

Abstract
Abnormal metabolism and sustained proliferation are hallmarks of cancer. Pyruvate kinase M2 (PKM2) is a metabolic enzyme that plays important roles in both processes. Recently, PKM2 was shown to have protein kinase activity phosphorylating histone H3 and promoting cancer cell proliferation. However, the mechanism and extent of this novel protein kinase in cancer cells remain unclear. Here, we report that binding of SAICAR, a metabolite abundant in proliferating cells, induces PKM2’s protein kinase activity in vitro and in cells. Protein microarray experiments revealed that more than 100 human proteins– mostly protein kinases– are phosphorylated by PKM2-SAICAR. In particular, PKM2-SAICAR phosphorylates and activates Erk1/2, which in turn sensitizes PKM2 for SAICAR-binding through phosphorylation. Additionally, PKM2-SAICAR was necessary to induce sustained Erk1/2 activation and mitogen-induced cell proliferation. Thus, the ligand-induced protein kinase activity from PKM2 is a mechanism that directly couples cell proliferation with intracellular metabolic status.
INTRODUCTION
Highly proliferative cells, such as cancer cells, display metabolic properties distinct from their neighboring normal cells (Vander Heiden, Cantley, Thompson, 2009). For example, cancer cells uptake more glucose than their neighboring normal cells and ferment lactate even when sufficient oxygen is supplied (Vander Heiden, Cantley, Thompson, 2009). The observed altered metabolism is regarded as a hallmark of cancer (Hanahan and Weinberg, 2011). However, the molecular basis connecting altered metabolic status to cell proliferation is still not completely understood.
Pyruvate kinase isoform M2 (PKM2) is a metabolic enzyme enriched in highly proliferating cells and most types of cancer cells (Mazurek et al., 2002; Christofk et al., 2008). PKM2 was recently identified as a major contributor for altered cellular metabolism and the growth of tumors, as replacing PKM2 with other isoforms of pyruvate kinases in cancer cells alleviates abnormal cellular metabolism, renders cells susceptible to stress in vitro, and limits the growth of tumor in xenografts (Christofk et al 2008). In addition, various pharmacological reagents targeting PKM2 in an isozyme-specific manner influence cell growth and proliferation (Chen et al., 2011; Anastasiou et al., 2012), suggesting that PKM2 may be a potential target of clinical applications (Vander Heiden, 2013).
Since the identification of PKM2 as a major contributor to cancer cell growth (Christofk et al 2008), research has focused on identifying the underlying mechanism of this isozyme-specific action to develop therapeutics and/or diagnostic tools (for review, see Gupta and Bamezai, 2010; Luo and Semenza, 2012; Mazurek, 2011). Recent results revealed that PKM2 in cancer cells has previously unrecognized properties distinct from other pyruvate kinase isoforms. First, PKM2 translocates from the cytoplasm to the nucleus of cancer cells upon stimulus, suggesting that PKM2 may have functions beyond glycolysis (Yang, et al., 2011). Indeed, PKM2 interacts with transcription factors and activates their functions (Luo et al., 2011).
Furthermore, PKM2 extracted from cancer cells shows protein kinase activity (Gao et al, 2012; Yang, et al., 2012a). Normally, pyruvate kinases catalyze the phosphotransfer reaction between phosphoenolpyruvate (PEP) and ADP, producing pyruvate and ATP. Surprisingly, PKM2 purified from cancer cell is capable of phosphorylating histone H3 threonine 11 (H3 T11) in vitro using PEP as a phospho-donor (Yang et al., 2012a). As expected, ADP inhibits the reaction in vitro (Yang et al., 2012a), suggesting that the same active site is used for both pyruvate kinase and protein kinase activity. Additionally, the phosphorylation of H3 by PKM2 was found in cells, and lead to increased cell proliferation by inducing expressions of several genes including MYC and CCND1 (Yang et al., 2012a).
Although this reaction occurs in cancer cells, the results observed by in vitro phosphorylation of H3 T11 by PKM2 purified from cancer cells implies that the reaction may not be robust enough to be significant in a biologically relevant context (Yang et al., 2012a). Additionally, recombinant PKM2 expressed and purified from E. coli did not display H3 T11 kinase activity (Yang et al., 2012a). Thus, protein kinase activity of PKM2 is dependent on alternative modifications orligand-binding, while pyruvate kinase activity is not. Indeed, phosphorylation of PKM2 by Erk1/2 appears to promote its protein kinase activity in cells (Yang et al., 2012b). However, the Erk1/2-mediated phosphorylation of PKM2 has not been shown to fully induce protein kinase activity in vitro.
To determine the mechanism defining how PKM2 contributes to cancer growth in an isozyme-specific manner, we previously searched for cellular metabolites that bind to PKM2 in an isozyme-selective manner (Keller et al., 2012). From our study, we found that SAICAR (succinyl-5-aminoimidazole-4-carboxamide-1-ribose-5′-phosphate), an intermediate of the de novo purine nucleotide biosynthesis process, accumulates in glucose-starved cancer cells and isozyme-selectively and directly activates pyruvate kinase activity of PKM2 in vitro and in cultured cancer cells, promoting survival in glucose-deprived conditions (Keller et al, 2012).
