

Abstract
Purpose
To investigate the multimodal morphological features in the different stages of Best vitelliform macular dystrophy (VMD) in subjects harboring mutations in the BEST1 gene, and their changes during the progression of the disease.
Methods
In this retrospective observational study performed between January 2007 and December 2012, 21 patients (42 eyes) with Best VMD from eight families with the BEST1 mutation were included. Best-corrected visual acuity (BCVA), fundus autofluorescence (FAF), and spectral domain optical coherence tomography (SDOCT) were evaluated at study entry and at last visit.
Results
The mean age of patients was 26.3±17.4 years. Seven new missense mutations in BEST1 were identified. Mean follow-up was 41.1±18.5 months. Mean BCVA was 0.34±0.34 LogMAR at study entry and 0.32±0.33 LogMAR at last follow-up visit (p = 0.2). The overall lesion area on FAF increased from 6.62±4.9 mm2 to 7.34±6.1 mm2 (p = 0.05). At study entry, on SD-OCT, photoreceptor inner segment ellipsoid portion (ellipsoid zone, EZ) was normal in 15 eyes, disrupted in 14 eyes, and absent in 13 eyes. In two eyes, EZ changed from normal to disrupted during follow-up. Three eyes of three patients showing pseudohypopyon lesions at study entry progressed to vitelliruptive lesions at the last follow-up visit. Three eyes of three patients showing vitelliruptive lesion at study entry reverted to pseudohypopyon lesion with overall enlargement of the lesion size.
Conclusions
Multimodal analysis allowed documenting a continuous material accumulation and reabsorption in Best VMD progression. Blue FAF and SD-OCT could represent noninvasive imaging techniques to monitor Best VMD.
Introduction
Vitelliform macular dystrophy (VMD; OMIM #153700), also called Best disease [1], is a clinically heterogeneous and pleomorphic disease, in most cases showing an autosomal dominant pattern of inheritance with extremely variable penetrance and expressivity. BEST1 (chromosome 11q12-q13) [2], the only gene virtually involved in all dominant Best VMD cases, encodes a 68 kDa protein called bestrophin-1 [3] that is localized to the basolateral plasma membrane of the retinal pigment epithelium (RPE) and appears to exhibit properties of Ca2+-activated Cl− channels [4]. Recently, several diseases have been linked to mutations in the BEST1 gene, including autosomal recessive VMD, autosomal recessive bestrophinopathy, adult-onset foveomacular vitelliform dystrophy, autosomal dominant vitreoretinochoroidopathy, and retinitis pigmentosa [5-11].
Autosomal dominant Best VMD has a bimodal onset distribution with one maximum peak before puberty and a second following puberty and extending through the fifth decade of life [12,13]. The previtelliform stage represents the first of five progressive stages defined based on fundus examination, and is characterized by absence of symptoms and normal macula or subtle RPE alterations [14,15]. The second stage of Best VMD, the vitelliform stage, is characterized by a well-circumscribed 0.5- to 2-disc-diameter “egg-yolk” lesion within the macula, and by symptoms of metamorphopsia, blurred vision, and a decrease of central vision [14,15]. This disease stage is followed by the third stage, which involves pseudohypopyon (the yellow material accumulated inferiorly), the vitelliruptive fourth stage (partial resorption of the material, scrambled-egg lesion) and the atrophic/fibrotic fifth stage (final macular atrophy or fibrosis) [14,15].
With the advent of optical coherence tomography (OCT), it has been possible to anatomically examine the vitelliform lesion in vivo. We recently reported on high-definition spectral-domain (SD)-OCT findings in all of the progressive stages of the disease, including the previtelliform stage [15], and proposed that early changes in Best VMD may involve the layer between the RPE and the inner segment and outer segment (IS/OS) interface, first with accumulation of material beneath the sensory retina, and then with disruption and loss of the IS and OS. The RPE is also affected in the disease course, with hypertrophy, disruption, and attenuation [15]. These findings are in agreement with a knock-in mouse model of Best VMD revealing accumulation of lipofuscin in the RPE and debris thought to be unphagocytosed photoreceptor OS and lipofuscin granules in the subretinal space [16]. Unfortunately, to date, no histopathological analyses have sampled the vitelliform lesion because patients examined postmortem had largely progressed beyond this stage. In fact, histopathological findings in Best VMD donor eyes only revealed abundant accumulation of lipofuscin in the RPE [17,18], and photoreceptor degeneration over a morphologically intact RPE layer [19,20]. For these reasons, in vivo OCT imaging currently represents the best way to assess the morphological changes in the different stages of Best VMD with a view to gaining insight into the physiopathology of disease progression.
Interestingly, histopathological findings suggest that Best VMD may affect a more diffuse region than that of the ophthalmoscopically visible lesion, as extrafoveal diffuse macular accumulation of lipofuscin has been observed in the RPE [18,19,21]. Blue fundus autofluorescence (FAF) is a useful tool to assess the extent of lipofuscin accumulation in the RPE in vivo [22-25]. However, in Best VMD, macular autofluorescence may originate not only from lipofuscin accumulation in the RPE, but also from debris (unphagocytosed photoreceptor OS) and lipofuscin accumulation in the subretinal space [26]. In this context, when associated with FAF imaging, OCT imaging may be extremely useful in determining the origin of macular autofluorescence in the different stages of Best VMD, together with the changes over time, by showing accumulation of material either in the RPE or in the subretinal space.
In this study, we investigated the multimodal morphological features of the different stages of Best VMD in subjects harboring mutations in BEST1 gene, and their changes during the progression of the disease. This analysis will give insights into the physiopathology of the disease by showing how the vitelliform material accumulates and reabsorbs, accompanied by changes in the outer and inner retinal layers.
Methods
We longitudinally reviewed FAF and SD-OCT images of 21 Best VMD subjects from eight families with BEST1 mutation followed at the Créteil University Eye Clinic between January 2007 and December 2012. At least one subject from each family was diagnosed with one of the progressive stages of Best VMD, on the basis of fundus examination and electrooculography, by two observers (GQ, EHS) [14,15,27-29]. In each family, affected subjects were screened for the BEST1 mutation identified in the Best VMD proband by exome sequencing [27-29]. Informed consent was obtained according to an approved protocol of the Paris Est Créteil University Institutional Review Board, in agreement with the Declaration of Helsinki. All patients underwent multiple ophthalmological examinations during follow-up, which included assessment of best-corrected visual acuity (BCVA) measured at 4 m with standard Early Treatment Diabetic Retinopathy Study charts, fundus biomicroscopy, blue FAF (Spectralis HRA+OCT, Heidelberg Engineering, Heidelberg, Germany), and SD-OCT (Spectralis HRA+OCT, Heidelberg Engineering).
Macular FAF findings and outer retinal layer structures observed in SD-OCT images were analyzed, interpreted, and measured independently GQ and EHS. The following features were recorded for each follow-up visit (from study entry to last follow-up visit): 1) stage of the disease [a) previtelliform stage; b) vitelliform stage; c) pseudohypopyon stage; d) vitelliruptive stage; e) atrophic or fibrotic stage] [14,15], as evaluated based on fundus biomicroscopy, FAF, and SD-OCT; 2) macular blue FAF features (hyperautofluorescence, hypoautofluorescence, or isoautofluorescence); 3) overall lesion area on FAF images measured using the Heidelberg software (Spectralis Acquisition and Viewing Modules; version 5.6.1.0, Heidelberg Engineering; Figure 1); 4) size of the inhomogeneously hyperautofluorescent and hypoautofluorescent components (areas) of the lesion; 5) status of the inner segment ellipsoid portion (ellipsoid zone, EZ), also known as the photoreceptor IS/OS interface [30,31] [a) almost normal—no detectable EZ changes; b) disrupted; c) absent] on central-fovea (within 500 μm from the foveal depression) SD-OCT scan; 6) reflectivity of the lesion [a) hyperreflective; b) hyporeflective; c) mixed hyperreflective/hyporeflective] as evaluated based on different SD-OCT scans (i.e. central-fovea, superior, and inferior horizontal scans, ± vertical scans passing through the fovea); 7) central macular thickness (CMT) measured using a 19-horizontal-lines protocol (6×6-mm area), each consisting of 1,024 A-scans per line (Spectralis Acquisition and Viewing Modules; version 5.6.1.0, Heidelberg Engineering); 8) thickness of the neurosensory retina at the fovea and maximal thickness and width of the lesion, measured using the caliper provided with Spectralis SD-OCT software (Spectralis Acquisition and Viewing Modules; version 5.6.1.0, Heidelberg Engineering). The values of the measurements were averaged for analysis. In case of disagreement regarding interpretation of the different features, or if the difference in the two readers’ (GQ, EHS) measurements was greater than 15% of the mean of two values, there was open adjudication.
Figure 1.
