

Abstract
Noninvasive, targeted drug delivery to the brain can be achieved using transcranial focused ultrasound (FUS), which transiently increases the permeability of the blood-brain barrier (BBB) for localized delivery of therapeutics from the blood to the brain. Previously, we have demonstrated that FUS can deliver intravenously-administered antibodies to the brain of a mouse model of Alzheimer’s disease (AD) and rapidly reduce plaques composed of amyloid-β peptides (Aβ). Here, we investigated two potential effects of transcranial FUS itself that could contribute to a reduction of plaque pathology, namely the delivery of endogenous antibodies to the brain and the activation of glial cells.
We demonstrate that transcranial FUS application leads to a significant reduction in plaque burden four days after a single treatment in the TgCRND8 mouse model of AD and that endogenous antibodies are found bound to Aβ plaques. Immunohistochemical and western blot analyses showed an increase in endogenous immunoglobulins within the FUS-targeted cortex. Subsequently, microglia and astrocytes in FUS-treated cortical regions show signs of activation through increases in protein expression and changes in glial size, without changes in glial cell numbers. Enhanced activation of glia correlated with increased internalization of Aβ in microglia and astrocytes.
Together these data demonstrate that FUS improved bioavailability of endogenous antibodies and a temporal activation of glial cells, providing evidence towards antibody- and glia-dependent mechanisms of FUS-mediated plaque reduction.
Keywords: amyloid-beta peptide, focused ultrasound, immunoglobulin, autoantibodies, microglia, astrocytes, Alzheimer’s disease, transgenic mice
Introduction
The blood-brain barrier (BBB) poses a challenge for the delivery of therapeutics to the brain for treatment of neurological diseases. Chemicals administered intravenously can facilitate the passage of therapeutics from the blood to the brain but produce variability in the extent and duration of BBB opening (Joshi et al., 2010; Patel et al., 2009; Salahuddin et al., 1988). Ideally, only brain areas affected by disease would be targeted for treatment, minimizing BBB disruption in other brain regions.
In Alzheimer’s disease (AD), amyloid-β peptides (Aβ) aggregate and form extracellular plaques. Animal studies delivering anti-Aβ antibodies directly to the cortex have demonstrated a rapid therapeutic response but employed invasive surgical techniques (Kotilinek et al., 2002; Wilcock et al., 2003). The use of transcranial focused ultrasound (FUS) guided by magnetic resonance imaging (MRI) to locally increase the permeability of the BBB has several advantages, including non-surgical application, targeting of specific brain regions, and control of the extent of BBB opening without damaging surrounding tissues when combined with an intravenous injection of microbubbles (Hynynen et al., 2001; Hynynen et al., 2005; McDannold et al., 2005; Sheikov et al., 2004). We previously demonstrated that MRI-guided FUS (MRIgFUS) efficiently delivered systemically administered anti-Aβ antibodies to targeted brain regions of the TgCRND8 mouse model of AD, reducing plaque load within 4 days (Jordão et al., 2010).
Here, we hypothesize that MRIgFUS alone reduces in Aβ pathology, considering that it promotes the entry of monomeric endogenous antibodies (Raymond et al., 2008; Sheikov et al., 2004). Previous studies have shown that endogenous antibodies present in the blood can bind to and disaggregate Aβ fibrils (Dodel et al., 2002; Du et al., 2003; Hyman et al., 2001). We first detected the entry of endogenous antibodies at the site of cortical Aβ plaques in MRIgFUS-treated TgCRND8 mice. Then, we investigated whether MRIgFUS allowed pentameric endogenous immunoglobulin M (IgM, ~900 kDa), in addition to monomeric immunoglobulin G (IgG, ~150 kDa), to pass from the blood to the brain in TgCRND8 and non-transgenic (non-Tg) mice. Finally, we evaluated whether MRI detection of FUS-mediated changes in BBB permeability can predict the amount of endogenous immunoglobulin entering the brain.
In addition, glia have been implicated in the mechanism of antibody-mediated Aβ clearance (Bard et al., 2000; Koenigsknecht-Talboo et al., 2008; Magga et al., 2010; Nicoll et al., 2006; Wilcock et al., 2003; Wilcock et al., 2004). Therefore, we investigated whether MRIgFUS-enhanced BBB permeability activates microglia and astrocytes, and whether these glia contain Aβ, which would suggest their contribution to Aβ internalization and clearance. Glial activation can be characterized by an increase in the expression of certain proteins, such as ionizing binding adaptor molecule 1 (Iba1) in phagocytic microglia (Ito et al., 2001; Ito et al., 1998) and glial fibrillary acidic protein (GFAP) in astrocytes (Pekny and Nilsson, 2005). Activated glial cells undergo morphological changes, including increased volume and surface area. We aimed to establish the glial activation response to MRIgFUS, and the potential role of glia in MRIgFUS-mediated plaque reduction. The temporal response of glial activation following MRIgFUS was characterized using Iba1 and GFAP expression in the cortex of TgCRND8 and non-Tg mice. Changes in glial volume and area, in addition to Aβ internalization by glia were also investigated.
MRIgFUS technology represents a major advance in the field of non-invasive drug delivery to the brain. For validation of this delivery technique for a wide range of applications, establishing the effects of MRIgFUS in animal models under normal and diseased conditions is important. Here, we show that under disease conditions, MRIgFUS alone reduces Aβ plaque load in the targeted cortex of TgCRND8 mice. Additionally, in both TgCRND8 and non-Tg mice, MRIgFUS delivers endogenous antibodies to the brain and activates glial cells.
Materials and Methods
Animals
Four month-old male and female non-Tg and TgCRND8 mice (Chishti et al., 2001) were used in this study. Mice at this age were chosen because they exhibit abundant plaque load and match our previous study (Jordão et al., 2010), for comparison purposes. Tissue for western blot analyses was collected from mice sacrificed at 4 hours (n=7 for non-Tg; n=6 for TgCRND8 mice), 4 days (n=6 for non-Tg; n=7 for TgCRND8 mice) and 15 days (n=7 for non-Tg and n=7 for TgCRND8 mice).
A separate cohort of mice were treated for immunohistochemistry analyses and sacrificed at 4 hours (n=6 for non-Tg; n=6 for TgCRND8 mice), 4 days (n=7 for non-Tg; n=9 for TgCRND8 mice) and 15 days (n=7 for non-Tg and n=6 for TgCRND8 mice). Of the 9 TgCRND8 mice used at 4 days post-MRIgFUS, 6 were treated as a MRIgFUS group in a previous study (Jordão et al., 2010) and 3 additional mice receiving the same treatment were added to complete the current study.
All animal procedures were conducted with the approval of the Animal Care Committee of Sunnybrook Research Institute and in compliance with the guidelines established by the Canadian Council on Animal Care and the Animals for Research Act of Ontario.
MRIgFUS treatment
Mice were anaesthetized with ketamine (150 mg/kg) and xylazine (10 mg/kg) and placed on a small animal MRIgFUS compatible positioning system (Chopra et al., 2009). MRI gadolinium contrast agent (Omniscan®, 0.2 ml/kg, 574 Da, GE Healthcare) and FUS microbubble contrast agent (Definity®, 80μl/kg for TgCRND8 and 40μl/kg for non-Tg mice; Lantheus Medical Imaging), were administered through a tail vein catheter at the onset of ultrasound treatment. Several studies have been conducted demonstrating that reproducible, reversible and non-damaging disruption of the BBB using FUS requires the administration of microbubbles (Hynynen et al., 2001; McDannold et al., 2006). As previously described (Jordão et al., 2010), focused ultrasound (0.3 MPa, 120 s, 10 ms bursts, 1 Hz repetition rate) was targeted to four foci, 1.5 mm apart, which creates approximately a 6.5 mm rosto-caudal line along the right cortex (Fig. 1A). The FUS transducer had a 10 cm diameter, 8 cm radius of curvature, and a frequency of 0.5 MHz). The positioning system was placed in a 1.5T MRI scanner (GE Healthcare) and standard T2- and T1-weighted imaging was performed. Anatomical mapping of target foci with MRI, prior to ultrasound, provided a baseline image (Fig. 1B) to compare changes in BBB permeability. These BBB changes were detected by an increase in intensity, also known as enhancement (Fig. 1B, black arrows), using an MRI gadolinium-based contrast agent that only enters the brain when the BBB is made permeable. A low enhancement (10–20%) was maintained to increase the permeability of the BBB for endogenous immunoglobulin entry, but limit the entry of larger macromolecules and prevent damage to the surrounding tissue (O’Reilly et al., 2011).