Here we report that the PKM2-SAICAR interaction is necessary and sufficient to induce robust protein kinase activity from PKM2 in vitro and in cancer cells. We also report that the PKM2-SAICAR complex phosphorylates over 100 human proteins – mostly protein kinases – that were previously unrecognized. In particular, PKM2-SAICAR directly activates Erk1 in vitro and within cells. As has been previously shown (Yang et al, 2012b), activated Erk1/2 phosphorylates PKM2. We found that the phosphorylation of PKM2 by Erk1/2 sensitizes PKM2 for SAICAR-binding, leading to a positive feedback loop. Additionally, upon EGFR activation, cellular SAICAR concentration is elevated, which is necessary to induce sustained activation of Erk1/2 and proliferative signaling via PKM2. These results provide a detailed molecular mechanism describing how two hallmarks – altered metabolism and sustained proliferative signaling –are interrelated in highly proliferating cells.
RESULTS
SAICAR-binding induces protein kinase activity of recombinant PKM2
Unlike purified recombinant PKM2, PKM2 extracted from cancer cells is capable of phosphorylating histone H3 threonine 11 (H3 T11) using PEP as a phosphate donor (Yang et al., 2012a). We attempted to reconstitute the activity in vitro using purified recombinant PKM2 (rPKM2) and PEP as the sole phosphate donor. First, we tested whether allosteric activators of PKM2, such as FBP (Mazurek et al., 2002) or SAICAR, can induce protein kinase activity from rPKM2 (Figure 1A). When the phosphorylation of recombinant histone H3 T11 by rPKM2 was probed in vitro, rPKM2 alone was not sufficient to efficiently phosphorylate H3 T11 as noted by Lu and coworkers (Yang, et al. 2012a; Figure 1B). The addition of FBP (Mazurek et al., 2002), slightly improved PKM2 protein kinase activity, while the addition of SAICAR drastically stimulated PKM2’s H3 T11 kinase activity (Figure 1B). In typical reactions, less than 10 nanomolar PKM2 was sufficient to completely phosphorylate excess (1- 2 μM) recombinant H3 T11 within 10 – 15 min, in the presence of SAICAR (kcat 6 s−1), suggesting that the PKM2-SAICAR complex is capable of carrying out a multiple turnover reaction. Michaelis-Menten constants (Km) of the complex were measured to be approximately 1–2 μM for monomeric histone H3.1 (Figure 1C) and 5 μM for PEP (Figure 1D). The low μM KmPEP of the PKM2-SAICAR complex indicates that a relaxed form of PKM2 is responsible for the phosphorylation of H3 T11. We would like to note that this reaction does not include any nucleotide and that PEP is likely to be the direct source of phosphate. To further ensure that PEP is the phosphate donor, we measured the production of pyruvate using lactate dehydrogenase varying protein substrate concentrations (Supplementary Figure S1A). We found that pyruvate is indeed formed upon the addition of histone H3.1. The rate of pyruvate formation was dependent on the concentration of H3.1 and SAICAR. The kcat (~ 5 s−1) and the Km (~ 1μM H3.1) values measured using this method (Figure S1A) were similar with values obtained by monitoring H3.1 phosphorylation (Figure 1C). The H3.1-dependent pyruvate production rate was stimulated by SAICAR (EC50 ~ 250 μM). The activation of protein kinase activity by SAICAR was barely observable when the SAICAR-insensitive mutant PKM2 Q393K was utilized (Keller et al., 2012; Supplementary Figure S1C). Together these results indicate that PEP is indeed a phosphate donor for the histone H3.1 mediated by the PKM2-SAICAR complex.
Figure 1. SAICAR induces protein kinase activity from recombinant PKM2.

(A) A schematic representation of the experiment. (B) Phosphorylation of recombinant histone H3.1 monomer (2 μM) by recombinant PKM2 (rPKM2, 10 nM) in the presence of PEP (0.10 mM) and ligands (FBP or SAICAR, 0.5 mM). The reaction mixture was incubated at 37°C for 5 min, quenched by the addition of SDS PAGE gel loading buffer, and subjected to SDS-PAGE. Phosphorylation of H3.1 T11 was monitored using anti-phospho-T11 H3 antibody as described in the Experimental Procedures. (C) Effect of H3.1 concentration on PKM2’s protein kinase activity. In the presence of SAICAR (0.5 mM), the reaction follows a typical Michaelis-Menten kinetics. FBP (0.5 mM) only marginally activates the kinase activity of PKM2. (D) Effect of PEP concentration on the phosphorylation of H3.1 T11 by the PKM2-SAICAR complex. (E and F) The inhibition of the PKM2-SAICAR mediated H3.1 T11 phosphorylation (E) by ADP and (F) by FBP. (G) Phosphorylation of purified and dephosphorylated Xenopus laevis nucleosome by PKM2-ligand complexes. (H) Effect of glucose and SAICAR concentration on cellular H3 T11 phosphorylation and the level of Myc. HeLa, adsl-kd, and paics-kd cells grown in DMEM (25 mM glucose) supplemented with dialyzed glucose were incubated in glucose-free DMEM medium supplemented with dialyzed FBS for 30 min before lysis and Western analysis. See also Figure S1, Figure S2, and Figure S3.