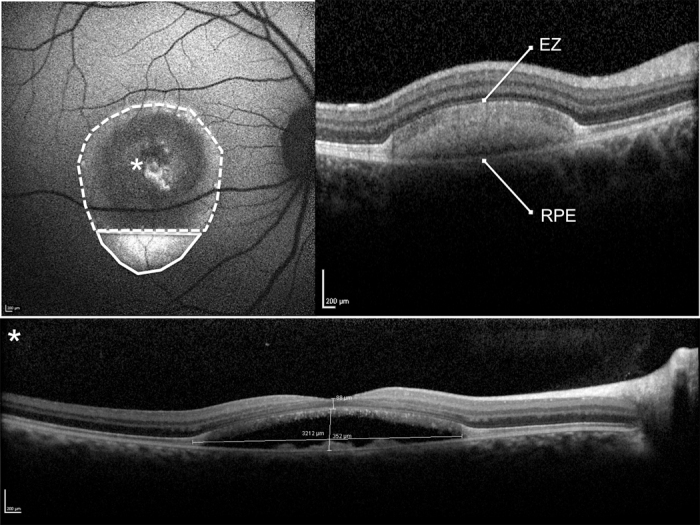
Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) of a pseudohypopyon lesion. Illustration of the per protocol evaluation of the hyperautofluorescent (area encircled by a continuous line) and hypoautofluorescent (area encircled by a dotted line) components of the lesion were measured using the measured using the Heidelberg software (top left panel). The overall lesion area is the sum of the hyperautofluorescent and hypoautofluorescent areas. The SD-OCT scan shows the hyper-reflective dome-shaped material located in the subretinal space, between the hyper-reflective photoreceptor inner segment (IS) ellipsoid portion (ellipsoid zone, EZ) and the hyperreflective retinal pigment epithelium (RPE)/Bruch’s membrane complex, matching with the hyperautofluorescent component of the lesion located inferiorly. The SD-OCT scan through the fovea (asterisk) shows the partial reabsorption of the hyperreflective material and replacement by a hyporeflective fluid component (bottom panel). The thickness of the neurosensory retina at the fovea and maximal thickness and width of the lesion was measured using the caliper provided with the Heidelberg software (bottom panel).
Statistical calculations were performed using Statistical Package for Social Sciences (version 17.0, SPSS Inc., Chicago, IL). The paired t test was used to assess changes in mean BCVA converted to the logarithm of the minimal angle of resolution (LogMAR), mean overall lesion area on FAF images, mean size of the hyper/hypoautofluorescent components (areas) of the lesion, mean CMT, mean maximal thickness and width of the lesion, and mean thickness of the neurosensory retina at the fovea. Changes in mean measurements and in specific features were analyzed as a whole and by categorizing the stage of the disease at study entry. Pearson’s coefficient was used to measure the strength of correlations among variables. The chosen level of statistical significance was p<0.05. For correlations with BCVA, the significance level was adjusted to take the number of correlations into account (α = 0.0167; Bonferroni’s adjustment for multiple correlations).
Results
Genetic analysis
The screening of the 11 exons encoding BEST1 in eight unrelated families resulted in the identification of seven previously reported different missense mutations clustered in exons 2, 4, and 7 [27-29] (Table 1).
Table 1. Clinical characteristics of 21 patients with Best vitelliform macular dystrophy at baseline and during the progression of the disease.
| Family | Case | Age | Gender | BEST1 mutation | Eye | Follow up (mo) | BCVA Baseline, ETDRS LogMAR | Disease stage Baseline | BCVA Follow-up, ETDRS LogMAR | Disease stage Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
Case1 |
18 |
F |
exon 2;
p.Ala10Thr |
R |
61 |
0.2 |
pseudohypopyon |
0.2 |
pseudohypopyon |
| |
|
|
|
|
L |
|
0.1 |
vitelliruptive |
0.1 |
vitelliruptive |
| 1 |
Case2 |
44 |
F |
exon 2;
p.Ala10Thr |
R |
61 |
0.4 |
vitelliform |
0.4 |
vitelliform |
| |
|
|
|
|
L |
|
1 |
pseudohypopyon |
1 |
vitelliruptive |
| 2 |
Case3 |
23 |
M |
exon 2
p.Ala10Val |
R |
38 |
0.7 |
fibrotic |
0.7 |
fibrotic |
| |
|
|
|
|
L |
|
0.8 |
fibrotic |
0.8 |
fibrotic |
| 2 |
Case4 |
50 |
M |
exon 2
p.Ala10Val |
R |
38 |
1 |
atrophic |
1 |
atrophic |
| |
|
|
|
|
L |
|
1 |
atrophic |
1 |
atrophic |
| 3 |
Case5 |
11 |
M |
exon 4
p.Thr91Ile |
R |
24 |
0.1 |
vitelliruptive |
0.1 |
pseudohypopyon |
| |
|
|
|
|
L |
|
0.1 |
pseudohypopyon |
0.1 |
vitelliruptive |
| 3 |
Case6 |
20 |
F |
exon 4
p.Thr91Ile |
R |
24 |
1 |
fibrotic |
1 |
fibrotic |
| |
|
|
|
|
L |
|
0.6 |
fibrotic |
0.6 |
fibrotic |
| 3 |
Case7 |
45 |
M |
exon 4
p.Thr91Ile |
R |
24 |
0.7 |
atrophic |
0.7 |
atrophic |
| |
|
|
|
|
L |
|
0.3 |
atrophic |
0.3 |
atrophic |
| 4 |
Case8 |
41 |
M |
exon 2
p.Val9Ala |
R |
50 |
0.4 |
fibrotic |
0.4 |
fibrotic |
| |
|
|
|
|
L |
|
1 |
fibrotic |
1 |
fibrotic |
| 4 |
Case9 |
12 |
F |
exon 2
p.Val9Ala |
R |
50 |
0 |
previtelliform |
0 |
previtelliform |
| |
|
|
|
|
L |
|
0 |
previtelliform |
0 |
previtelliform |
| 5 |
Case10 |
36 |
F |
exon 2
p.Arg25Trp |
R |
51 |
0.5 |
vitelliruptive |
0.5 |
vitelliruptive |
| |
|
|
|
|
L |
|
0.5 |
vitelliruptive |
0.5 |
vitelliruptive |
| 5 |
Case11 |
11 |
F |
exon 2
p.Arg25Trp |
R |
51 |
0.1 |
vitelliruptive |
0 |
vitelliruptive |
| |
|
|
|
|
L |
|
1 |
fibrotic |
0.4 |
fibrotic |
| 5 |
Case12 |
71 |
M |
exon 2
p.Arg25Trp |
R |
12 |
0.4 |
pseudohypopyon |
0.4 |
pseudohypopyon |
| |
|
|
|
|
L |
|
0 |
previtelliform |
0 |
previtelliform |
| 6 |
Case13 |
14 |
M |
exon 4
p.Arg92Cys |
R |
72 |
0.4 |
fibrotic |
0.4 |
fibrotic |
| |
|
|
|
|
L |
|
0.3 |
vitelliruptive |
0.3 |
pseudohypopyon |
| 6 |
Case14 |
9 |
M |
exon 4
p.Arg92Cys |
R |
35 |
0 |
previtelliform |
0 |
previtelliform |
| |
|
|
|
|
L |
|
0 |
previtelliform |
0 |
previtelliform |
| 6 |
Case15 |
17 |
M |
exon 4
p.Arg92Cys |
R |
12 |
0 |
previtelliform |
0 |
previtelliform |
| |
|
|
|
|
L |
|
0 |
previtelliform |
0 |
previtelliform |
| 6 |
Case16 |
20 |
F |
exon 4
p.Arg92Cys |
R |
12 |
0 |
fibrotic |
0 |
fibrotic |
| |
|
|
|
|
L |
|
0 |
pseudohypopyon |
0 |
vitelliruptive |
| 7 |
Case17 |
47 |
F |
exon 7
p.Ile230Thr |
R |
63 |
0.2 |
previtelliform
+ multifocal |
0.2 |
previtelliform
+ multifocal |
| |
|
|
|
|
L |
|
0.1 |
previtelliform
+ multifocal |
0.1 |
previtelliform
+ multifocal |
| 7 |
Case18 |
8 |
M |
exon 7
p.Ile230Thr |
R |
63 |
0.2 |
pseudohypopyon |
0.2 |
pseudohypopyon |
| |
|
|
|
|
L |
|
0.2 |
vitelliruptive |
0.2 |
pseudohypopyon |
| 7 |
Case19 |
6 |
F |
exon 7
p.Ile230Thr |
R |
23 |
0 |
previtelliform |
0 |
previtelliform |
| |
|
|
|
|
L |
|
0 |
previtelliform |
0 |
previtelliform |
| 8 |
Case20 |
27 |
F |
exon 2
p.Thr4Ala |
R |
50 |
0.4 |
fibrotic |
0.4 |
fibrotic |
| |
|
|
|
|
L |
|
0.1 |
none |
0.1 |
none |
| 8 |
Case21 |
23 |
F |
exon 2
p.Thr4Ala |
R |
50 |
0.2 |
vitelliruptive |
0.2 |
vitelliruptive |
| L | 0.4 | fibrotic | 0.4 | fibrotic |
M: male; F: female; R: right; L: left; BCVA: best corrected visual acuity; ETDRS: Early Treatment Diabetic Retinopathy Study
Clinical data and multimodal imaging at study entry
We examined 42 eyes of 21 patients harboring BEST1 mutations (10 male, 11 female; Table 1). The mean age of patients was 26.3±17.4 years. Age of onset (at time of diagnosis) varied between 2 and 71 years (median = 20). At study entry, LogMAR BCVA ranged between 0 and 1 LogMAR (mean, 0.34±0.34 LogMAR); significant correlation was found between BCVA and EZ status at fovea (Pearson’s correlation −0.4, p<0.001). All patients had typical bilateral macular lesions, except one who had unilateral macular lesion (Case 20), and one that presented with multifocal bilateral lesions (Table 1).