Fig. 1.

MRIgFUS setup and confirmation of BBB disruption. A, Mice were placed in a supine position over a transducer, generating FUS waves upwards to target locations in the brain. B, Target locations along the right hemisphere were chosen using MRI images taken before MRIgFUS treatment (left). Effectiveness of BBB opening along the right hemisphere was confirmed from MRI contrast-enhanced images and the entry of gadolinium agent after MRIgFUS treatment (right hemisphere, black arrows). The image chosen here for representation of gadolinium entry in the brain was taken from a mouse displaying high enhancement levels. MRI intensities were detected and compared between the untreated (C, left hemisphere) and MRIgFUS-treated region with influx of gadolinium (D, right hemisphere). E, The mean intensity in the MRIgFUS-treated cortex was significantly higher than in the untreated contralateral cortex. F, The amount of gadolinium agent entering the MRIgFUS-treated region over time was calculated by subtracting the increase in enhancement seen in the MRIgFUS-treated region to the background intensity in the untreated region. Scale bar: B = 0.5 cm. Data is presented as paired values between left, untreated and right, MRIgFUS-treated, ***p<0.0001.
MRI enhancement analysis
A time series of post-FUS MRI T1-weighted images were analyzed with MatLab software (version R2008b, MathWorks) for determination of intensity differences between the left, untreated (Fig. 1C) and right, treated (Fig. 1D) regions of the brain within boxes of 10×30 pixels. Comparing the mean intensity of the right and left regions (at ~ 6 minutes after FUS) demonstrated that there is a significant influx of gadolinium contrast agent into MRIgFUS-targeted areas (Fig. 1E, ***p<0.0001, paired t-test, n=15). This provided confirmation that the permeability of the BBB was increased following FUS treatment.
Changes in BBB permeability over time were evaluated by collecting temporally sequential mean intensities of the right and left regions, which were subtracted from the image taken prior to MRIgFUS treatment (1B, left panel). The intensities of the untreated cortex were subtracted from the treated cortex to plot enhancement over time (Fig. 1F) in GraphPad Prism 5 (GraphPad Software Inc.). A one phase exponential association [Y=Ymax(1−e−kx)] was used to fit the data points. From these curves, the maximum enhancement (Ymax) was estimated, and the rate of enhancement was determined by the slope at the time of half the estimated maximum enhancement (see example: Fig. 4A). The maximum enhancement and rate of enhancement are related, but the rate is determined from within the initial linear portion of the curve; whereas the maximum enhancement is an extrapolation of the data, projecting the expected maximum gadolinium contrast agent delivered. Both the intensity and relative enhancement are expressed in arbitrary units (a.u.).
Fig. 4.
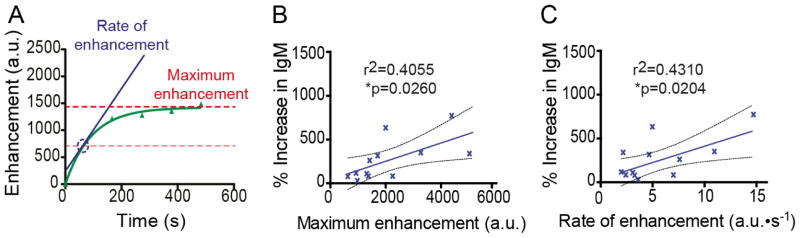
Increases in IgM are proportional to the enhancement in MRIgFUS-treated cortical regions. A, Using enhancement data gathered from MRI post-treatment scans, the estimated maximum enhancement (red) and rate of enhancement (blue) can be determined for all MRIgFUS-treated mice (representative enhancement graph for one mouse). B, The maximum enhancement, representative of the maximum amount of gadolinium able to enter the brain after BBB opening, was positively correlated to the increase in IgM levels measured in the MRIgFUS-treated cortex of the same mice (r2=0.4055, p=0.026, n=12). C, Similarly, the increase of endogenous IgM detected within the MRIgFUS-treated cortex was correlated to the rate of enhancement (r2=0.4310 p=0.020, n=12), which is indicative of the speed at which gadolinium enters the brain. Dashed lines indicate a 95% confidence intervals (B–C).
Immunohistochemistry
Mice were transcardially perfused with phosphate-buffered saline and 4% paraformaldehyde (PFA). Brains were removed, post-fixed in 4% PFA overnight, and saturated in a 30% sucrose solution. Coronal brain sections were cut at 40 μm.
For brightfield staining of Aβ plaques, IgG and IgM, sections were pre-treated with 3% hydrogen peroxide in methanol. Aβ plaques were stained using 6F3D anti-Aβ antibody (1:400; Dako North American Inc.) and biotinylated donkey anti-mouse IgG. IgG and IgM were stained using biotinylated donkey anti-mouse IgG and IgM, respectively. Staining was revealed using streptivadin-conjugated horseradish peroxidase (HRP) (Vectastain Elite, Vector Laboratories) and 3,3′-diaminobenzidine (DAB; Sigma).
Fluorescent detection of IgG and IgM with Aβ plaques was performed using donkey anti-mouse IgG Cy5 to detect IgG, donkey anti-mouse IgM biotin and strepavidin-Dylight 488 for IgM, and rabbit anti-Aβ1–42 (1:100; Millipore Corp.) and donkey anti-rabbit Cy3 for Aβ.
Microglia were stained with rabbit anti-Iba1 (1:1000; Wako Chemicals) and astrocytes with goat anti-GFAP (1:500; AbD Serotec). For the glial activation time course, glial markers were revealed on separate brain sections using donkey anti-rabbit Cy3 or donkey anti-goat Cy3. For analysis of changes in volume and size of glia surrounding Aβ plaques, donkey anti-goat Cy5 and donkey anti-rabbit Cy3 were used, with 0.01% Thioflavin S to stain for Aβ plaques.
To detect intracellular Aβ, primary antibodies for Aβ (6E10, mouse anti-Aβ, 1:100; Sigma), Iba1 and GFAP were used. Sections were then incubated with donkey anti-mouse Cy3, donkey anti-rabbit Dylight 488 and donkey anti-goat Cy5. All secondary antibodies were incubated at 1:200 (Jackson ImmunoResearch Laboratories, Inc.).
Aβ plaque quantification
A series of one in nine sections (between 1.94 mm anterior of Bregma to 0.34 mm posterior of Bregma) were randomly selected to stain and quantify plaque load as previously described (Jordão et al., 2010). Six coronal sections were analyzed per mouse. Stereo Investigator software (version 5.05.4; MBF Bioscience) was used to estimate plaque number (optical fractionator) and size (nucleator probe) in left and right cortical regions. The surface area of Aβ was generated from the product of the estimated total number of plaques and the mean plaque size.
Immunoblots
Following perfusion with saline, brains were removed and frozen in liquid nitrogen. The left (untreated) and right (MRIgFUS-treated) cortices were dissected and separately homogenized in lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 10% glycerol) containing a protease inhibitor cocktail (Calbiochem). Protein samples (20 μg for IgG, IgM, and GFAP blots; 50 μg for Iba1 blots) were separated by 12% SDS-PAGE (except for Iba1 blots which were run on 18% SDS-PAGE) and transferred to PVDF membranes (Sigma). For detection of immunoglobulins, immunoblots were incubated with mouse anti-GAPDH antibody (1:1000; Meridian Life Science, Inc.). The HRP-conjugated antibodies used were donkey anti-mouse IgM HRP, goat anti-mouse IgG-Fc-© HRP and donkey anti-mouse IgG HRP (1:1000, 1:1000 and 1:2000 respectively, Jackson ImmunoResearch Laboratories, Inc.).