The protein kinase activity of the PKM2-SAICAR complex was inhibited by ADP (Figure 1E) in a manner similar to PKM2 purified from cancer cell nucleoplasm (Yang et al., 2012a) suggesting that the PKM2-SAICAR complex uses an identical active site for both protein and pyruvate kinase activities. The protein kinase activity of the SAICAR-PKM2 complex was inhibited by FBP, a glycolysis intermediate that activates PKM2’s pyruvate kinase activity (Figure 1F). Suppression of protein kinase activity of PKM2-SAICAR by FBP indicates that these PKM2-activating metabolites regulate PKM2 in two distinct mechanisms: FBP primarily induces PKM2’s pyruvate kinase activity while SAICAR activates both pyruvate (Keller et al., 2012) and protein kinase activities of PKM2.
Because there are three different subtypes of H3 and each plays distinct roles in mammals (Hake and Allis, 2006), we asked whether the PKM2-SAICAR complex displays H3 subtype specificity (Supplementary Figure S1D and S1E). Among H3 subtypes, only H3.1 was robustly phosphorylated by the PKM2-SAICAR complex, while phosphorylation of other H3 subtypes (H3.2 and H3.3) was not as efficient in vitro (Supplementary Figure S1D). These findings suggest that PKM2-SAICAR may distinguish between H3 subtypes.
Histone H3.1 is incorporated into nucleosome in a replication-dependent manner and mostly found as a component of nucleosome (Hake and Allis, 2006). Thus, we tested whether PKM2-SAICAR phosphorylates H3 when it is a part of a nucleosome. When dephosphorylated nucleosome purified from Xenopus laevis was used as a substrate, H3.1 T11 in the nucleosome was phosphorylated by PKM2-SAICAR as efficiently as the H3.1 monomer (Figure 1G).
Next, we asked if SAICAR induces the phosphorylation of histone H3 T11 in cancer cells. Using HeLa cells with altered SAICAR metabolism (Keller et al., 2012) we monitored the phosphorylation of H3 T11 and subsequent accumulation of c-Myc (Figure 1H). Previously, we reported that the cellular concentration of SAICAR is elevated when HeLa and other cancer cells were deprived of glucose (Keller et al, 2012). In addition, we also reported that knocking down the expression of SAICAR cleavage enzyme adenylosuccinate lyase (adsl-kd) constitutively elevates the cellular concentration of SAICAR while knocking down SAICAR synthase (paics-kd) prevents the accumulation of SAICAR (Keller et al, 2012). When HeLa cells were subjected to a SAICAR-accumulating condition (30 min in glucose-free medium), the phosphorylation of H3 T11 and the level of Myc protein were both elevated (Figure 1H). In cells constitutively accumulating SAICAR (adsl-kd cells), H3 T11 phosphorylation and Myc expression were both constitutively elevated (Figure 1H). Meanwhile, cells in which the SAICAR synthase was knocked down (paics-kd) did not show any evidence of H3 T11 phosphorylation nor Myc expression regardless of glucose conditions (Figure 1H). These results indicate that the elevation of SAICAR concentration is necessary to induce H3 T11 phosphorylation and Myc expression in HeLa cells.
In accordance with our previous in vitro result describing the H3-subtype specificity of the PKM2-SAICAR complex (Supplementary Figure S1D), the SAICAR-dependent phosphorylation of H3 T11 in HeLa cells was also H3-subtype specific (Supplementary Figure S1E). When HeLa, adsl-kd, and paics-kd cells were transfected with plasmids encoding hemagglutinin (HA)-tagged H3.1 or H3.3 (Kim et al., 2011), we observed subtype specific phosphorylation of H3 T11, which also correlates with cellular level of SAICAR. In HeLa cells, HA-tagged H3.1 T11 was phosphorylated in conditions accumulating SAICAR while phosphorylation of HA-tagged H3.3 T11 was never observed. In adsl-kd cells, H3.1 T11 was constitutively phosphorylated while the phosphorylation could not be induced in paics-kd cells. Phosphorylation of H3.3 T11 was not detectable in either HeLa or paics-kd cells. Marginal H3.3 phosphorylation was detectable in adsl-kd cells, albeit at far lower levels than H3.1. The level of Myc correlated with the phosphorylation of H3.1 T11 in all conditions tested. These results indicate that SAICAR accumulation is necessary to induce H3.1 T11 phosphorylation and Myc expression in HeLa cells.