Early stage lesions were characterized by the accumulation of yellowish material within the macula or outside the macular area (multifocal lesions), giving an aspect of foveal granularity (previtelliform lesions) or a typical, well-circumscribed yellow “egg yolk” (vitelliform lesions). This material showed no or only slight autofluorescence in the previtelliform stage (Figure 2), but was highly autofluorescent in the vitelliform stage (Figure 3). On SD-OCT scans, the previtelliform stage was characterized by a slight thickening of the hyperreflective band located between the hyperreflective EZ and the hyperreflective RPE/Bruch’s membrane complex (representing a thickening of the RPE/OS junction, also known as the interdigitation zone, IZ) [30,31] (Figure 2), while it appeared as a hyperreflective dome-shaped lesion located in the subretinal space between the EZ and the RPE/Bruch’s membrane complex (representing a further thickening of the IZ; Figure 3).
Figure 2.
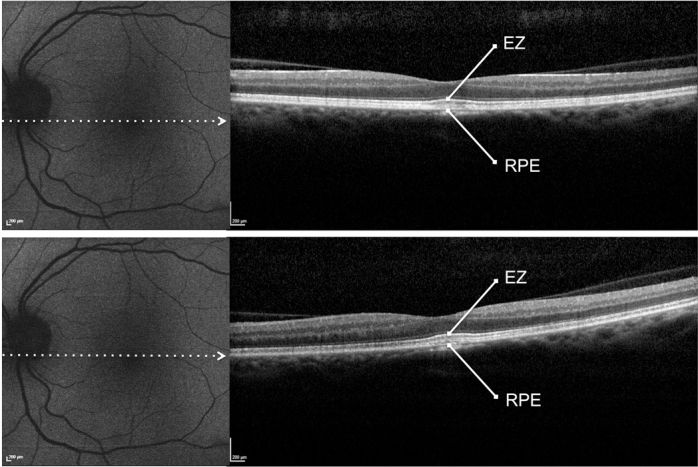
Patient #9. Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the left eye affected with previtelliform lesion at both study entry and last follow-up visit (50 months later). Blue FAF frames show no increased macular autofluorescence at either study entry (top left panel) or the last follow-up visit (bottom left panel). SD-OCT scans show a slight thickening of the hyperreflective band located between the hyperreflective photoreceptor inner segment (IS) ellipsoid portion (ellipsoid zone, EZ) and the hyperreflective retinal pigment epithelium (RPE)/Bruch’s membrane complex at study entry (top right panel) and the last follow-up visit (bottom right panel).
Figure 3.
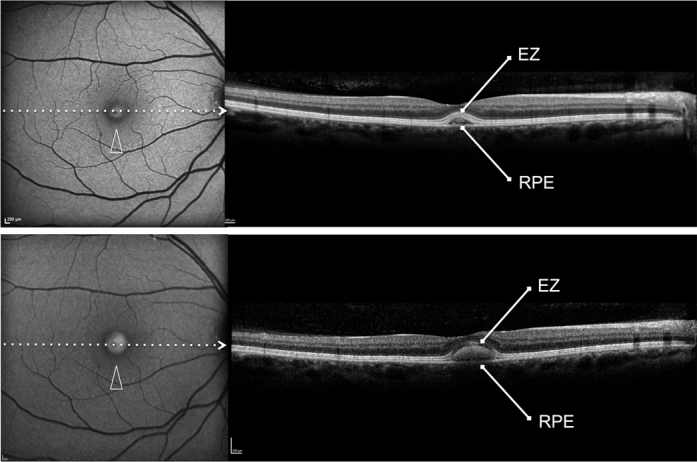
Patient #2. Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the right eye affected with vitelliform lesion at both study entry and last follow-up visit (61 months later). Blue FAF frames show a highly autofluorescent macular lesion at study entry (top left panel, arrowhead), which was enlarged at the last follow-up visit (bottom left panel, arrowhead). SD-OCT scans show a hyperreflective dome-shaped lesion located in the subretinal space, between the hyperreflective photoreceptor inner segment (IS) ellipsoid portion (ellipsoid zone, EZ) and the hyperreflective retinal pigment epithelium (RPE)/Bruch’s membrane complex at study entry (top right panel), which had increased at the last follow-up visit (bottom right panel).
Later stages included pseudohypopyon (Figure 4, Figure 5, and Figure 6) and vitelliruptive lesions (Figure 7; scrambled-egg aspect with dispersion of the vitelliform material without sign of atrophy or fibrosis), characterized by partial/complete reabsorption of the yellowish material and replacement by a fluid component showing no increased fluorescence on FAF or reflectivity on OCT examination (Figure 4, Figure 5, Figure 6, and Figure 7). Late lesions were characterized by reduced fluorescence on FAF due to partial/complete atrophy (with or without residual dispersed material; Figure 8) or fibrosis (with no detectable active choroid neovascularization) within the area previously occupied by the yellowish material (Figure 9). It is noteworthy that all of these late lesions were characterized by the presence of a hyperautofluorescent ring.
Figure 4.
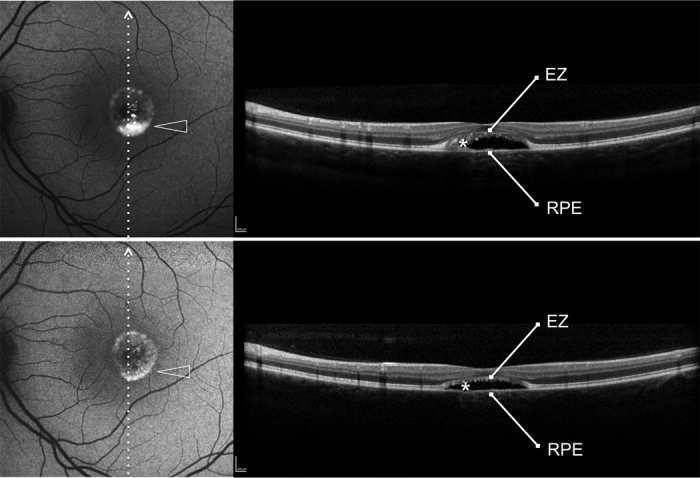
Patient #16.Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the left eye affected with pseudohypopyon lesion at study entry and vitelliruptive lesion last follow-up visit (12 months later). Blue FAF frames and SD-OCT scans at study entry (top left and top right panels) show a partial reabsorption of the hyperautofluorescent (arrowhead)/hyperreflective material (asterisk) located between the hyperreflective photoreceptor inner segment (IS) ellipsoid portion (ellipsoid zone, EZ) and the hyperreflective retinal pigment epithelium (RPE)/Bruch’s membrane complex, and replacement by a fluid component. At the last follow-up visit, blue FAF frames and SD-OCT scans (bottom left and bottom right panels) show further reabsorption of the hyperautofluorescent (arrowhead)/hyperreflective material (asterisk).
Figure 5.
