelho-Cerqueira E, Carmo-Goncalves P, Pinheiro AS, Cortines J, Follmer C. alpha-Synuclein as an intrinsically disordered monomer--fact or artefact? FEBS J. 2013;280(19):4915–4927. doi: 10.1111/febs.12471. [DOI] [PubMed] [Google Scholar]
- 13.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39(10):2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 14.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein locus triplication casues Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 15.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167–1169. doi: 10.1016/s0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 16.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 17.Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 18.Narayanan V, Guo Y, Scarlata S. Fluorescence studies suggest a role for α-synuclein in the phosphatdiylinositol lipid signaling pathway. Biochem. 2005;44:462–470. doi: 10.1021/bi0487140. [DOI] [PubMed] [Google Scholar]
- 19.Middleton ER, Rhoades E. Effects of Curvature and Composition on ±-Synuclein Binding to Lipid Vesicles. Biophysical Journal. 2010;99(7):2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15(2):361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 21.Stefanis L, Kholodilov N, Rideout HG, Burke RE, Greene LA. Synuclein-1 is selectively up-regulated in response to nerve growth factor treatment in PC12 cells. J Neurochem. 2001;76:1165–1176. doi: 10.1046/j.1471-4159.2001.00114.x. [DOI] [PubMed] [Google Scholar]
- 22.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25(1):239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 23.Wan OW, Chung KKK. The Role of Alpha-Synuclein Oligomerization and Aggregation in Cellular and Animal Models of Parkinson’s Disease. PLoS ONE. 2012;7(6):e38545. doi: 10.1371/journal.pone.0038545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Golovko MY, Rosenberger TA, Feddersen S, Færgeman NJ, Murphy EJ. α-Synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. Journal of Neurochemistry. 2007;101(1):201–211. doi: 10.1111/j.1471-4159.2006.04357.x. [DOI] [PubMed] [Google Scholar]
- 25.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22(1):110–114. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 26.Iwata A, Miura S, Kanazawa I, Sawada M, Nukina N. α-Synuclein forms a complex with transcription factor Elk-1. J Neurochem. 2001;77:239–252. doi: 10.1046/j.1471-4159.2001.t01-1-00232.x. [DOI] [PubMed] [Google Scholar]
- 27.Ghee M, Fournier A, Mallet J. Rat alpha-synuclein interacts with tat binding protein 1, a component of the 26S proteasomal complex [In Process Citation] J Neurochem. 2000;75(5):2221–2224. doi: 10.1046/j.1471-4159.2000.0752221.x. [DOI] [PubMed] [Google Scholar]
- 28.Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, et al. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol Cell Proteomics. 2007;6(5):845–859. doi: 10.1074/mcp.M600182-MCP200. [DOI] [PubMed] [Google Scholar]
- 29.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20(9):3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37(14):4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 31.Pronin AN, Morris AJ, Surguchov A, Benovic JL. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem. 2000;275(34):26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- 32.Lee FJ, et al. Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 33.Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866(1–2):33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]
- 34.Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutation may contribute to the pathogensis of Parkinson’s disease. HumanMolec Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- 35.Tabrizi SJ, Orth M, Wilkinson JM, Taanman JW, Warner TT, Cooper JM, et al. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity [In Process Citation] Hum Mol Genet. 2000;9(18):2683–2689. doi: 10.1093/hmg/9.18.2683. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E. Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J Biol Chem. 1999;274(41):28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]
- 37.van Hemert MJ, Steensma HY, van Heusden GPH. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. BioEssays. 2001;23(10):936–946. doi: 10.1002/bies.1134. [DOI] [PubMed] [Google Scholar]
- 38.Ostrerova N, Petrucelli L, Farrer M, Mehta N, Choi P, Hardy J, et al. α-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci. 1999;19(14):5782–5791. doi: 10.1523/JNEUROSCI.19-14-05782.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 40.Suh P, Park J, Manzoli L, Cocco L, Peak J, Katan M, et al. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB reports. 2008;41:415–434. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- 41.Guo Y, Rosati B, Scarlata S. α-synuclein increases the cellular level of phospholipase Cβ1. Cellular Signalling. 2012;24(5):1109–1114. doi: 10.1016/j.cellsig.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Exton JH. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annual Review of Pharmacology & Toxicology. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 43.Hepler JR, Gilman AG. G-proteins. Trends Biochem Sciences. 1992;17:383–387. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- 44.Guo Y, Scarlata S. A Loss in Cellular Protein Partners Promotes α-Synuclein Aggregation in Cells Resulting from Oxidative Stress. Biochemistry. 2013;52(22):3913–3920. doi: 10.1021/bi4002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leng Y, Chase TN, Bennett MC. Muscarininc receptor stimulation induces translocation of an alpha-synuclein oligomer from plasma membrane to a light vesicles fraction in cytoplasm. J Biol Chem. 2001;276:28212–28218. doi: 10.1074/jbc.M011121200. [DOI] [PubMed] [Google Scholar]
- 46.Philip F, Guo Y, Aisiku O, Scarlata S. Phospholipase Cβ1 is linked to RNA interference of specific genes through translin-associated factor X. The FASEB Journal. 2012 doi: 10.1096/fj.12-213934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 48.Mizuno Y, Hattori N, Kubo S-i, Sato S, Nishioka K, Hatano T, et al. Progress in the pathogenesis and genetics of Parkinson’s disease. Philosophical Transactions of the Royal Society B: Biological Sciences. 2008;363(1500):2215–2227. doi: 10.1098/rstb.2008.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goodwin J, Nath S, Engelborghs Y, Pountney DL. Raised calcium and oxidative stress cooperatively promote alpha-synuclein aggregate formation. Neurochem Int. 2013;62(5):703–711. doi: 10.1016/j.neuint.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. The American journal of pathology. 2000;157(2):401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abeywardana T, Lin Yu H, Rott R, Engelender S, Pratt Matthew R. Site-Specific Differences in Proteasome-Dependent Degradation of Monoubiquitinated α-Synuclein. Chemistry & Biology. 2013;20(10):1207–1213. doi: 10.1016/j.chembiol.2013.09.009. http://dx.doi.org/10.1016/j.chembiol.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ancolio K, Alves da Costa C, Ueda K, Checler F. Alpha-synuclein and the Parkinson’s disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett. 2000;285(2):79–82. doi: 10.1016/s0304-3940(00)01049-1. [DOI] [PubMed] [Google Scholar]
- 53.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science. 2004;305(5688):1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 54.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cellular and Molecular Life Sciences. 2011;68(5):749–763. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]