SAICAR accumulation induces nuclear localization of PKM2
Most pyruvate kinases are localized in the cellular cytoplasm where glycolysis occurs. In normal conditions, PKM2 is also localized in cytoplasm and is excluded from the nucleus (Yang et al., 2011). However, stimulation of cells with epidermal growth factor (EGF) induces partial nuclear localization of PKM2 in glioblastoma cells (Yang et al., 2011).
As we found that SAICAR stimulates PKM2-mediated phosphorylation of a nuclear protein (histone H3.1) both in vitro and in cells (Figure 1), we speculated that SAICAR accumulation might be connected with the nuclear localization of PKM2. To test this possibility, cellular localization of endogenous PKM1/2 was probed in various conditions (Supplementary Figure S2). In HeLa cells maintained in glucose-rich medium in the absence of EGF, PKM2 was excluded from nucleus as expected (Supplementary Figure S2A). However, upon glucose depletion, PKM2 localized partially to the cell nucleus (Supplementary Figure S2A) in a manner similar to PKM2 in EGF-treated glioblastoma cells (Yang et al., 2011). Similar to glucose-deprivation, EGF-treatment also produced a similar localization pattern of endogenous PKM2 in HeLa cells (Supplementary Figure S2B). Cells constitutively accumulating SAICAR (adsl-kd cells) showed nuclear localization of PKM2 regardless of condition (Supplementary Figure S2C). When endogenous PKM2 was replaced with a SAICAR-insensitive PKM2 Q393K mutant (Keller et al., 2012) in HeLa cells, the subcellular localization change was not inducible by EGF, glucose withdrawal, or adsl-kd (Supplementary Figure S2D-E). These results indicate that the interaction between PKM2 and SAICAR is necessary for the nuclear localization of PKM2.
Due to the observed role of SAICAR in the nuclear localization of PKM2, we decided to examine the effect of SAICAR on the constitutively nuclear mutant PKM2 R399E (Gao et al. 2012; Supplementary Figure S3). Recombinant PKM2 R399E was purified from E. coli and the kinetic parameters measured. PKM2 R399E had a significantly lower Km for PEP than wild type PKM2 (20 μM), indicating that this mutant exists in the relaxed form (Supplementary Figure S3A). PKM2 R399E is hypersensitive to SAICAR both in pyruvate kinase activity (Supplementary Figure S3B; R399E EC50 SAICAR: 12 μM, PKM2 wild-type EC50 SAICAR: 300 μM) and in protein kinase activity using H3.1 as a substrate (Supplementary Figure S3C; R399E EC50 SAICAR: ~30 μM, PKM2 EC50 SAICAR: ~200–300 μM).
PKM2-SAICAR phosphorylates many proteins involved in cell proliferation
Our results show that SAICAR induces protein kinase activity of PKM2 in vitro, and that this activity is necessary for histone H3.1 T11 phosphorylation in HeLa cells. Next, to better understand the extent of this novel protein kinase activity, we searched for additional human proteins phosphorylated by PKM2-SAICAR. For the purpose, a human proteome microarray representing 9,479 recombinant human proteins was incubated in a solution containing PEP and PKM2 in the presence or the absence of SAICAR (Figure 2). Proteins phosphorylated were then visualized using ProQ Diamond stain. Because PEP is the only phosphate donor in this experiment, it is unlikely that the results will be influenced by the presence of immobilized protein kinases, which use ATP.
Figure 2. Protein microarray analysis revealed proteins phosphorylation by the PKM2-SAICAR complex.
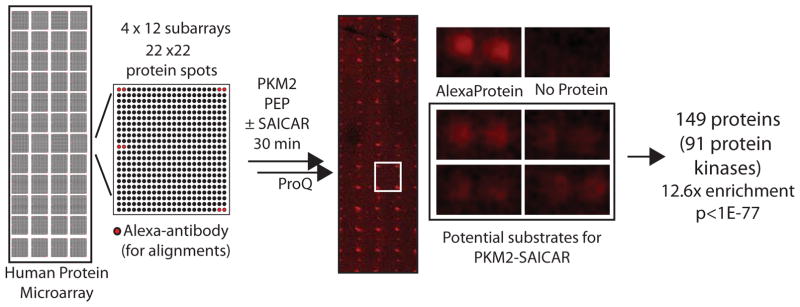
Schematic overview of the protein microarray used is shown. The array contains 48 (4 × 12) sub-arrays. In each sub-array, there are 484 (22 × 22) spots, including Alexa-labeled antibody spots used to align the array images. Proteins or controls were spotted in doublets. Most proteins were also immobilized in another subarray (total 4 spots). The protein array was incubated with 5 mL solution containing rPKM2 with or without SAICAR for 30 min followed by the addition of PEP. After a 30 min reaction, phosphorylated proteins were detected with ProQ Diamond staining as described in detail in the Supplementary Information. A representative protein microarray result is shown along with zoomed images. The list of potential PKM2-SAICAR substrates identified from this experiment is available in the Supplementary Table S1.