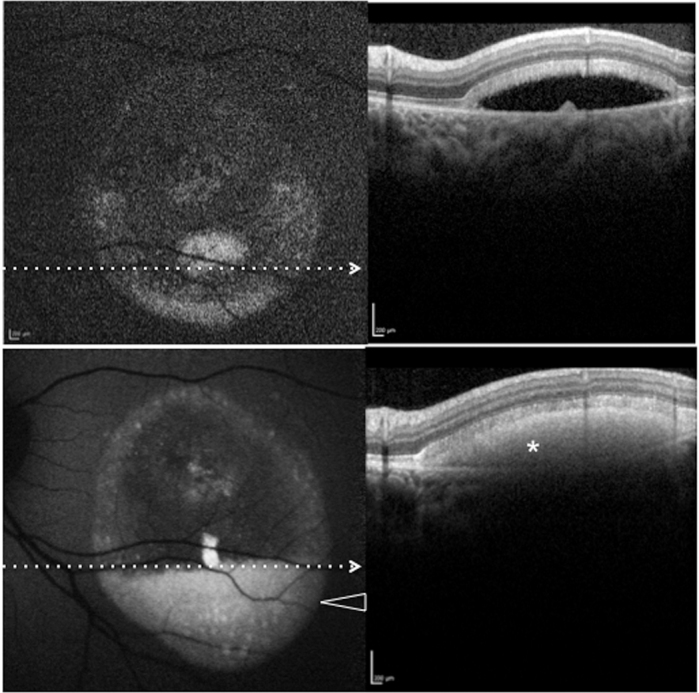
Patient #18. Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the left eye affected with pseudohypopyon lesion at study entry and vitelliruptive lesion last follow-up visit (63 months later). Blue FAF frames and SD-OCT scans at study entry (top left and top right panels) show almost complete absence (reabsorption) of the autofluorescent/hyperreflective material, which has been replacement by a fluid component. At the last follow-up visit, blue FAF frames and SD-OCT scans (bottom left and bottom right panels) show development of the hyperautofluorescent (arrowhead)/hyperreflective material (asterisk).
Figure 6.
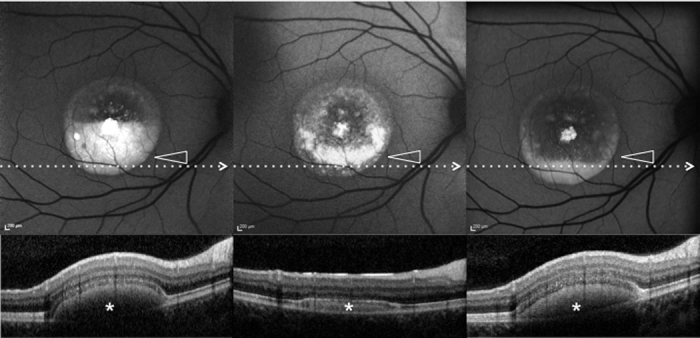
Patient #1.Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the right eye affected with pseudohypopyon lesion at both study entry and last follow-up visit (61 months later). Blue FAF frames and SD-OCT scans at study entry (top left and bottom right panels) show a partial reabsorption of the hyperautofluorescent (arrowhead)/hyperreflective material (asterisk) and replacement by a fluid component. During follow-up, blue FAF frames and SD-OCT scans (top middle and bottom middle panels) show further reabsorption of the hyperautofluorescent (arrowhead)/hyperreflective material (asterisk). At the last follow-up visit, blue FAF frames and SD-OCT scans (top left and bottom right panels) show development of the hyperautofluorescent (arrowhead)/hyper-reflective material (asterisk).
Figure 7.
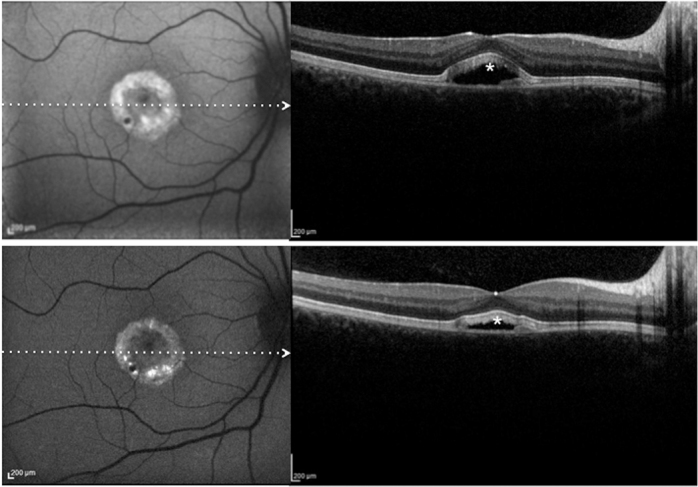
Patient #21.Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the right eye affected with vitelliruptive lesion at both study entry and last follow-up visit (50 months later). Blue FAF frames and SD-OCT scans at study entry (top left and bottom right panels) show reabsorption of the hyperautofluorescent/hyperreflective subretinal material (asterisk) and replacement by a fluid component. At the last follow-up visit, blue FAF remained almost unchanged (top right panel), while SD-OCT showed a decrease in subretinal fluid (asterisk; bottom right panel).
Figure 8.
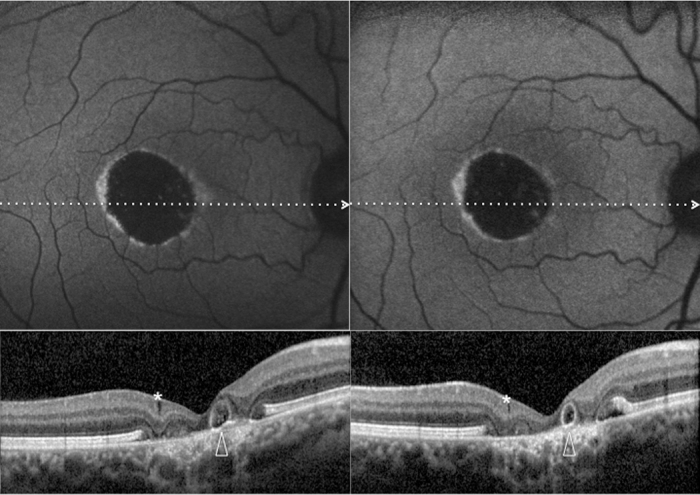
Patient #5.Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the right eye affected with atrophic lesion at both study entry and last follow-up visit (38 months later). At study entry (top left and bottom left panels), blue FAF frames show reduced autofluorescence (with some residual dispersed autofluorescent material), and SD-OCT shows atrophic changes (diffuse loss of photoreceptor and other sensory retina layers, with retinal pseudocysts [asterisk] and outer retinal tabulation [arrowhead]) within the area previously occupied by the yellowish material. Blue FAF and SD-OCT findings appear unchanged at the last follow-up visit (top right and bottom right panels). Note the presence of a hyperautofluorescent ring.
Figure 9.
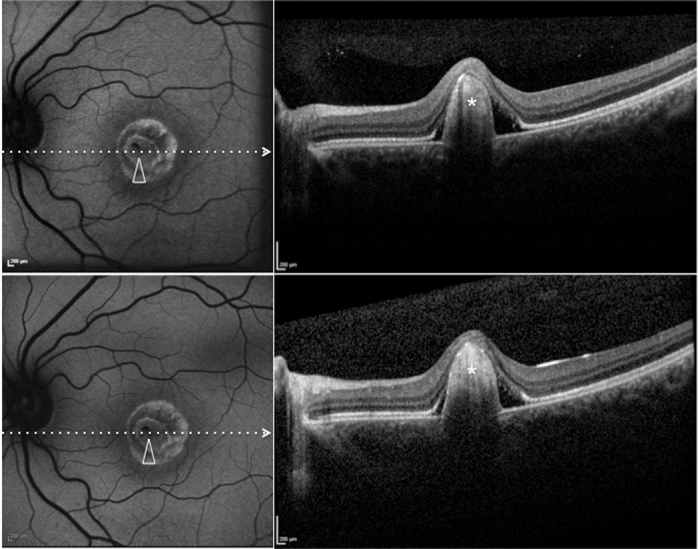
Patient #6. Blue fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) reveal the right eye affected with fibrotic lesion at both study entry and last follow-up visit (24 months later). At study entry (top left and top right panels), blue FAF frames show central (arrowhead) reduced autofluorescence (with some residual dispersed autofluorescent material), and SD-OCT shows a prominent highly hyperreflective thickening at retinal pigment epithelium level, inducing marked anterior bulging, accompanied by diffuse loss and thinning of the sensory retina (asterisk). Blue FAF and SD-OCT findings appear unchanged at the last follow-up visit (bottom left and bottom right panels). Note the presence of a hyperautofluorescent ring.
Based on fundus biomicroscopy, blue FAF, and SD-OCT at study entry, the macular lesions were counted as follows (Table 1): 1) no lesions: one eye of one patient; 2) previtelliform lesions: 11 eyes of six affected patients; 3) vitelliform lesions: one eye of one affected patient; 4) pseudohypopyon: six eyes of six affected patients; 5) vitelliruptive lesions (scrambled-egg aspect with dispersion of the vitelliform material without sign of atrophy or fibrosis): eight eyes of seven affected patients; 6) atrophic lesions (atrophy with or without residual dispersed material): four eyes of two patients; and 7) Fibrotic lesions: 11 eyes of eight patients.