For Iba1 and GFAP quantification, immunoblots were incubated with primary antibodies, anti-Iba1 (Abcam) or anti-GFAP (1:1000; Dako), and anti-GAPDH (1:5000 for Iba1 blots; 1:2000 for GFAP blots; Meridian Life Science, Inc.). The peroxidase-conjugated antibodies used were donkey anti-mouse IgG (Jackson Immuno Research Laboratories, Inc.) and donkey anti-rabbit IgG (Jackson Immuno Research Laboratories, Inc.).
Immunodetection was performed using the SuperSignal chemiluminescent reagent (Thermo Scientific Pierce). Quantification was carried out after acquisition with a CCD camera (SynGene) and analysis with adapted software (GeneSnap and GeneTools; SynGene). Each protein of interest (IgG, IgM, Iba1 and GFAP) was individually normalized to GAPDH levels and to an internal control present in each blot. This procedure allows for comparison of the levels of a given protein in the untreated and the MRIgFUS treated hemisphere and across different time points.
To determine the concentration of IgG and IgM in the brain, a concentration curve was created with the IgG 6F3D antibody (Dako North American Inc.) and the IgM anti-A2B5 antibody (Millipore). From the curves, the concentration of endogenous IgG and IgM were determined for the mouse used as the internal control, which was then used to determine the concentrations of IgG and IgM in all other animals.
Confocal analysis
Co-localization of immunoglobulins with Aβ plaques was detected by confocal microscopy (Zeiss 18 Axiovert 100M, LSM 510; Carl Zeiss). Images from right, treated cortex and left, untreated cortex were collected using the same exposure settings. Confocal microscopy was also used to acquire Z-stack images (optical slices of 0.5 μm) of plaques and surrounding glia. Three images were acquired from both the MRIgFUS-treated and untreated cortex of two sections per animal (n=6–7 TgCRND8 mice).
Imaris Analysis
Confocal z-stack images were reconstructed in three dimensions using Imaris software (Bitplane). Briefly, the surface function was used to create three-dimensional structures of Aβ plaques, microglia and astrocytes (surface area detail level = 0.3 μm, threshold = 80) within each picture. The volume of Aβ plaques were determined and used to set the volume of two equal-volume cylindrical regions of interest (ROI). The volume of the ROIs was ten-fold greater than the volume of the plaque. The ROI closest to the plaque is referred to the proximal ROI, whereas the distal ROI is the volume just outside of the proximal ROI and further away from the plaque. To detect Aβ within cells, the Cy3 staining for the Aβ plaque is subtracted from the image to create a new Cy3 channel that only shows smaller Aβ-positive staining clusters. This was done to ensure glial projections into the plaque would not be considered as co-localizations. Then, only staining in the new Cy3 channel that colocalized within the 488 channel staining (for microglia) or Cy5 staining (for astrocytes) was isolated and a surface was created for Aβ (surface area detail level = 0.25 μm, threshold = 90).
Based on the surfaces created, the software was used to quantify the number of Iba1-, GFAP- or Aβ-positive entities, which provided an estimate of the number of microglia, astrocytes and Aβ co-localizations. The number of entities of microglia, astrocytes and Aβ are referred to herein as “counts”.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software Inc.). Paired t-test analyses (two-tailed unless otherwise indicated, α=0.05) were used to compare equivalent regions of the left (untreated) and right (MRIgFUS-treated) cortex of mice. In cases of randomized ROIs (with varying plaque sizes), and to compare differences between untreated and MRIgFUS-treated mice, unpaired t-tests or U-tests were used (two-tailed, α=0.05). Correlation analyses were performed using linear regression analysis with 95% confidence intervals (α=0.05).
Results
Plaque burden is reduced in cortical brain regions targeted with focused ultrasound
TgCRND8 mice treated with MRIgFUS along the right cortex were sacrificed at 4 days post-treatment. Brains were sectioned and stained for plaques with an anti-Aβ antibody specific for the N-terminus (residues 8–17). Contours were drawn outlining the MRIgFUS-targeted cortical region (Fig. 2A, right) and an equivalent region on the contralateral side (Fig. 2A, left). Plaques were quantified at 40X magnification within each of these contours. After a single treatment, plaque size (Fig. 2B) and total surface area (Fig. 2C) were significantly reduced by 20% and 13%, respectively, in the cortex targeted with MRIgFUS compared to an untreated equivalent cortical region of the left hemisphere (B, *p=0.016, paired t-test; C, **p=0.003, paired t-test, n=9). The number of Aβ plaques in the MRIgFUS-targeted compared to the untreated cortex show a trend of reduction (9%, Fig. 2D, p=0.061, two-tailed paired t-test, n=9) and a one-tailed t-test, appropriate for our hypothesis that MRIgFUS will reduce plaque pathology, reached significance at *p=0.03 (n=9).
Fig. 2.

MRIgFUS treatment causes a reduction in Aβ plaque pathology. Plaque load was assessed 4 days following MRIgFUS treatment using immunohistochemistry and stereology. A, Plaques were quantified in MRIgFUS-treated (right) and an equivalent untreated (left) region of the cortex. Significant reductions were observed in mean plaque size (B) and total Aβ surface area (C) in the MRIgFUS-treated compared to untreated cortex. D, Plaque counts within the MRIgFUS-targeted region demonstrated a trend of reduction compared to the untreated region. Within the MRIgFUS-treated cortex, endogenous IgG (F) and IgM (G), were found to be co-localized with plaques (E). At the site of plaques in the untreated cortex (H), a small amount of IgG (I) but no IgM (J) was found. White arrows indicate the location of Aβ plaques (E–J). Scale bar: A=0.5 mm, E–J = 50 μm. Data is presented as paired values between left, untreated and right, MRIgFUS-treated, *p<0.05, **p<0.01.
To examine the potential contribution of endogenous antibodies, found in the blood, to the MRIgFUS-mediated reduction in Aβ pathology, immunohistochemistry and confocal microscopy were used to determine whether endogenous antibodies could be found decorating Aβ plaques. Analysis of MRIgFUS-targeted cortex revealed that endogenous IgG (Fig. 2E) and IgM (Fig. 2F) were bound to plaques (Fig. 2G, white arrows indicate the location of plaques) at 4 days post-treatment. In the untreated cortex, plaques (Fig. 2H) were decorated only with trace amounts of mouse IgG (Fig. 2I) and not mouse IgM (Fig. 2J).
MRIgFUS-dependent BBB opening allows endogenous immunoglobulin to enter the brain
MRIgFUS is known to have the capacity to deliver systemic IgG antibodies to the brain (Jordão et al., 2010; Kinoshita et al., 2006a, b; Raymond et al., 2008; Sheikov et al., 2004). However, to date MRIgFUS delivery of IgM antibodies has not been investigated. In comparison to monomeric IgGs, IgMs have a molecular weight 6-times larger and can have a greater avidity than IgG for the same antigen. Quantification of the increase of endogenous IgG and IgM in the brain following MRIgFUS treatment is important, for the characterization of the biological effects of this treatment technique, as well as for determining its contribution to the mechanism of Aβ clearance.
Blood was removed from the brain by transcardial perfusion. Using immunohistochemistry, we detected greater levels of endogenous IgG (Fig. 3A–B) and IgM (Fig. 3C–D) in brain regions targeted by MRIgFUS compared to the untreated hemisphere in TgCRND8 and non-Tg mice at 4 days post-MRIgFUS treatment. Quantitative immunoblotting was performed using a separate cohort of TgCRND8 and non-Tg mice treated with MRIgFUS on the right hemisphere and sacrificed 4 days following treatment. IgG levels were significantly increased in the MRIgFUS-treated cortex, compared to untreated, in TgCRND8 (Fig. 3E, 1.8-fold increase, *p=0.047, paired t-test, n=7) and non-Tg mice (Fig. 3F, 1.5-fold increase, *p=0.031, paired t-test, n=6). The increase in IgG levels was not significantly different between MRIgFUS-treated TgCRND8 and non-Tg mice (p=0.73, unpaired t-test). In TgCRND8 and non-Tg mice, the estimated mean concentration of IgG in the MRIgFUS-treated cortex after 4 days was 400 pg/μg of protein.