Using this approach, more than 150 proteins were identified as potential substrates for PKM2-SAICAR (Supplementary Table S1). To our surprise, the vast majority of these proteins were protein kinases (more than 80 human protein kinases; Table S1; Gene ontology (GO) term enrichment: p < 1e-77; 12.6-fold enrichment of GO:0004672, “protein kinase activity”). These potential PKM2-SAICAR substrates include both receptor kinases such as EGFR and non-receptor protein kinases. In particular, many protein kinases involved in the mitogen-activated protein kinase (MAPK) pathway (MAPK1, MAPK3, MAP2K1, MAP2K2, MAP3K11, MAP3K2, MAP3K3) (Chang and Karin, 2001) were identified as potential substrates of PKM2-SAICAR. Among the proteins phosphorylated by the PKM2-SAICAR complex, at least 23 (ABL1, ABL2, ALK, CCDC6, CHEK2, EGFR, ERBB2, FGFR1, FGFR2, FGFR3, JAK2, JAK3, KDR, KIT, MAP2K1, MAP2K2, MET, NTRK3, PCM1, PDGFRA, RET, ROS1, and SYK) have been implicated in cancer at the genetic level, according to the Cancer Gene Atlas database (http://cancergenome.nih.gov/). Several additional proteins (e.g. Cdc25A, Cdk2, Cdk4) involved in cell cycle progression (Nilsson and Hoffmann, 2000; Bartek et al., 2004) were also identified as potential substrates for PKM2-SAICAR. Interestingly, recent work has identified Bub3, a spindle assembly checkpoint point, as a substrate for PKM2 (Jiang et al., 2013). It is not yet clear what features of these proteins are recognized by PKM2-SAICAR. However, these results clearly indicate that preferred substrates for PKM2-SAICAR are protein kinases and other proteins involved in cell proliferation.
Next, we individually tested phosphorylation of several selected potential substrates (Figure 3). Because proteins involved in the MAPK pathway (Pearson et al., 2001; Chang and Karin, 2001) were enriched in our protein microarray results, we focused on validating these proteins as substrates of PKM2-SAICAR. As Erk1/2 (also known as MAPK3 and MAPK1, respectively; Roskoski, 2012) has been reported to be involved in the regulation of PKM2 (Yang et al., 2012b), we first tested whether PKM2-SAICAR phosphorylates Erk1 using PEP as a phosphate donor (Figure 3A). Indeed, PKM2-SAICAR phosphorylates recombinant Erk1 expressed and purified from E. coli, when the reaction product was analyzed using ProQ Diamond phosphoprotein staining. The phosphorylation was inhibited by ADP and, to a lesser degree, by FBP, in a manner similar to the phosphorylation of H3.1 T11 by PKM2-SAICAR. When the reaction product was probed with an antibody specific to Erk1/2 phosphorylated at its activation loop threonine (T202 in human Erk1) and/or tyrosine (Y204 in human Erk1) residues, the antibody effectively recognized Erk1 phosphorylated by PKM2-SAICAR. This suggests that PKM2-SAICAR may phosphorylate either T202 or Y204 of Erk1. As discussed later, we indeed found that T202 is the likely site of phosphorylation. Similar to histone H3.1, the activation of protein kinase activity by SAICAR abolished when the SAICAR-insensitive PKM2 Q393K mutant is utilized (Supplementary Figure S3D).
Figure 3. Phosphorylation of EGFR/MAPK signaling proteins by PKM2-SAICAR complex.

(A) Phosphorylation of recombinant Erk1 by the PKM2-SAICAR complex. Recombinant Erk1 (1 μM) was subjected to the phosphorylation by PKM2 in the presence or absence of various ligands (0.5 mM each) and probed with SDS-PAGE and (from top to bottom) ProQ Diamond phosphoprotein staining, Western blotting with anti-active Erk1/2 antibody, with anti-Erk1/2 antibody, and with anti-PKM2 antibody. (B) Effect of PKM2-SAICAR on the activity of Erk1. Protein kinase activity of Erk1, after treatment with PKM2 in the absence or in the presence of 0.50 mM SAICAR, was monitored using a fluorogenic Erk1/2 activity assay kit as described in detail in the Experimental Procedures. ‘A’ denotes the use of commercial active (MEK-phosphorylated) Erk2 instead of Erk1. Means +/- SEM. are noted (n = 3). (C) Identification of residue pphosphorylated by PKM2-SAICAR. Recombinant Erk1, Erk1 T202A, and Erk1 T204F were subjected to phosphorylation in the presence or absence of 0.5 mM SAICAR and probed with anti-active Erk1/2 antibody. (D) Phosphorylation of other proteins by the PKM2-SAICAR complex. A few positive hits (EGFR, MEK1, PAK2) and negative hits (B-Raf and Grb2) from Figure 2 are individually tested for the phosphorylation by PKM2 in the presence of various ligands. Detailed experimental procedures are described in the Experimental Procedures and in the Supplementary Information. See also Figure S4.