Clinical and multimodal imaging changes during follow-up
A patient with unilateral macular lesion at study entry (Case 20) still showed no lesion in the fellow eye at last follow-up visit. Eleven eyes of six affected patients (Cases 9, 12, 14, 15, 17, and 19) showing previtelliform lesions still exhibited previtelliform lesions at the last follow-up visit (Figure 2). One eye of one patient (Case 2) showing vitelliform lesions at study entry still showed vitelliform lesions at the last follow-up visit (Figure 3). Three eyes of three patients (Cases 1, 12, and 18) showing pseudohypopyon lesions at study entry still showed pseudohypopyon lesions at the last follow-up visit (Figure 6). Five eyes of four patients (Cases 1, 10, 11, and 21) showing vitelliruptive lesions at study entry still showed vitelliruptive lesions at the last follow-up visit (Figure 7). Four eyes of two patients (Cases 4 and 7) showing atrophic lesions at study entry still showed atrophic lesions at the last follow-up visit (Figure 8). Eleven eyes of eight patients (Cases 3, 6, 8, 11, 13, 16, 20, and 21) showing fibrotic lesions at study entry still showed fibrotic lesions at the last follow-up visit (Figure 9).
Three eyes of three patients showing pseudohypopyon lesions at study entry progressed to vitelliruptive lesions at the last follow-up visit (Figure 4). Interestingly, three eyes of three patients showing vitelliruptive lesion at study entry reverted to pseudohypopyon lesion with overall enlargement of the lesion size (Figure 5). Moreover, one eye of one patient with pseudohypopyon lesion at study entry, and pseudohypopyon lesion at the last follow-up visit, reverted to vitelliruptive lesion during follow-up (Figure 6).
Clinical data and multimodal imaging at the last follow-up visit
Mean follow-up for the overall study population (42 eyes of 21 patients) was 41.1±18.5 months. At last follow-up visit, LogMAR BCVA ranged between 0 and 1 LogMAR (mean, 0.02±0.06 LogMAR); a significant correlation was found between BCVA and EZ status at the fovea (Pearson’s correlation −0.6, p<0.001).
Based on fundus biomicroscopy, blue FAF, and SD-OCT at study entry, the macular lesions were counted as follows (Table 1): 1) no lesions: one eye of one patient; 2) previtelliform lesions: 11 eyes of six affected patients; 3) vitelliform lesions: one eye of one affected patient; 4) pseudohypopyon: six eyes of five affected patients; 5) vitelliruptive lesions: eight eyes of seven affected patients; 6) atrophic lesions: four eyes of two patients; and 7) fibrotic lesions: 11 eyes of eight patients.
Changes in mean measurements (mean BCVA, mean overall lesion area on FAF images, mean size of the hyper/hypoautofluorescent components of the lesion, mean CMT, mean maximal thickness and width of the lesion, and mean thickness of the neurosensory retina at the fovea) and in specific features (macular FAF features, status of the EZ, reflectivity of the lesion on SD-OCT scans, presence/absence of focal RPE thickenings (“RPE bumps”), and presence/absence of retinal pseudocysts), evaluated as a whole, and by categorizing the stage of the disease at study entry are reported in Table 2, Table 3, Table 4, and Table 5.
Table 2. Changes in mean measurements and in specific features in patients with Best vitelliform macular dystrophy.
| Mean measurements and specific features | Study entry | Last visit (41.14±18.5 months) | P(1) | P(2) | P(3) |
|---|---|---|---|---|---|
|
BCVA (LogMAR) |
0.34±0.34 |
0.32±0.33 |
0.2 |
|
- |
|
Overall lesion area (mm2) |
6.62±4.9 |
7.34±6.1 |
0.05 |
0.03 |
0.04 |
|
Hyperautofluorescent component of the lesion (mm2) |
4.10±3.7 |
4.16±3.7 |
0.3 |
0.06 |
0.05 |
|
Hypoautofluorescent component of the lesion (mm2) |
2.94±2.7 |
3.20±2.8 |
0.7 |
0.07 |
0.06 |
|
FAF fovea (n) |
Hypo=29 / Hyper=3 / Iso=10 |
Hypo=29 / Hyper=3
/ Iso=10 |
- |
0.7 |
0.7 |
|
EZ status fovea (n) |
Normal=15 / Disrupted=14/
Absent=13 |
Normal=13 / Disrupted=16 /Absent=13 |
- |
<0.001 |
<0.001 |
|
OCT reflectivity (n) |
Mixed=22 / Hyper=12 /
Hypo=3 |
Mixed=22 / Hyper=12 / Hypo=3 |
- |
0.2 |
0.4 |
|
Maximal OCT lesion thickness (μm) |
299.87±187.8 |
315.21±161.0 |
0.5 |
0.03 |
0.03 |
|
Maximal OCT lesion width (μm) |
2503.41±975.0 |
2546.07±1125.2 |
0.1 |
0.4 |
0.6 |
|
OCT neurosensory retinal thickness fovea (μm) |
89.25±28.5 |
90.85±40.9 |
0.9 |
0.03 |
0.02 |
| CMT (μm) | 341.08±178.5 | 351.17±156.7 | 0.8 | 0.1 | 0.2 |
BCVA: best-corrected visual acuity; CMT: central macular thickness; FAF: fundus autofluorescence; EZ: Ellipsoid Zone; LogMAR: logarithm of the minimal angle of resolution; n: number of eyes; OCT: optical coherence tomography; RPE: retinal pigment epithelium; P (1) statistical significance of change between baseline and last visit; P (2) statistical significance of correlation with BCVA at baseline; P (3) statistical significance of correlation with BCVA at last follow up visit.
Table 3. Changes in mean measurements and in specific features in patients with Best vitelliform macular dystrophy by categorizing the stage of the disease at study entry.
| Previtelliform (n=11) FU: 34.3±19.7 months | Study entry | Last visit | p |
|---|---|---|---|
| BCVA (LogMAR) |
0.02±0.06 |
0.02±0.06 |
1 |
| FAF fovea (n) |
Iso=9 Hyper=2 |
Iso=9 Hyper=2 |
1 |
| EZ status (n) |
Normal=11 |
Normal=11 |
1 |
| OCT reflectivity (n) |
Hyper=11 |
Hyper=11 |
1 |
| CMT (μm) |
256.4±26.6 |
247.5±21.8 |
0.3 |
|
Vitelliform (n=1) FU: 61 months |
Study entry |
Last visit |
|
| BCVA (LogMAR) |
0.4 |
0.4 |
1 |
| Overall lesion area (mm2) |
0.28 |
0.36 |
0.9 |
| Hyperautofluorescent component of the lesion (mm2) |
0.28 |
0.36 |
0.9 |
| Hypoautofluorescent component of the lesion (mm2) |
0 |
0 |
1 |
| FAF fovea |
Hyper |
Hyper |
1 |
| EZ status fovea |
Normal |
Normal |
1 |
| OCT reflectivity |
Hyper |
Hyper |
1 |
| Maximal OCT lesion thickness (μm) |
175 |
210 |
0.8 |
| Maximal OCT lesion width (μm) |
601 |
687 |
0.8 |
| OCT neurosensory retinal thickness fovea (μm) |
88 |
90 |
0.9 |
| CMT (μm) |
278 |
310 |
0.8 |
|
Pseudohypopyon (n=6) FU: 49.1±24.7 months |
Study entry |
Last visit |
p |
| BCVA (LogMAR) |
0.31±0.3 |
0.27±0.1 |
0.6 |
| Overall lesion area (mm2) |
5.96±2.9 |
6.30±3.3 |
0.7 |
| Hyperautofluorescent component of the lesion (mm2) |
2.82±2.6 |
2.37±2.1 |
0.009 |
| Hypoautofluorescent component of the lesion (mm2) |
3.22±1.8 |
4.81±2.0 |
0.01 |
| FAF fovea (n) |
Hypo=6 |
Hypo=6 |
1 |
| EZ status fovea (n) |
Normal=2 / Disrupted=4 |
Disrupted=6 |
0.8 |
| OCT reflectivity (n) |
Mixed=6 |
Mixed=3
Hypo=3 |
0.7 |
| Maximal OCT lesion thickness (μm) |
288.5±57.2 |
243.6±60.4 |
0.09 |
| Maximal OCT lesion width (μm) |
2510±895.9 |