Fig. 3.

MRIgFUS facilitates the entry of IgG and IgM into the brain. Using immunohistochemistry, high levels of IgG were detected in MRIgFUS-treated compared to untreated cortex of TgCRND8 (A) and non-Tg (B) mice. Similarly, IgM observed in MRIgFUS-treated cortical regions was greater than that detected in contralateral cortex of TgCRND8 (C) and non-Tg mice (D). Quantitative western blot analysis revealed that compared to untreated cortex, the cortex treated with MRIgFUS had greater levels of IgG in TgCRND8 (E) and non-Tg (F) mice; and of IgM in TgCRND8 (G) and non-Tg (H) mice. Scale bar: A–D= 500 μm. Data are presented as the mean ± SEM, *p<0.05.
IgM levels were increased in the cortex targeted by MRIgFUS compared to the untreated contralateral cortex by 4.6-fold in TgCRND8 (Fig. 3G, *p=0.031, paired t-test, n=6) and 6.7-fold in non-Tg mice (Fig. 3H, *p=0.031, paired t-test, n=6). These IgM level increases were not statistically different between MRIgFUS-treated TgCRND8 and non-Tg mice (p=0.39, unpaired t-test, n=6 per group). The estimated mean concentration of IgM present in the targeted cortex of all MRIgFUS-treated mice was 200 pg/μg of protein.
Quantification of IgG and IgM levels 4 hours after MRIgFUS treatment showed consistently higher levels of immunoglobulin in the right, MRIgFUS-targeted cortex compared to the untreated cortex (1.9- and 2.8-fold increase respectively, n=3, data not shown). These data from 4 hours and 4 days post-treatment demonstrate that endogenous immunoglobulins enter the brain following MRIgFUS treatment and remain in the brain for up to 4 days.
IgM levels correlate with MRI detected increases in BBB permeability
To further characterize MRIgFUS-mediated effects on endogenous immunoglobulin delivery through the BBB, we investigated whether the levels of endogenous immunoglobulin present in the brain correlated with the degree of BBB opening following MRIgFUS. As we found no statistical difference in the increase in immunoglobulin between TgCRND8 mice and non-Tg littermates, we combined the groups to examine the relationship between BBB opening and immunoglobulin found in the brain. We used the mean intensities over time to plot the curve for the enhancement (or difference in intensity) seen on the right, MRIgFUS-treated hemisphere over time for each animal (Fig. 4A). From each curve, the maximum enhancement was estimated and represents the maximum amount of gadolinium agent that enters the brain (Fig. 4A, bold red dashed line). To indicate the speed at which gadolinium enters the brain, the rate of enhancement (Fig. 4A, blue circle) was determined at the time it takes to reach half of the maximum enhancement (faded red dashed line). Both the maximum enhancement (Fig. 4B) and rate of enhancement (Fig. 4C) for each mouse was positively correlated with the levels of IgM detected in the brain 4 days post-treatment (*p=0.026 and *p=0.020 respectively, n=12). In contrast, IgG levels did not correlate with rate of enhancement or maximum enhancement (data not shown, p=0.081 and p=0.531 respectively, n=13).
Time-dependent Increases in Glial Activation Markers Occur Following MRIgFUS Treatment
In addition to the delivery of endogenous immunoglobulin, we considered the response of glia cells to MRIgFUS as a possible contributor to the mechanism of Aβ clearance. We treated TgCRND8 mice with MRIgFUS and sacrificed them at 4 hours, 4 days and 15 days post-treatment. Iba1 was used to detect phagocytic microglia and GFAP was used for astrocytes.
As early as 4 hours following MRIgFUS treatment, there appeared to be a slight increase in Iba1-positive staining in the right, MRIgFUS-targeted cortex (Fig. 5A′) compared to the contralateral side (Fig. 5A). The increase in Iba1-positive staining in the MRIgFUS-treated cortex (Fig. 5B′), compared to the untreated cortex (Fig. B), was most apparent at 4 days and it resolved by 15 days (Fig. 5C, C′). Increases in GFAP staining, particularly in the MRIgFUS-treated cortex compared to the untreated cortex were detected at 4 and 15 days, but not at 4 hours (Fig. 5D–F′). These qualitative findings suggest MRIgFUS treatment has an effect on glia, and that Iba1-positive microglia respond more rapidly than GFAP-positive astrocytes. This glial response was found to be similar in the cortex of non-Tg mice treated with MRIgFUS (data not shown).
Fig. 5.

A time-dependent increase in Iba1 and GFAP staining in TgCRND8 mice treated with MRIgFUS. At 4 hours post-MRIgFUS treatment, Iba1 staining appears to be slightly increased in the MRIgFUS-treated cortex (A′) compared to the untreated cortex (A). Iba1-immunostaining intensity appeared to be elevated at 4 days (B, B′) and dampened by 15 days (C, C′). D, D′, GFAP expression did not seem affected by the MRIgFUS treatment at 4 hours. In contrast, at 4 (E, E′) and 15 (F, F′) days post-treatment, GFAP-positive staining was found to be increased on the right, MRIgFUS-targeted side compared to the left, untreated side. Scale bar: A–F′ = 200 μm.
MRIgFUS Induced Protein Expression of Iba1 and GFAP
The relative expression levels of Iba1 and GFAP in MRIgFUS-treated cortex compared to untreated cortex were quantified by western blot in a new cohort of TgCRND8 mice and non-Tg littermates. Shortly after MRIgFUS treatment (4 hours) there was an increase in Iba1 protein levels in TgCRND8 (Fig. 6A, 2.1-fold, *p=0.012, paired t-test, n=6) and non-Tg mice (Fig. 6B, 5.2-fold, *p=0.019, paired t-test, n=6). At 4 days post-treatment, Iba1 levels were 2.8-fold and 2.9-fold greater in the MRIgFUS-treated cortex compared to the untreated cortex of transgenic (Fig. 6C, *p=0.047, paired t-test, n=6) and non-transgenic mice (Fig. 6D, *p=0.032, paired t-test, n=6) respectively. By 15 days after MRIgFUS treatment, targeted cortical regions did not exhibit a significant difference in Iba1 levels compared to untreated regions (TgCRND8: Fig. 6E, p=0.18, paired t-test, n=7; non-Tg: Fig. 6F, p=0.16, paired t-test, n=7). The increases in Iba1 levels were not significantly different between TgCRND8 and non-Tg mice (p>0.05 for all time points, unpaired t-test).
Fig. 6.

Levels of Iba1 and GFAP protein expression in response to MRIgFUS in TgCRND8 and non-transgenic mice. Iba1 protein levels were significantly increased at 4 hours post-treatment in the MRIgFUS-treated cortex (FUS, grey column) relative to the left, untreated cortex (UT, white column) in TgCRND8 (A) and non-Tg (B) mice. At 4 days, Iba1 levels remained elevated in the MRIgFUS-treated cortex of transgenic (C) and non-Tg mice (D). E–F, At 15 days, no statistical difference was found between treated and untreated cortical levels of Iba1. GFAP protein levels were not different between MRIgFUS-treated and untreated cortex at 4 hours (G–H), but they were significantly increased in the MRIgFUS-targeted cortex at 4 days in TgCRND8 (I) and non-Tg (J) mice. At 15 days post-MRIgFUS treatment, TgCRND8 mice had a significant increase in GFAP levels in the MRIgFUS-treated cortex (K), whereas GFAP levels in the MRIgFUS-treated cortex of non-Tg mice were not statistically different from the untreated cortex (L). Mean ± SEM shown, *p<0.05, **p<0.01.
GFAP levels at 4 hours post-treatment were not increased in the MRIgFUS-treated cortex of TgCRND8 (Fig. 2G, p=0.65, paired t-test, n=6) and non-Tg mice (Fig. 6H, p=0.93, paired t-test, n=7), compared to the untreated hemisphere. At 4 days after treatment, GFAP levels were approximately two-fold greater in the MRIgFUS-treated cortex than in the untreated cortex for both TgCRND8 (Fig. 6I, *p=0.043, paired t-test, n=7) and non-Tg mice (Fig. 6J, *p=0.045, paired t-test, n=6). At 15 days, GFAP levels remained significantly elevated in the MRIgFUS-targeted cortex, relative to the untreated cortex, in TgCRND8 mice (Fig. 6K, 2.6-fold, **p=0.006, paired t-test, n=7) but not in non-Tg mice (Fig. 6L, p=0.094, paired t-test, n=6).