As PKM2-SAICAR phosphorylates the activation loop of Erk1 in vitro, we tested the effect of this phosphorylation on its enzymatic activity. Erk1, like other canonical MAPKs, is an inefficient catalyst without activation. Full activation of Erk1 requires two phosphorylation reactions of the activation loop T202 and Y204 (Roskoski, 2012). The phosphorylation by PKM2-SAICAR activated Erk1 more than 150-fold (Figure 3B). In comparison, dually phosphorylated Erk2 (denoted with an ‘A’ in Figure 3B) was even more active than Erk1 phosphorylated with PKM2-SAICAR. This result is consistent with the reported kcat value of Erk1/2 phosphorylated at T202 alone (reported kcat values for non-phosphorylated, pT, pY, and pT-pY Erk2 are 1, 227, 620, and 8940 s−1, respectively; Sugden et al., 2011). SAICAR-insensitive PKM2 Q393K mutant did not phosphorylate Erk1 even in the presence of SAICAR (Supplementary Figure S3D). Thus, Erk1 was indeed phosphorylated and partially activated by PKM2-SAICAR, at least in vitro.
Typically, in MEK-mediated phosphorylation of Erk1/2, the phosphorylation of the activation loop tyrosine precedes the phosphorylation of the activation loop threonine (Haystead et al., 1992). However, threonine mono-phosphorylated Erk1/2 was also has been detected in cells (Sugden et al., 2011). To further locate the phosphorylation site, recombinant Erk1 mutants whose activation loop residues were mutated (T202A and Y204F). PKM2-SAICAR was unable to phosphorylate Erk1 T202A, but it was still able to phosphorylate the Y204F mutant (Figure 3C). These data show that the T202 is the likely site of phosphorylation, and the phosphorylation of Erk1 by PKM2-SAICAR appears distinct from the MEK-mediated Erk1/2 activation.
In addition to Erk1, we also tested the phosphorylation by PKM2-SAICAR of several additional selected recombinant proteins found to be phosphorylated by PKM2-SAICAR in the protein microarray experiment (Figure 2 and 3D). When commercial recombinant epidermal growth factor receptor (EGFR; Carpenter, 1987) expressed and purified from insect cells was tested as a substrate, it was phosphorylated in a manner dependent on PKM2, SAICAR, and PEP. Similar results were observed with other recombinant proteins (MEK1 and PAK2) identified as potential PKM2 substrates from the microarray experiment MAPK pathway proteins (B-Raf and Grb2) not phosphorylated by PKM2-SAICAR in the microarray experiment were subjected to individual tests; none were phosphorylated by PKM2-SAICAR.
PKM2-SAICAR complex phosphorylates protein substrates in cells
Our results indicate that there is an intimate relationship between PKM2-SAICAR and EGF/MAPK signaling. Next, we asked if SAICAR is functionally involved in the EGF/MAPK signaling in cancer cells. We first measured cellular levels of SAICAR in HeLa and in H1299 cells before and after EGF addition (Figure 4A). Cellular SAICAR levels were elevated rapidly in these cells upon EGF addition (Figure 4A and Supplementary Figure S4) to a level sufficient to activate PKM2 (EC50 300 μM; Keller et al. 2012). Thus, SAICAR may be a relevant ligand for PKM2 upon EGFR activation in addition to previously reported glucose-deprived cells.
Figure 4. SAICAR-PKM2 interaction is necessary and sufficient to induce H3 T11 and Erk1/2 phosphorylation.

(A) Addition of EGF (10 ng/mL; 2 hrs) is sufficient to induce cellular concentration of SAICAR in HeLa and in H1299 cells. A time-course change of cellular SAICAR concentration upon EGF addition to HeLa cells is available in the Supplementary Information. Mean +/- SEM are noted (n = 3). (B and C) Effect of PKM2-SAICAR interaction on EGF-dependent phosphorylation of H3 T11 and MYC expression. Cells were incubated in EGF-free DMEM (DMEM, 25 g/L glucose, 10% dialyzed FBS) or for 3 hrs in medium supplemented with 10 ng/mL EGF. ‘M1*’ and ‘QK’ denote cells in which endogenous PKM1 and PKM2 were knocked down, and PKM1 or SAICAR-insensitive PKM2 Q393K mutant from plasmids were expressed, respectively. (D) Effect of PKM2-SAICAR interaction on Erk1/2 activation.
Next, we asked if PKM2-SAICAR is involved in EGF-induced protein phosphorylation in cells. We first tested the phosphorylation of H3 T11 in cells (Figure 4B and 4C). H3 pT11 and Myc levels were both elevated upon EGFR activation in HeLa cells as previously reported (Yang et al., 2012a). In cells constitutively accumulating SAICAR (adsl-kd; Keller et al, 2012), both H3 pT11 and Myc levels were constitutively elevated (Figure 4B and 4C). When endogenous PKM2 was replaced with SAICAR-insensitive PKM1 (Figure 4B) or PKM2 mutant (Q393K; Keller et al., 2012; Figure 4C), EGF addition was not able to induce H3 T11 phosphorylation or Myc expression (Figure 4B and 4C) regardless of cellular SAICAR level. Our data suggests that PKM2-SAICAR is necessary for EGF-induced phosphorylation of H3 T11 and the subsequent accumulation of Myc. The phosphorylation of H3 T11 by PKM2-SAICAR in EGF-treated cells was also specific to H3.1 subtype (Supplementary Figure 2).