2381.4±780.9 |
0.2 |
| OCT neurosensory retinal thickness fovea (μm) |
93.5±30.4 |
89.2±8.6 |
0.09 |
| CMT (μm) |
392.0±53.7 |
362.0±70.0 |
0.1 |
|
Vitelliruptive (n=8)
FU: 45.1±17.6 months |
Study entry |
Last visit |
p |
| BCVA (LogMAR) |
0.25±0.16 |
0.24±0.34 |
0.6 |
| Overall lesion area (mm2) |
4.99±3.5 |
5.38±3.5 |
0.7 |
| Hyperautofluorescent component of the lesion (mm2) |
0 |
1.20±2.4 |
0.02 |
| Hypoautofluorescent component of the lesion (mm2) |
4.99±3.5 |
4.28±1.8 |
0.6 |
| FAF fovea (n) |
Hypo=8 |
Hypo=8 |
1 |
| EZ status fovea (n) |
Disrupted=8 |
Disrupted=8 |
1 |
| OCT reflectivity (n) |
Hypo=3 / Mixed=5 |
Mixed=8 |
0.7 |
| Maximal OCT lesion thickness (μm) |
238.8±89.9 |
198.8±102.3 |
0.5 |
| Maximal OCT lesion width (μm) |
2285.33±737.5 |
1907.75±579.2 |
0.3 |
| OCT neurosensory retinal thickness fovea (μm) |
104.16±45.6 |
84.75±17.3 |
0.4 |
| CMT (μm) |
365.0±47.5 |
379.62±55.8 |
0.5 |
|
Atrophic (n=4)
FU: 31.0±8.0 months |
Study entry |
Last visit |
p |
| BCVA (LogMAR) |
0.75±0.33 |
0.75±0.33 |
1 |
| Overall lesion area (mm2) |
3.72±2.5 |
3.70±2.9 |
0.2 |
| Hyperautofluorescent component of the lesion (mm2) |
1.49±2.1 |
1.49±2.1 |
1 |
| Hypoautofluorescent component of the lesion (mm2) |
2.23±2.2 |
2.21±2.8 |
0.3 |
| FAF fovea (n) |
Hypo 4 |
Hypo 4 |
1 |
| EZ status fovea (n) |
Absent 4 |
Absent 4 |
1 |
| Maximal OCT lesion width (μm) |
1966.75±596.9 |
2012.75±528.4 |
0.5 |
| OCT neurosensory retinal thickness fovea (μm) |
60.2±5.6 |
54.5±12.8 |
0.6 |
| CMT (μm) |
168.0±34.5 |
163.5±39.5 |
0.5 |
|
Fibrotic (n=11)
FU: 41.7±16.8 months |
Study entry |
Last visit |
p |
| BCVA (LogMAR) |
0.60±0.32 |
0.55±0.30 |
0.3 |
| Overall lesion area (mm2) |
8.59±5.3 |
8.57±6.2 |
0.7 |
| Hyperautofluorescent component of the lesion (mm2) |
5.41±2.3 |
5.42±2.0 |
0.9 |
| Hypoautofluorescent component of the lesion (mm2) |
3.18±1.0 |
3.15±1.2 |
0.8 |
| FAF fovea (n) |
Hypo=11 |
Hypo=11 |
1 |
| EZ status fovea (n) |
Absent=9 /Disrupted=2 |
Absent=9 /Disrupted=2 |
1 |
| OCT reflectivity (n) |
Mixed=11 |
Mixed=11 |
1 |
| Maximal OCT lesion thickness (μm) |
354.6±191.6 |
348.8±190.9 |
0.8 |
| Maximal OCT lesion width (μm) |
2788.4±832.4 |
2721.2±864.0 |
0.7 |
| OCT neurosensory retinal thickness fovea (μm) |
104.7±63.8 |
98.8±41.9 |
0.1 |
| CMT (μm) | 458.3±249.7 | 457.2±245.2 | 0.2 |
BCVA: best-corrected visual acuity; CMT: central macular thickness; FAF: fundus autofluorescence; FU: follow up; EZ: Ellipsoid Zone; LogMAR: logarithm of the minimal angle of resolution; n: number of eyes; OCT: optical coherence tomography; RPE: retinal pigment epithelium.
Table 4. Changes in mean measurements and in specific features in patients with Best vitelliform macular dystrophy progressing or not from pseudohypopyon stage to vitelliruptive stage.
| Pseudohypopyon to vitelliruptive (n=3) FU: 32.3±25.5 months | Study entry | Last visit | P value |
|---|---|---|---|
| BCVA (LogMAR) |
0.36±0.5 |
0.36±0.5 |
- |
| Overall lesion area (mm2) |
5.32±2.9 |
5.16±3.3 |
0.6 |
| Hyperautofluorescent component of the lesion (mm2) |
2.41±1.8 |
0 |
0.01 |
| Hypoautofluorescent component of the lesion (mm2) |
2.91±2.7 |
5.16±3.3 |
0.01 |
| FAF fovea (n) |
Hypo=3 |
Hypo=3 |
|
| EZ status fovea (n) |
Disrupted=2/ Normal=1 |
Disrupted=3 |
|
| OCT reflectivity (n) |
Mixed=3 |
Hypo=3 |
|
| Maximal OCT lesion thickness (μm) |
226.5±13.2 |
215.6±15.4 |
0.1 |
| Maximal OCT lesion width (μm) |
2110.5±895.9 |
2056.0±648.4 |
0.3 |
| OCT neurosensory retinal thickness fovea (μm) |
92.5±16.4 |
90.7 14.5 |
0.1 |
| CMT (μm) |
332.0±13.7 |
323.0±21.1 |
0.1 |
|
Pseudohypopyon not progressing to vitelliruptive (n=3)
FU: 45.3±28.9 months |
Study entry |
Last visit |
P value |
| BCVA (LogMAR) |
0.22±0.11 |
0.26±0.1 |
0.1 |
| Overall lesion area (mm2) |
6.93±3.8 |
7.24±4.6 |
0.6 |
| Hyperautofluorescent component of the lesion (mm2) |
3.19±1.2 |
2.64±1.6 |
0.07 |
| Hypoautofluorescent component of the lesion (mm2) |
3.74±1.8 |
4.60±1.5 |
0.07 |
| FAF fovea (n) |
Hypo=3 |
Hypo=3 |
- |
| EZ status fovea (n) |
Disrupted=2/ Normal=1 |
Disrupted=3 |
|
| OCT reflectivity (n) |
Mixed=3 |
Mixed=3 |
- |
| Maximal OCT lesion thickness (μm) |
321.4±71.8 |
293.5±88.4 |
0.2 |
| Maximal OCT lesion width (μm) |
2925.4±320.6 |
2815.0±331.8 |
0.1 |
| OCT neurosensory retinal thickness fovea (μm) |
94.6±15.3 |
87.5±14.8 |
0.1 |
| CMT (μm) | 450.4±41.4 | 423.0±60.8 | 0.1 |
BCVA: best-corrected visual acuity; CMT: central macular thickness; FAF: fundus autofluorescence; FU: follow up; EZ: Ellipsoid Zone; LogMAR: logarithm of the minimal angle of resolution; n: number of eyes; OCT: optical coherence tomography; RPE: retinal pigment epithelium.
Table 5. Changes in mean measurements and in specific features in patients with Best vitelliform macular dystrophy reverting or not from vitelliruptive stage to pseudohypopyon stage.
| Vitelliruptive to pseudohypopyon (n=3) FU: 53.0±25.5 months | Study entry | Last visit | P value |
|---|---|---|---|
| BCVA (LogMAR) |
0.2±0.1 |
0.2±0.1 |
- |
| Overall lesion area (mm2) |
4.57±2.7 |
5.36±3.0 |
0.1 |
| Hyperautofluorescent component of the lesion (mm2) |
0 |
1.87±1.5 |
0.01 |
| Hypoautofluorescent component of the lesion (mm2) |
4.57±2.7 |
3.49±1.3 |
0.03 |
| FAF fovea (n) |
Hypo=3 |
Hypo=3 |
- |
| EZ status fovea (n) |
Disrupted=3 |
Disrupted=3 |
|
| OCT reflectivity (n) |
Hypo=3 |
Mixed=3 |
- |
| Maximal OCT lesion thickness (μm) |
285.2±44.2 |
291.5±51.6 |
0.1 |
| Maximal OCT lesion width (μm) |
2289.2±382.1 |
2329.5±457.5 |
0.3 |
| OCT neurosensory retinal thickness fovea (μm) |
92.4±4.3 |
89.0±5.6 |
0.1 |
| CMT (μm) |
397.2±61.4 |
417.0±52.3 |
0.1 |
|
Vitelliruptive not progressing to pseudohypopyon (n=5)
FU: 52.8±4.6months |
Study entry |
Last visit |
P value |
| BCVA (LogMAR) |
0.28±0.2 |
0.26±0.2 |
- |
| Overall lesion area (mm2) |
5.15±4.0 |
5.5±4.0 |
0.1 |
| Hyperautofluorescent component of the lesion (mm2) |
0 |
0 |
- |
| Hypoautofluorescent component of the lesion (mm2) |
5.15±4.0 |
5.5±4.0 |
0.1 |
| FAF fovea (n) |
Hypo=5 |
Hypo=5 |
- |
| EZ status fovea (n) |
Disrupted=5 |
Disrupted=5 |
|
| OCT reflectivity (n) |
Mixed=5 |
Mixed=5 |
- |
| Maximal OCT lesion thickness (μm) |
233.6±99.6 |
188.8±120.4 |
0.02 |
| Maximal OCT lesion width (μm) |
2283.0±822.6 |
1818.8±592.0 |
0.07 |
| OCT neurosensory retinal thickness fovea (μm) |
105.2±50.9 |
81.2±19.4 |
0.04 |
| CMT (μm) | 361.4±52.2 | 365.6±33.9 | 0.8 |
BCVA: best-corrected visual acuity; CMT: central macular thickness; FAF: fundus autofluorescence; FU: follow up; EZ: Ellipsoid Zone; LogMAR: logarithm of the minimal angle of resolution; n: number of eyes; OCT: optical coherence tomography; RPE: retinal pigment epithelium.