In summary, these data indicate that Iba1-positive microglia, but not GFAP-positive astrocytes, are activated 4 hours following MRIgFUS treatment. At 4 days, when plaque burden was reduced, both Iba-1 positive microglia and GFAP-positive astrocytes are activated. By 15 days, activation of microglia has subsided but activation of astrocytes remains elevated in the TgCRND8 mice.
Quantitative Volume and Area Analysis of Glia Surrounding Aβ Plaques
In addition to increases in protein expression, changes in glial cell size can indicate activation of glial cells. To investigate glial cell changes and help elucidate a possible relationship with MRIgFUS-mediated Aβ plaque reduction, we compared the volumes of microglia and astrocytes surrounding Aβ plaques in TgCRND8 mice at 4 days post-MRIgFUS, when plaque load was reduced (Fig. 2). Coronal brain sections were stained for Aβ plaques, microglia and astrocytes (Fig. 7A, plaques in red, microglia in green, astrocytes in blue). Plaques within the cortex were randomly selected for Z-stack imaging with confocal microscopy. Imaris software was used to create a surface structure for plaques, microglia and astrocytes (Fig. 7B). The area immediately surrounding the Aβ plaque (Fig. 7C, dark grey) was labeled as the proximal ROI and the area just further from the plaque was referred to as the distal ROI (Fig. 7C, light grey). Microglia and astrocytes within these two different ROIs were analyzed separately to evaluate possible differences between the MRIgFUS-induced responses of glia that are immediately adjacent to, and at a distance from, Aβ plaques.
Fig. 7.

Iba1- and GFAP-positive cells located distally to plaques increase in size following MRIgFUS treatment. A, Iba1-positive (green) and GFAP-positive (blue) cell volume surrounding plaques (red) were detected by immunohistochemistry. B, Optical sections were obtained using confocal microscopy and used to create three-dimensional reconstructions. C, Aβ plaque volumes were determined using the surface function and then regions of interest (ROIs) were created, one in contact with the plaque (proximal ROI, dark grey) and second, further from the plaque (distal ROI, light grey). The volume of Iba1-positive cells (D) and GFAP-positive cells (E) were significantly increased in the distal ROI of the MRIgFUS-targeted cortex, compared to the untreated cortex. Similarly, the surface area of microglia (G) and astrocytes (H) were also increased in the distal ROI following MRIgFUS treatment. Scale bar: C = 30μm. Mean ± SEM shown, *p<0.05, **p<0.01.
Glia surrounding plaques within MRIgFUS-treated and untreated cortex were analyzed and their volumes were normalized to the volume of their respective ROIs. In the MRIgFUS-treated cortex, compared to the contralateral, the microglia volume distal to plaques was significantly increased, by 65%, in the MRIgFUS-treated cortex compared to the contralateral (Fig. 7D, **p=0.004, Mann-Whitney test, n=42), but was not affected in the proximal ROI (Fig. 7D, p=0.65, Mann-Whitney test, n=42). Similarly, astrocyte volume located proximally to plaques was not significantly changed following MRIgFUS treatment (Fig.7E, p=0.58, Mann-Whitney test, n=42), but did increase in the region distal to plaques by 36% in MRIgFUS-treated cortex (Fig. 7E, *p=0.025, Mann-Whitney test, n=42).
In addition to increases in volume, the three-dimensional glial surface area (normalized to ROI volume) also changed in response to MRIgFUS in ROI distal to plaques. Specifically, microglia surface area was elevated by 29% in the distal ROI (Fig. 7F, **p=0.004, Mann-Whitney test, n=42) while it remained not significantly affected by MRIgFUS in the proximal ROI (Fig. 7F, p=0.77, Mann-Whitney test, n=42). Astrocytes also responded to MRIgFUS treatment in the cortex by a 50% increase in surface area in the distal ROI (Fig. 7G, *p=0.014, Mann-Whitney test, n=42). The surface area of astrocytes was not changed by MRIgFUS treatment in the proximal ROI (Fig. 7G, p=0.36, Mann-Whitney test, n=42). The observed increases in microglial and astrocyte volume and surface area provide evidence that MRIgFUS treatment is activating glial cells within brain-targeted regions.
Glia Count is Not Affected by MRIgFUS Treatment
Increases in total glial volume and surface area may be due to either the increases of resident cells or an increase in the number of glial cells in response to MRIgFUS. To differentiate these possibilities, we estimated the number of Iba1-positive and GFAP-positive cells within each ROI in MRIgFUS-treated and untreated cortices. We found that there was no statistical difference in the number of Iba1-positive counts for the proximal and distal ROIs (Fig. 8A, p=0.61 and p=0.83 respectively, Mann-Whitney test, n=36). Similarly, there was no difference in GFAP-positive counts in the proximal and distal ROIs of the MRIgFUS-treated hemisphere compared to the contralateral (Fig. 8B, p=0.99 and p=0.22 respectively, Mann-Whitney test, n=36). These data indicate that the increase in glia volume and surface is not due to an increase in cell number.
Fig. 8.

MRIgFUS treatment does not change the number of glial cells but increases their volume per cell. Representative images of microglia (A) and astrocytes (B) found in the proximal (dark grey) and distal (light grey) ROIs surrounding a single plaque in the MRIgFUS-treated cortex of a TgCRND8 mouse are shown. The number of Iba1- and GFAP-positive counts were not different between MRIgFUS-treated and untreated cortex (C, D respectively). E, The volume per Iba1-positive cell count, however, was increased significantly in the distal ROI of plaques in the MRIgFUS-targeted cortex compared to the untreated cortex. F, GFAP-positive volume per cell was significantly increased both in proximal and distal ROIs within the cortex treated with MRIgFUS. Mean ± SEM shown, *p<0.05, **p<0.01, ****p<0.0001.
To further characterize glial cell volume, we quantified the volume per glial-positive count. There was no difference between Iba1-positive volume per count in the MRIgFUS-treated region compared to the untreated region proximal to the plaque (Fig. 8C, p=0.91, Mann-Whitney test, n=36). In contrast, there was a 40% increase in Iba1-positive volume per count distal to plaques in MRIgFUS-targeted areas (Fig. 8C, *p=0.016, Mann-Whitney test, n=36). GFAP-positive volume per count was increased by 113% in the proximal ROI and 230% in the distal ROI of the MRIgFUS-treated cortex (Fig. 8D, **p=0.001 and ****p<0.0001 respectively, Mann-Whitney test, n=36).
In summary, microglial volume per cell count was elevated in the ROI distal to plaques in the MRIgFUS-treated cortex. MRIgFUS did not significantly increase the microglia volume per count in the proximal ROI, where the population density of microglia is typically greater. Astrocyte volume per count was increased both proximal and distal to plaques. Astrocytes in the distal ROI exhibited a greater increase in volume compared to astrocytes in the proximal ROI in response to MRIgFUS.
Greater Aβ Co-localization in Glial Cells of the MRIgFUS-Treated Cortex
Finally, we investigated the co-localization of Aβ with microglia and astrocytes, as an indicator of Aβ internalization by glial cells. Aβ was found within microglia (Fig. 9A) and astrocytes (Fig. 9B) of TgCRND8 mice in the untreated and MRIgFUS-treated cortex. In order to evaluate how activation of glia by MRIgFUS treatment may contribute to Aβ plaque reduction by MRIgFUS treatment alone, Aβ within glia in the proximal and distal ROIs were identified (Fig. 9C) and the number of Aβ-positive counts within each glial cell were quantified (Fig. 9D, representative internalized Aβ within a single microglial cell). Aβ counts within microglia proximally located to plaques was 52% greater in the MRIgFUS-treated cortex, compared to the untreated cortex (Fig. 9E, *p=0.036, Mann-Whitney test, n=36). Microglia found distal to plaques in the MRIgFUS-treated cortex contained 159% more internalized Aβ counts than those in the untreated cortex (Fig. 9E, ***p=0.0006, Mann-Whitney test, n=36).