In a similar manner, we probed the activation of Erk1/2 in HeLa cells (Figure 4D). In HeLa cells, addition of EGF induces activation of Erk1/2 as expected. Cells with constitutively elevated levels of SAICAR (adsl-kd) showed constitutive activation of Erk1/2, while paics-kd cells were unable to activate Erk1/2 in response to EGF (Figure 4D). Additionally, the PKM2-SAICAR interaction was required for Erk1/2 activation in cells, as replacing endogenous PKM2 with the SAICAR-insensitive PKM2 Q393K mutant (QK in Figure 4D) was sufficient to prevent Erk1/2 activation in adsl-kd cells. Overall, our results show that PKM2-SAICAR phosphorylates protein substrates (histone H3 and Erk1/2) in cancer cells.
Positive feedback regulation between PKM2-SAICAR and Erk1/2
Recently, Erk2 was reported to phosphorylate PKM2, which is required for PKM2’s nuclear translocation and subsequent protein kinase activity (Yang et al., 2012b). Along with our data, these results indicate that PKM2-SAICAR and Erk1/2 may form a positive feedback regulation loop, where the activation of one protein renders the other more likely to be activated. To test this possibility, we phosphorylated recombinant PKM2 with constitutively active Erk2. After phosphorylation, recombinant PKM2 was repurified and subjected to pyruvate kinase (Figure 5A) and histone H3 T11 kinase (Figure 5B) activity assays. Upon the Erk2-mediated phosphorylation, PKM2 became far more sensitive to SAICAR (EC50 shifts from ~ 0.3 mM to ~ 10 μM SAICAR). Considering that the cellular concentration of SAICAR in non-stimulated cancer cells ranges between 10–100 μM (Keller et al, 2012), our results indicate that Erk-mediated phosphorylation sensitizes PKM2 to SAICAR, even when the cellular concentration of SAICAR is restored to the original level. Our results, thus, support the previous finding that the phosphorylation of PKM2 alone induces nuclear localization and cell proliferation in cells (Yang et al, 2012b). Together, these results indicate that PKM2-SAICAR and Erk1/2 are mutually sensitizing each other to promote the MAPK protein kinase signaling (Figure 5C).
Figure 5. SAICAR-PKM2 interaction is necessary and sufficient for the sustained proliferative signaling.

br>(A and B) Phosphorylation of PKM2 by active Erk1 sensitizes PKM2 for SAICAR binding. Recombinant PKM2 was phosphorylated by active GST-tagged Erk1. After re-purification of PKM2, the effect of the phosphorylation on PKM2’s sensitivity was measured (A) by monitoring pyruvate kinase activity (mean +/- SEM. are noted; n =3) and (B) by monitoring H3.1 T11 kinase activity. Upon phosphorylation by Erk1/2, PKM2 becomes more sensitive (EC50 0.30 mM and 12 μM SAICAR before and after the phosphorylation by Erk1/2, respectively). (C) A schematic representation summarizing the data shown in this manuscript. (D) Effect of SAICAR-PKM2 on the activation of Erk1/2. HeLa, adsl-kd, QK, and adsl-kd QK, SA (cells whose PKM2 is replaced with PKM2 S37A mutant), and adsl-kd SA cells were grown in DMEM (25 mM glucose) supplemented with dialyzed FBS, briefly (30 min) incubated in medium containing 10 ng/mL recombinant EGF, and then incubated in EGF-free medium. Cells were harvested at noted time points, and Erk1/2 activation was monitored by Western blot, probing with antibody specific to activated Erk1/2. (E) Effect of SAICAR-PKM2 on cell proliferation. HeLa cells and their derivatives (105 cells) were seeded in a well (6-well plates) for 24 hours, followed by a 24-hour incubation in medium with or without 10 ng/mL EGF. Cell number was then measured. See also Figure S5.
PKM2-SAICAR is necessary and sufficient for proliferation signaling
A common consequence of a positive feedback loop is a switch-like behavior (Mitrophanov and Groisman, 2008). Because PKM2-SAICAR and Erk1/2 form a positive feedback loop in vitro, PKM2-SAICAR may contribute to the sustained proliferative signaling, a hallmark of cancer cells (Hanahan and Weinberg, 2012). To test this possibility, HeLa cells and their derivatives were briefly (30 min) incubated with a medium containing EGF. The medium was then replaced with medium lacking EGF, and the activation of Erk1/2 over time was monitored (Figure 5D). In comparison with non-transfected HeLa cells, adsl-kd cells showed a more rapid activation of Erk1/2, which supports our view that PKM2-SAICAR primes Erk1/2 activation (Figure 4A–B). In addition, the activation of Erk1/2 in adsl-kd cells was sustained for a longer period of time (Figure 5D). Replacing PKM2 with the SAICAR-insensitive PKM2 Q393K (or QK) mutant in wild-type or adsl-kd cells showed only marginal activation of Erk1/2 in comparison to HeLa cells, indicating that PKM2-SAICAR is a core component required to sustain Erk1/2 activation (Figure 5D).