Discussion
In this study, we investigated the multimodal morphological features of Best VMD in subjects harboring mutations in BEST1 gene, and their changes during the progression of the disease. Due to the paucity of histopathological data on the natural course of the disease, this analysis represents a unique opportunity to gather insights into the physiopathology of Best VMD.
Overall, we found that the previtelliform lesions (characterized on blue FAF by absence or only slight autofluorescence and on SD-OCT by a slight thickening of the hyperreflective band located between the hyperreflective EZ and the hyperreflective RPE/Bruch’s membrane complex [i.e., the IZ]) did not progress to a different disease stage over a mean period of 34.3±19.7 months in all 11 eyes (six patients; Table 1 and Table 3). Patients presenting the previtelliform stage of the disease were generally younger than patients presenting more advanced stages (Table 1). In the current series, this may have accounted for the absence of progression of all previtelliform lesions and does not exclude the possibility that this stage might progress to advanced stages later in life.
The vitelliform lesion (characterized on blue FAF by well-circumscribed high autofluorescence within the macula and on SD-OCT by a dome-shaped hyperreflectivity located in the subretinal space [between the EZ and the RPE/Bruch’s membrane complex]) was found in only one eye (one patient; Table 1), and after 61 months, we did not observe a progression to a more advanced stage of the disease (Table 1 and Table 3). However, on FAF, the vitelliform lesion showed an enlargement of the hyperautofluorescent lesion (from 0.28 mm2 to 0.36 mm2), and on SD-OCT, an increase of both CMT (from 278 μm to 310 μm) and the hyperreflective subretinal lesion was observed (thickness: from 175 μm to 210 μm; width: from 601 μm to 687 μm; Table 3).
Pseudohypopyon lesions (characterized on blue FAF by a well-circumscribed high autofluorescence within the inferior-macula and on SD-OCT by a hyperreflectivity located in the inferior-macula subretinal space [between the EZ and the RPE/Bruch’s membrane complex]) were possibly due to partial reabsorption of the hyperautofluorescent (FAF) and hyperreflective (SD-OCT) material (replaced by a fluid component, showing no increased fluorescence on FAF or reflectivity on SD-OCT examination) and sedimentation of residual material according to the laws of gravity. Pseudohypopyon lesions did not progress to a different disease stage over a mean period of 45.3±28.9 months in three eyes (three patients; Table 1 and Table 3). In these eyes, the pseudohypopyon lesion showed a slight enlargement of the overall lesion area (from 6.93±3.8 mm2 to 7.24±4.6 mm2, p = 0.6) and a decrease of the hyperautofluorescent lesion (from 3.19±1.2 mm2 to 2.64±1.6 mm2, p = 0.07; Table 3–Table 4) on FAF. On SD-OCT, a slight decrease of both CMT (from 450.4±41.4 μm to 423.0±60.8 μm, p = 0.1) and the mixed hyper/hyporeflective subretinal lesion (thickness: from 321.4±71.8 μm to 293.5±88.4 μm, p = 0.2; width: from 2,925.4±320.6 μm to 2,815.0±331.8 μm, p = 0.1) was recorded (Table 3–Table 4). On the other hand, pseudohypopyon lesions progressed to vitelliruptive lesions in three eyes (three patients) after 32.3±25.5 months (Table 1 and Table 3). In these eyes, the overall lesion area decreased slightly (from 5.32±2.9 mm2 to 5.16±3.3 mm2, p = 0.6) and the hyperautofluorescent lesion disappeared (Table 3–Table 4). On SD-OCT, a modest decrease of both CMT (from 332.0±13.7 μm to 323.0±21.1 μm, p = 0.1) and the hyper/hypo-reflective subretinal lesion (thickness: from 226.5±13.2 μm to 215.6±15.4 μm, p = 0.1; width: from 2,110.5±895.9 μm 2056.0±648.4 μm, p = 0.3) was recorded (Table 3–Table 4).
Vitelliruptive lesions, characterized by no increased autofluorescence and reflectivity on blue FAF and SD-OCT, respectively, were possibly due to complete reabsorption of the material and replacement by a fluid component. Vitelliruptive lesions did not progress to a different disease stage over a mean period of 52.8±4.6 months in five eyes (four patients; Table 1 and Table 3). In these eyes, the vitelliruptive lesion showed a minor enlargement of the hypoautofluorescent lesion (from 5.15±4.0 mm2 to 5.5±4.0 mm2, p = 0.1; Table 3 and Table 5). On SD-OCT, a slight increase of CMT (from 361.4±52.2 μm to 365.6±33.9 μm, p = 0.8) and a modest decrease of the hyporeflective subretinal lesion (thickness: from 233.6±99.6 μm to 188.8±120.4 μm, p = 0.02; width: from 2283.0±822.6 μm 1818.8±592.0 μm, p = 0.07) were recorded (Table 3 and Table 5). On the other hand, after a mean of 45.1±17.6 months, we did not find a progression to a more advanced stage of the disease in any case (Table 1 and Table 3). Interestingly, vitelliruptive lesions reverted to pseudohypopyon lesions in three eyes (three patients) after 53.0±25.5 months (Table 1 and Table 3). In these eyes, the overall lesion area showed a modest increase (from 4.57±2.7 mm2 to 5.36±3.0 mm2, p = 0.1) and a hyperautofluorescent lesion appeared (mean 1.87±1.5 mm2; Table 3 and Table 5). On SD-OCT, a slight increase of both CMT (from 397.2±61.4 μm to 417.0±52.3, p = 0.1) and the hyporeflective subretinal lesion (thickness: from 285.2±44.2 μm to 291.5±51.6 μm, p = 0.1; width: from 2,289.2±382.1 μm to 2,329.5±457.5 μm, p = 0.3) was recorded (Table 3 and Table 5). It is noteworthy that one eye of one patient with a pseudohypopyon lesion at study entry first reverted to vitelliruptive lesion and then progressed to pseudohypopyon lesion once more (Figure 6).
Atrophic lesions, characterized by decreased autofluorescence on blue FAF and by diffuse loss of photoreceptor and other sensory retina layers on SD-OCT, did not progress to a different disease stage over a mean period of 31.0±8.0 months in all four eyes (two patients; Table 1 and Table 3). In these eyes, we did not find an enlargement of either the overall lesion area (from 3.72±2.5 mm2 to 3.70±2.9 mm2, p = 0.2) or the hypoautofluorescent lesion (from 2.23±2.2 μm to 2.21±2.8 μm, p = 0.3; Table 3). On SD-OCT, no change of CMT (from 168.0±34.5 μm to 163.5±39.5 μm, p = 0.5) was recorded (Table 3).
Fibrotic lesions were characterized by inhomogeneous areas of absolute hypoautofluorescence mixed with hyperautofluorescence (due to some residual dispersed autofluorescent material) on blue FAF and by a prominent, highly hyperreflective thickening at the RPE level. This induced a marked anterior bulging, accompanied by diffuse loss and thinning of the sensory retina on SD-OCT, and did not progress to a different disease stage over a mean period of 41.7±16.8 months in any of 11 eyes (eight patients; Table 1 and Table 3). In these eyes, we did not find an enlargement of either the overall lesion area (from 8.59±5.3 mm2 to 8.57±6.2 mm2, p = 0.7) or the hypoautofluorescent lesion (from 3.18±1.0 μm to 3.15±1.2 μm, p = 0.8; Table 3). On SD-OCT, no change in CMT (from 458.3±249.7 μm to 457.2±245.2 μm, p = 0.2) was recorded (Table 3).
Taken together, the current findings suggest that Best VMD should be considered as a dynamic disease-alternating phases of material accumulation and reabsorption in its progression: This is characterized by centrifugal expansion (mainly downward due to gravitation) and contraction, at least until development of the end-stage form of the disease (atrophic lesions do not appear to expand over time). All of these elements together favor the concept that RPE-mediated changes in the ionic environment of the subretinal space may lead to aberrant interaction between photoreceptors and the RPE, resulting in the continuous accumulation of fluid and OS debris in the subretinal space [15,26-29,32,33]. In turn, multimodal imaging features in the different stages of Best VMD, and their changes during the progression of the disease, suggest that although the abnormal protein encoded by BEST1 gene is expressed in the RPE, its primary anatomic impact should be at the photoreceptor level [15,26-29,32,33].