Fig. 9.

Aβ internalization by Iba1- and GFAP-positive cells is increased in MRIgFUS-treated cortex. Brain sections were stained for Aβ (red), Iba1 (green) and GFAP (blue). Orthogonal views of confocal imaging show Aβ within Iba1-positive (A, representative Iba1-Aβ co-localization encircled) and GFAP-positive cells (B, representative GFAP-Aβ co-localization encircled) indicating internalization of Aβ at 4 days following MRIgFUS treatment. Imaris software was used to create three-dimensional surfaces of glial cells and detect Aβ within the glia found proximal (Aβ in red) and distal (Aβ in purple) to plaques (C, Aβ within a single cell shown in D). E, Aβ counts within Iba1-positive cells was greater within MRIgFUS-treated cortex in both proximal and distal regions surrounding plaques. F, Similarly, GFAP-positive cells within the MRIgFUS-treated cortex also contained a greater number of Aβ counts than those in the untreated, contralateral cortex. In addition, the total volume of internalized Aβ was greater in Iba1-positive (G) and GFAP-positive (H) cells within MRIgFUS-treated regions, proximal and distal to plaques. Scale bar: A–B = 25 μm. Mean ± SEM shown, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Although not the predominant phagocytes in the brain, astrocytes can also internalize Aβ and following MRIgFUS treatment, astrocytes within the proximal ROI had 79% increase of internalized Aβ counts (Fig. 9F, *p=0.019, Mann-Whitney test, n=36). Astrocytes distal to plaques in the MRIgFUS-treated cortex showed an even greater increase in Aβ counts by 270% compared to the distal astrocytes in untreated cortex (Fig. 9F, ****p<0.0001, Mann-Whitney test, n=36).
In addition to Aβ count, we also quantified the total Aβ volume present within glia. The total volume of Aβ found in microglia in the MRIgFUS-treated cortex compared to the untreated cortex was increased by 239% in the proximal and 386% in the distal ROI (Fig. 9G, ****p<0.0001 for both proximal and distal ROIs, Mann-Whitney test, n=36). Astrocytes proximal to plaques in the MRIgFUS-treated cortex contained 584% greater volume of Aβ than those in the untreated cortex (Fig. 9H, **p=0.006, Mann-Whitney test, n=36). Astrocytes distal to plaques internalized an Aβ volume 414% greater in the MRIgFUS-targeted hemisphere than in the contralateral hemisphere (Fig. 9H, ****p<0.0001, Mann-Whitney test, n=36).
These analyses demonstrate that MRIgFUS has the potential to increase Aβ phagocytosis by microglia and astrocytes in the MRIgFUS-treated cortex proximal and distal to Aβ plaques.
Discussion
MRIgFUS is a promising strategy to enhance the delivery of therapeutics to the brain for treatment of neurological disorders (Burgess et al., 2012; Burgess et al., 2011b; Huang et al., 2012; Jordão et al., 2010; Kinoshita et al., 2006b; Raymond et al., 2008; Thévenot et al., 2012; Treat et al., 2007). Although disrupting the BBB can allow potentially harmful cells or toxins into the brain (Miller, 2002), unlike osmotic or chemical BBB disruption, MRIgFUS can target permeabilization to relatively small areas of the BBB at a time (Burgess et al., 2011b; Jordão et al., 2010; Thévenot et al., 2012). Additionally, in a previous study, we demonstrated that by maintaining an enhancement of < 20% prevents the extravasation of red blood cells (Jordao et al., 2010) and by extension, also other similarly-sized blood-borne cells. The application of MRIgFUS modifies the BBB in a reversible and controlled manner (Burgess et al., 2011a; O’Reilly and Hynynen, 2012). The MRIgFUS parameters used here are below the threshold of damage and are not likely to cause acidophilia or neuronal atrophy (Hynynen et al., 2003; Hynynen et al., 2005; Kinoshita et al., 2006a). Ultrasound technology is being adapted to propagate safely through the skull, with controlled effects in several species, including non-human primates and humans (McDannold et al., 2012; McDannold et al., 2010). From these studies, MRI and post-mortem histology confirmed that brain integrity was preserved and no adverse effects were observed in behavior of treated animals or humans.
These previous studies are very promising. Nonetheless, further investigations are required to discover potential mechanisms, and establish safety and efficacy of MRIgFUS treatments. Here, we found that MRIgFUS itself, without the administration of exogenous therapeutics, reduces Aβ pathology, facilitates the entry of endogenous antibodies and activates glial cells in the brain. We propose that antibodies invading the MRIgFUS-targeted area contribute to plaque reduction. Furthermore, Aβ was found to colocalize with activated microglia and astrocytes, suggesting that these glial cells can internalize Aβ and facilitate its clearance.
In a previous study, we have shown that the delivery of peripherally-administered, exogenous anti-Aβ IgG antibody, BAM-10, to MRIgFUS-targeted brain areas significantly reduced plaque number (12%), mean plaque size (12%) and total Aβ surface area (23%) in TgCRND8 mice within four days (Jordão et al., 2010). Here, using the same MRIgFUS parameters, but without the intravenous supplementation of exogenous antibodies, we found that MRIgFUS alone significantly reduces mean plaque size (20%) and total Aβ surface area (13%) within four days. A trend of 9% reduction in plaque number was noted in MRIgFUS-targeted cortex. Both studies may involve similar mechanisms of Aβ clearance, although the peripheral sink mechanism, which employs intravenously-administered antibodies to draw Aβ out of the brain (DeMattos et al., 2001), is likely to play a greater role in the previous study where antibodies were delivered to the bloodstream, in comparison to MRIgFUS treatment alone. In addition, considering the data obtained in the current study, the reduction in plaque pathology observed in Jordão et al. (2010) likely represents the combined effects of exogenous BAM-10 and endogenous antibodies being delivered to the brain by MRIgFUS. Nonetheless, the effects of MRIgFUS on amelioration of plaque pathology emphasizes the benefit of using MRIgFUS as a delivery method to facilitate anti-Aβ treatment. However, this effect is modest and complementing MRIgFUS treatment with the intravenous administration of anti-Aβ antibodies may be required for treatment efficacy that is therapeutically relevant,
The finding that MRIgFUS alone reduces plaque load is unprecedented and therefore the mechanism by which this occurs has not previously been explored. In this study, we explored two probable contributing factors, namely the delivery of blood-borne endogenous antibodies to the brain and glial activation.
Increased levels of endogenous immunoglobulin in the cortex of TgCRND8 mice treated with MRIgFUS were observed within the same region and time-frame that decreased Aβ deposition was found. Previous studies have shown that small amounts of systemic IgG can cross the BBB (Banks et al., 2002; Bard et al., 2000; Deane et al., 2005). Due to their larger size, IgM are less likely to cross the BBB (Sigurdsson et al., 2004). Our data are in support of greater amounts of IgG entering the brain compared to IgM, with increased bioavailability to targeted brain regions following MRIgFUS treatment. The co-localization of endogenous mouse IgG and IgM with plaques in the MRIgFUS-treated cortex suggests that these delivered endogenous antibodies facilitate plaque reduction.
The demonstration of a correlation between MRI enhancement and the levels of endogenous IgM entering the brain is relevant to the application of MRIgFUS as a method of drug delivery to the brain in general, and in cases of immunotherapy for AD in particular. Previous BBB permeability studies have been conducted indicating size limits of molecules that can enter the brain following FUS treatment using dextrans of different molecular weights (Choi et al., 2011), HRP and IgG (Sheikov et al., 2004). However, size is not the only variable determining passage across the BBB, and delivery across the BBB following MRIgFUS may occur by more than one mechanism, including both paracellular and transcellular routes (Cho et al., 2011; Deng et al., 2012; Hynynen et al., 2005; Sheikov et al., 2008; Sheikov et al., 2004). This highlights the benefit of determining MRIgFUS delivery of different molecules empirically. Future studies evaluating correlations between enhancement and the amount of therapeutics entering the brain will be helpful to establish the use of MRI data during FUS treatment to estimate how much of a given drug enters specific brain regions. Based on this study, enhancement data correlates best with the entry of molecules that are above the threshold of bypassing the BBB, such as IgM. After MRIgFUS is applied, the BBB continuously diminishes in permeability and returns to its normal state of homeostasis within about 6 hours (Sheikov et al., 2004). Hence, larger molecules (such as IgM compared to IgG) are likely to be more affected by this change over time and their concentration will be more easily predicted by enhancement data.
Antibodies entering the brain can contribute to Aβ clearance by solubilization of Aβ (Bard et al., 2000; McLaurin et al., 2002; Solomon et al., 1997), facilitating transport of Aβ:antibody complexes (Deane et al., 2005), opsonizing Aβ for microglial phagocytosis (Bard et al., 2000; Wilcock et al., 2003), or a combination of the aforementioned mechanisms (Wilcock et al., 2003; Wilcock et al., 2004). In the current study, to gain a better understanding of MRIgFUS effects in both diseased and non-diseased cases, we characterized the response of microglia and astrocytes to MRIgFUS treatment in TgCRND8 and non-Tg mice, and we examined the potential involvement of glia in MRIgFUS-mediated Aβ plaque reduction in TgCRND8 mice.
We found that MRIgFUS increased the immuno-positive signal and protein expression of glial cell markers, specifically Iba1 for microglia and GFAP for astrocytes, in both TgCRND8 and non-Tg mice. Although the level of glial activation in untreated AD mice is already elevated in comparison to non-Tg mice (Chen et al., 1998; Chishti et al., 2001; Frautschy et al., 1998; Masliah et al., 1996), the overall increase in Iba1 and GFAP protein expression, due to MRIgFUS treatment, was similar in transgenic and non-transgenic mice compared to their respective basal levels. MRIgFUS activation of astrocytes may occur due to effects on astrocytic endfeet in contact with the BBB during disruption or by an imbalance in water or ions resulting from a breach in the BBB. Disruption of the BBB is also known to cause activation of nearby microglia, which can be stimulated by serum products such as fibrinogen (Adams et al., 2007; Ransohoff and Perry, 2009). The entry of endogenous immunoglobulin into brain can also activate microglia, as expected based on immunotherapy studies (Bard et al., 2000; Magga et al., 2010; Wilcock et al., 2003).
The volume and surface area of glial cells in the brain of MRIgFUS-treated TgCRND8 mice was increased, with no apparent change in glia counts. These data suggest that MRIgFUS enhances the activation of glial cells within targeted brain regions without leading to glial cell proliferation or recruitment to the treatment site. By 15 days following MRIgFUS, astrocyte activation was lower compared to 4 days and microglia activation had subsided to untreated levels. Our findings indicate that activation of glial cells by MRIgFUS is likely to be transient.
Activation of glial cells at 4 days following MRIgFUS treatment, when Aβ plaque load was reduced, was accompanied by a significant increase in the amount of Aβ found within glial cells in the MRIgFUS-targeted region. These findings are consistent with a role for microglia and astrocytes in the internalization and clearance of Aβ. Counts and total volume of Aβ were increased within microglia in close contact with, and distal to, Aβ plaques. These data agree with other studies demonstrating microglia phagocytosis of fibrillar Aβ can occur as early as a few days following immunization (Bacskai et al., 2001; Schenk et al., 1999; Wilcock et al., 2003).
The contribution of astrocytes to Aβ clearance is often considered negligible compared to that of microglia (Nicoll et al., 2006). In the present study, we found that although microglia contained a greater volume of Aβ, astrocytes in MRIgFUS-treated regions demonstrated a significant increase in Aβ uptake compared to astrocytes in the untreated cortex. These data suggested that astrocytes could also play an important role in Aβ clearance following MRIgFUS treatment. Activation of astrocytes due to MRIgFUS-microbubble effects at the cerebrovascular level may stimulate these cells to internalize Aβ. Alternatively, the delivery of endogenous immunoglobulin may opsonize Aβ and stimulate Aβ phagocytosis.
Clearance of Aβ is greatly impaired during the course of AD and this could be related to a dysfunction of glial cells occurring early in the progression of the disease (Yeh et al., 2011). Additionally, proteins associated with Aβ may contribute to the impairment of glial cells to internalize Aβ (Familian et al., 2007; Takata et al., 2012), rendering clearance of Aβ more difficult. In the current study, analysis of glial cells in regions of close proximity and distal to plaques demonstrated that distal glial cells have greater potential to increase in size and to take up Aβ after treatment. Under a different treatment paradigm, microglia in non-plaque areas of TgCRND8 mice demonstrated increased uptake of Aβ (Hawkes et al., 2012). Furthermore, glia in close proximity to plaques are already activated and may not have the capacity to respond further, whereas glia further from plaques may show greater changes in activation due to lower initial activation levels.
Conclusions
Our study demonstrates that Aβ plaque size is significantly reduced within 4 days of treating TgCRND8 mice, using MRIgFUS as the only external therapeutic intervention. At the same time, MRIgFUS increases endogenous immunoglobulin levels and glial activation. The decoration of Aβ plaques with IgG and IgM in the MRIgFUS-treated cortex, when plaque load was reduced, suggests a potential role for endogenous antibodies in the mechanism of MRIgFUS-mediated amelioration of Aβ pathology. Furthermore, activated glial cells in the MRIgFUS-treated cortex contained increased amounts of Aβ, indicating that glial cells may contribute to Aβ uptake and clearance following MRIgFUS treatment. These possible mechanisms of MRIgFUS-induced Aβ plaque reduction emphasize the advantage of this delivery strategy for AD, and for other neurological disorders benefiting from treatment with endogenous antibodies or the activation of glial cells.
Highlights.
Focused ultrasound alone reduces amyloid-β plaque pathology 4 days post-treatment
Host immunoglobulins enter the ultrasound-treated cortex where they bind to plaques
Microglia and astrocyte markers are increased in the ultrasound-treated cortex
Volumes of microglia and astrocytes are increased in the ultrasound-treated cortex
Aβ within microglia and astrocytes is increased in the ultrasound-treated cortex
Acknowledgments
We thank Drs. Paul Fraser, David Westaway, and Peter St George-Hyslop for their contributions in creating the TgCRND8 mice. The authors are grateful to Dr. Stefanovic for the use of Imaris software used for the glial analysis. We are also grateful to Nicholas Ellens and Ping Wu for their technical assistance with MRIgFUS experiments. Shawna Rideout-Gros, Alex Garces, and Stephanie Bell helped with animal care. We thank Rosemary Ahrens and Mary Hill for genotyping and animal care. Funding was provided by Ontario Mental Health Foundation (IA), National Institutes of Health (KH), and Canadian Institutes of Health Research (JFJ, IA: FRN 93603, JM: FRN 10246).