In the current series, we were not able to identify any factors related either with the bimodal onset of autosomal dominant Best VMD [12,13] or with the disease progression through different stages. Particularly, if we look at Family 1 (Table 1), while Case 1 presented as early as at the age of 18 years old with pseudohypopyon and vitelliruptive lesions in the right eye (RE) and left eye (LE) respectively (third and fourth stage, respectively) [14,15], while Case 2 presented at the older age of 44 years with vitelliform and pseudohypopyon lesions in the RE and LE, respectively (second and third stage, respectively) [14,15]. Interestingly, while in the older family member(Case 2), the vitelliform lesion did not progress over 61 months, the pseudohypopyon lesion (a more advanced stage) did progress to the vitelliruptive lesion; on the other hand, in the younger family member (Case 1), neither the pseudohypopyon nor the vitelliruptive lesions (i.e., more advanced stages) progressed over 61 months.
This study had several limitations, including its retrospective design, the relatively short duration of follow-up, and the small number of patients included. However, our analysis provides valuable information on the modalities of Best VMD progression, and on the rate of material/fluid accumulation until the development of the end-stage atrophic form of the disease, which may be used not only in the counseling affected patients, but also in the planning of therapeutic clinical trials for this dominantly inherited macular dystrophy. In conclusion, we documented a continuous material accumulation and reabsorption in Best VMD progression. Blue FAF and SD-OCT are important noninvasive imaging techniques to monitor Best VMD.
References
- 1.Best F. Uber eine hereditare maculaafektion; Beitrage zur verergslehre. Zschr Augenheilkunde. 1905;13:199–212. [Google Scholar]
- 2.Stone EM, Nichols BE, Streb LM, Kimura AE, Sheffield VC. Genetic linkage of vitelliform macular degeneration Best's disease to chromosome 11q13. Nat Genet. 1992;1:246–50. doi: 10.1038/ng0792-246. [DOI] [PubMed] [Google Scholar]
- 3.Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AA, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–7. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 4.Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA. 2002;99:4008–13. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufman SJ, Goldberg MF, Orth DH, Fishman GA, Tessler H, Mizuno K. Autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol. 1982;100:272–8. doi: 10.1001/archopht.1982.01030030274008. [DOI] [PubMed] [Google Scholar]
- 6.Lotery AJ, Munier FL, Fishman GA, Weleber RG, Jacobson SG, Affatigato LM, Nichols BE, Schorderet DF, Sheffield VC, Stone EM. Allelic variation in the VMD2 gene in Best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41:1291–6. [PubMed] [Google Scholar]
- 7.Bakall B, Radu RA, Stanton JB, Burke JM, McKay BS, Wadelius C, Mullins RF, Stone EM, Travis GH, Marmorstein AD. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp Eye Res. 2007;85:34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Gass JD. A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc. 1974;72:139–56. [PMC free article] [PubMed] [Google Scholar]
- 9.Patrinely JR, Lewis RA, Font RL. Foveomacular vitelliform dystrophy, adult type: a clinicopathologic study including electron microscopic observations. Ophthalmology. 1985;92:1712–8. doi: 10.1016/s0161-6420(85)34097-6. [DOI] [PubMed] [Google Scholar]
- 10.Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O'Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85:581–92. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E, Brown PD, Robson AG, Wright GA, Kestelyn P, Holder GE, Webster AR, Manson FD, Black GC. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordström S, Barkman Y. Hereditary maculardegeneration (HMD) in 246 cases traced to one gene-source in central Sweden. Hereditas. 1977;84:163–76. doi: 10.1111/j.1601-5223.1977.tb01394.x. [DOI] [PubMed] [Google Scholar]
- 13.Deutman AF, Hoyng C. Macular. Dystrophies. Retina. Ed. SJ Ryan, 3rd ed. Ed. Mosby, St. Louis, Missouri. 2001;70:1210-57. [Google Scholar]
- 14.Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88:688–92. doi: 10.1016/s0161-6420(81)34965-3. [DOI] [PubMed] [Google Scholar]
- 15.Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G, Souied EH. High definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol. 2008;146:501–7. doi: 10.1016/j.ajo.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Stanton JB, Wu J, Yu K, Hartzell HC, Peachey NS, Marmorstein LY, Marmorstein AD. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum Mol Genet. 2010;19:1108–18. doi: 10.1093/hmg/ddp583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weingeist TA, Kobrin JL, Watzke RC. Histopathology of Best’s macular dystrophy. Arch Ophthalmol. 1982;100:1108–14. doi: 10.1001/archopht.1982.01030040086016. [DOI] [PubMed] [Google Scholar]
- 18.Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differential macular and peripheral expression of bestrophin in human eyes and its implication for Best disease. Invest Ophthalmol Vis Sci. 2007;48:3372–80. doi: 10.1167/iovs.06-0868. [DOI] [PubMed] [Google Scholar]
- 19.Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Late development of vi- telliform lesions and flecks in a patient with Best disease: clinicopathologic correlation. Arch Ophthalmol. 2005;123:1588–94. doi: 10.1001/archopht.123.11.1588. [DOI] [PubMed] [Google Scholar]
- 20.Frangieh GT, Green WR, Fine SL. A histopathologic study of Best’s macular dystrophy. Arch Ophthalmol. 1982;100:1115–21. doi: 10.1001/archopht.1982.01030040093017. [DOI] [PubMed] [Google Scholar]
- 21.O’Gorman S, Flaherty WA, Fishman GA, Berson EL. Histopathologic findings in Best's vitelliform macular dystrophy. Arch Ophthalmol. 1988;106:1261–8. doi: 10.1001/archopht.1988.01060140421045. [DOI] [PubMed] [Google Scholar]
- 22.Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci. 1995;36:718–29. [PubMed] [Google Scholar]
- 23.von Rückmann A, Fitzke FW, Fan J, Halfyard A, Bird AC. Abnormalities of fundus autofluorescence in central serous retinopathy. Am J Ophthalmol. 2002;133:780–6. doi: 10.1016/s0002-9394(02)01428-9. [DOI] [PubMed] [Google Scholar]
- 24.von Rückmann A, Fitzke FW, Bird AC. In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol. 1997;115:609–15. doi: 10.1001/archopht.1997.01100150611006. [DOI] [PubMed] [Google Scholar]
- 25.von Rückmann A, Fitzke FW, Bird AC. Distribution of pigment epithelium autofluorescence in retinal disease state recorded in vivo and its change over time. Graefes Arch Clin Exp Ophthalmol. 1999;237:1–9. doi: 10.1007/s004170050186. [DOI] [PubMed] [Google Scholar]
- 26.Arnold JJ, Sarks JP, Killingsworth MC, Kettle EK, Sarks SH. Adult vitelliform macular degeneration: a clinicopathological study. Eye (Lond) 2003;17:717–26. doi: 10.1038/sj.eye.6700460. [DOI] [PubMed] [Google Scholar]
- 27.Querques G, Zerbib J, Santacroce R, Margaglione M, Delphin N, Rozet JM, Kaplan J, Martinelli D, Delle Noci N, Soubrane G, Souied EH. Functional and clinical data of Best Vitelliform Macular Dystrophy patients with mutations in BEST1 gene. Mol Vis. 2009;15:2960–72. [PMC free article] [PubMed] [Google Scholar]
- 28.Querques G, Atmani K, Bouzitou-Mfoumou R, Leveziel N, Massamba N, Souied EH. Preferential hyperacuity perimeter in best vitelliform macular dystrophy. Retina. 2011;31:959–66. doi: 10.1097/IAE.0b013e3181f441c1. [DOI] [PubMed] [Google Scholar]
- 29.Querques G, Zerbib J, Santacroce R, Margaglione M, Delphin N, Querques L, Rozet JM, Kaplan J, Souied EH. The spectrum of subclinical Best vitelliform macular dystrophy in subjects with mutations in BEST1 gene. Invest Ophthalmol Vis Sci. 2011;52:4678–84. doi: 10.1167/iovs.10-6500. [DOI] [PubMed] [Google Scholar]
- 30.Spaide RF, Curcio CA. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: literature review and model. Retina. 2011;31:1609–19. doi: 10.1097/IAE.0b013e3182247535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spaide RF. Questioning optical coherence tomography. Ophthalmology. 2012;119:2203–4. doi: 10.1016/j.ophtha.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Spaide RF, Noble K, Morgan A, Freund KB. Vitelliform macular dystrophy. Ophthalmology. 2006;113:1392–400. doi: 10.1016/j.ophtha.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 33.Kay CN, Abramoff MD, Mullins RF, Kinnick TR, Lee K, Eyestone ME, Chung MM, Sohn EH, Stone EM. Three-dimensional distribution of the vitelliform lesion, photoreceptors, and retinal pigment epithelium in the macula of patients with best vitelliform macular dystrophy. Arch Ophthalmol. 2012;130:357–64. doi: 10.1001/archophthalmol.2011.363. [DOI] [PMC free article] [PubMed] [Google Scholar]