Footnotes
Disclosure Statement
Dr. Kullervo Hynynen has stock in FUS Instruments, from which he receives non-study related support.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams RA, et al. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–582. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides. 2002;23:2223–2226. doi: 10.1016/s0196-9781(02)00261-9. [DOI] [PubMed] [Google Scholar]
- Bard F, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Burgess A, Aubert I, Hynynen K. Focused ultrasound: crossing barriers to treat Alzheimer’s disease. Therapeutic Delivery. 2011a;2:281–286. doi: 10.4155/tde.11.5. [DOI] [PubMed] [Google Scholar]
- Burgess A, Huang Y, Querbes W, Sah DW, Hynynen K. Focused ultrasound for targeted delivery of siRNA and efficient knockdown of Htt expression. J Control Release. 2012;163:125–129. doi: 10.1016/j.jconrel.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Ayala-Grosso CA, Ganguly M, Jordão JF, Aubert I, Hynynen K. Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PLoS One. 2011b;6:e27877. doi: 10.1371/journal.pone.0027877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KS, et al. Neurodegenerative Alzheimer-like pathology in PDAPP 717V-->F transgenic mice. Prog Brain Res. 1998;117:327–334. [PubMed] [Google Scholar]
- Chishti MA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Cho EE, Drazic J, Ganguly M, Stefanovic B, Hynynen K. Two-photon fluorescence microscopy study of cerebrovascular dynamics in ultrasound-induced blood-brain barrier opening. J Cereb Blood Flow Metab. 2011;31:1852–1862. doi: 10.1038/jcbfm.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JJ, Selert K, Gao Z, Samiotaki G, Baseri B, Konofagou EE. Noninvasive and localized blood-brain barrier disruption using focused ultrasound can be achieved at short pulse lengths and low pulse repetition frequencies. J Cereb Blood Flow Metab. 2011;31:725–737. doi: 10.1038/jcbfm.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra R, Curiel L, Staruch R, Morrison L, Hynynen K. An MRI-compatible system for focused ultrasound experiments in small animal models. Med Phys. 2009;36:1867–1874. doi: 10.1118/1.3115680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, et al. IgG-assisted age-dependent clearance of Alzheimer’s amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;25:11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, et al. The role of caveolin-1 in blood-brain barrier disruption induced by focused ultrasound combined with microbubbles. J Mol Neurosci. 2012;46:677–687. doi: 10.1007/s12031-011-9629-9. [DOI] [PubMed] [Google Scholar]
- Dodel R, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer’s disease. Ann Neurol. 2002;52:253–256. doi: 10.1002/ana.10253. [DOI] [PubMed] [Google Scholar]
- Du Y, et al. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- Familian A, Eikelenboom P, Veerhuis R. Minocycline does not affect amyloid beta phagocytosis by human microglial cells. Neurosci Lett. 2007;416:87–91. doi: 10.1016/j.neulet.2007.01.052. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, Deng L, Fenili D, Nitz M, McLaurin J. In Vivo Uptake of beta-Amyloid by Non-Plaque Associated Microglia. Curr Alzheimer Res. 2012;9:890–901. doi: 10.2174/156720512803251084. [DOI] [PubMed] [Google Scholar]
- Huang Q, Deng J, Wang F, Chen S, Liu Y, Wang Z, Cheng Y. Targeted gene delivery to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Exp Neurol. 2012;233:350–356. doi: 10.1016/j.expneurol.2011.10.027. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R. Autoantibodies to amyloid-beta and Alzheimer’s disease. Ann Neurol. 2001;49:808–810. doi: 10.1002/ana.1061. [DOI] [PubMed] [Google Scholar]
- Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology. 2001;220:640–646. doi: 10.1148/radiol.2202001804. [DOI] [PubMed] [Google Scholar]
- Hynynen K, McDannold N, Martin H, Jolesz FA, Vykhodtseva N. The threshold for brain damage in rabbits induced by bursts of ultrasound in the presence of an ultrasound contrast agent (Optison) Ultrasound Med Biol. 2003;29:473–481. doi: 10.1016/s0301-5629(02)00741-x. [DOI] [PubMed] [Google Scholar]
- Hynynen K, McDannold N, Sheikov NA, Jolesz FA, Vykhodtseva N. Local and reversible blood-brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage. 2005;24:12–20. doi: 10.1016/j.neuroimage.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jordao JF, et al. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-beta plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLoS One. 2010;5:e10549. doi: 10.1371/journal.pone.0010549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordão JF, et al. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-beta plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PLoS One. 2010;5:e10549. doi: 10.1371/journal.pone.0010549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Ergin A, Wang M, Reif R, Zhang J, Bruce JN, Bigio IJ. Inconsistent blood brain barrier disruption by intraarterial mannitol in rabbits: implications for chemotherapy. J Neurooncol. 2010;104:11–19. doi: 10.1007/s11060-010-0466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proc Natl Acad Sci U S A. 2006a;103:11719–11723. doi: 10.1073/pnas.0604318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Targeted delivery of antibodies through the blood-brain barrier by MRI-guided focused ultrasound. Biochem Biophys Res Commun. 2006b;340:1085–1090. doi: 10.1016/j.bbrc.2005.12.112. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, et al. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotilinek LA, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magga J, et al. Human intravenous immunoglobulin provides protection against Abeta toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer’s disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDannold N, Vykhodtseva N, Hynynen K. Targeted disruption of the blood-brain barrier with focused ultrasound: association with cavitation activity. Phys Med Biol. 2006;51:793–807. doi: 10.1088/0031-9155/51/4/003. [DOI] [PubMed] [Google Scholar]
- McDannold N, Arvanitis CD, Vykhodtseva N, Livingstone MS. Temporary disruption of the blood-brain barrier by use of ultrasound and microbubbles: safety and efficacy evaluation in rhesus macaques. Cancer Res. 2012;72:3652–3663. doi: 10.1158/0008-5472.CAN-12-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDannold N, Vykhodtseva N, Raymond S, Jolesz FA, Hynynen K. MRI-guided targeted blood-brain barrier disruption with focused ultrasound: histological findings in rabbits. Ultrasound Med Biol. 2005;31:1527–1537. doi: 10.1016/j.ultrasmedbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- McDannold N, Clement GT, Black P, Jolesz F, Hynynen K. Transcranial magnetic resonance imaging- guided focused ultrasound surgery of brain tumors: initial findings in 3 patients. Neurosurgery. 2010;66:323–332. doi: 10.1227/01.NEU.0000360379.95800.2F. discussion 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin J, et al. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- Miller G. Drug targeting. Breaking down barriers. Science. 2002;297:1116–1118. doi: 10.1126/science.297.5584.1116. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- O’Reilly MA, Hynynen K. Ultrasound enhanced drug delivery to the brain and central nervous system. Int J Hyperthermia. 2012;28:386–396. doi: 10.3109/02656736.2012.666709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly MA, Waspe AC, Ganguly M, Hynynen K. Focused-ultrasound disruption of the blood-brain barrier using closely-timed short pulses: influence of sonication parameters and injection rate. Ultrasound Med Biol. 2011;37:587–594. doi: 10.1016/j.ultrasmedbio.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs. 2009;23:35–58. doi: 10.2165/0023210-200923010-00003. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Raymond SB, Treat LH, Dewey JD, McDannold NJ, Hynynen K, Bacskai BJ. Ultrasound enhanced delivery of molecular imaging and therapeutic agents in Alzheimer’s disease mouse models. PLoS One. 2008;3:e2175. doi: 10.1371/journal.pone.0002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahuddin TS, Johansson BB, Kalimo H, Olsson Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. An electron microscopic study. Acta Neuropathol. 1988;77:5–13. doi: 10.1007/BF00688236. [DOI] [PubMed] [Google Scholar]
- Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Sheikov N, McDannold N, Sharma S, Hynynen K. Effect of focused ultrasound applied with an ultrasound contrast agent on the tight junctional integrity of the brain microvascular endothelium. Ultrasound Med Biol. 2008;34:1093–1104. doi: 10.1016/j.ultrasmedbio.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikov N, McDannold N, Vykhodtseva N, Jolesz F, Hynynen K. Cellular mechanisms of the blood-brain barrier opening induced by ultrasound in presence of microbubbles. Ultrasound Med Biol. 2004;30:979–989. doi: 10.1016/j.ultrasmedbio.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, et al. An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-beta derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, et al. Microglial Amyloid-beta1-40 Phagocytosis Dysfunction Is Caused by High-Mobility Group Box Protein-1: Implications for the Pathological Progression of Alzheimer’s Disease. Int J Alzheimers Dis. 2012;2012:685739. doi: 10.1155/2012/685739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenot E, et al. Targeted Delivery of Self-Complementary Adeno-Associated Virus Serotype 9 to the Brain, Using Magnetic Resonance Imaging-Guided Focused Ultrasound. Hum Gene Ther. 2012 doi: 10.1089/hum.2012.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treat LH, McDannold N, Vykhodtseva N, Zhang Y, Tam K, Hynynen K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int J Cancer. 2007;121:901–907. doi: 10.1002/ijc.22732. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, et al. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CY, Vadhwana B, Verkhratsky A, Rodriguez JJ. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro. 2011;3:271–279. doi: 10.1042/AN20110025. [DOI] [PMC free article] [PubMed] [Google Scholar]