To further examine our proposed positive feedback loop, we next replaced PKM2 with the PKM2 S37A mutant insensitive to Erk1/2 (Yang et al. 2012b). First, we purified recombinant PKM2 S37A and determined the kinetic parameters. In our hands, the activity of PKM2 S37A is slightly lower than wild type PKM2 (Supplementary Figure S5A, kcat: 0.5 s−1, wild type kcat: 2–4 s−1). PKM2 S37A is still sensitive to SAICAR in both pyruvate kinase activity (Supplementary Figure S5B, EC50 ~ 250 μM SAICAR) and protein kinase activity (Supplementary Figure S5C) assays. We then replaced wild type PKM2 with PKM2 S37A mutant in HeLa and adsl-kd cells (‘SA’ and ‘SA adsl-kd’ in Figure 5D). When Erk1/2 signalings in these cells were monitored, the activation of Erk1/2 in PKM2 S37A cells was more rapidly diminished (Figure 5D). This result is consistent with the mutual sensitization model (Figure 5C).
We next sought to determine whether the PKM2-SAICAR interaction is necessary for mitogen-induced cell proliferation (Figure 5E). HeLa cells and their derivatives were incubated for one day in the presence and in the absence of EGF, and the population size increase was monitored. As expected, EGF stimulated the proliferation of HeLa cells, while adsl-kd cells showed constitutive hyper-proliferation. Conversely, EGF only marginally stimulated the proliferation of paics-kd cells. Additionally, cells where endogenous PKM2 was replaced with the PKM2 Q393K mutant were also nearly insensitive to EGF. Thus, our results supports that SAICAR and its interaction with PKM2 are necessary for the mitogen-induced proliferation of cancer cells. Finally, we replaced endogenous PKM2 is replaced with the non-phosphorylatable S37A mutant, which is reported to be excluded from the nucleus (Yang et al. 2012b). EGF did not significantly stimulate proliferation in HeLa cells with PKM2 S37A mutant (Figure 5E). Cells over-accumulating SAICAR (adsl-kd) were also affected by the replacement of PKM2 with PKM2 S37A mutant. These results show that the sustained Erk signaling mediated by the mutual sensitization of PKM2-SAICAR and Erk is a necessary component for the mitogen-induced proliferation.
DISCUSSION
Protein kinases are known to play important regulatory roles in many biological processes. In eukaryotic cells, well-defined families of protein kinases catalyze protein phosphorylation. These typical protein kinases use ATP as a phosphate donor, and share common structural and sequence features. In addition to these classical protein kinases, there has been speculation that other energetic molecules, like PEP, may play a role in protein phosphorylation in mammalian cells, but an enzyme catalyzing this reaction was not identified (Jeyasingham and Carlson, 1998; Khandelwal et al., 1983). In bacteria, PEP can be used for protein phosphorylatione, as shown in the PEP-dependent sugar phosphotransferase system of Bacillus (Postma et al., 1993). PKM2 is the first identified mammalian protein, to our knowledge, that phosphorylates protein substrates using PEP as a phosphate donor. However, because recombinant PKM2 alone was unable to reproduce the results obtained with PKM2 purified from cancer cells, investigating the molecular basis of this novel protein kinase activity was difficult. In this paper, we showed that metabolite binding is sufficient to induce the protein kinase activity of PKM2. Our results can expand our understanding of protein kinases by identifying a new dimension of regulation.
In addition to previously known histone H3 (Yang et al., 2012a), we also identified many human proteins as potential substrates for PKM2-SAICAR, with the vast majority of proteins involved in the regulation of cell proliferation. Further investigation on how PKM2-SAICAR recognizes its substrates may reveal a previously unrecognized feature common to these proteins involved in cell cycle regulation.
Previous work on PKM2 has revealed an interaction between MAPK activity and PKM2, showing that Erk1/2 phosphorylates PKM2 (Yang et al., 2012b). In our work, the reciprocal reaction – phosphorylation of MAPK protein kinases by PKM2 – also occurs in vitro and in cells upon the binding of SAICAR to PKM2. Our result challenges the view that PKM2 is a passive recipient of MAPK protein kinase signaling and suggests that PKM2 is a more active component that is necessary for the MAPK protein kinase signaling.
Intricately related to MAPK activity is cell cycle control and proliferation. As has been well documented, malignant tumor cells frequently display sustained proliferation. Many different mechanisms such as mutations in genes encoding MAPK signaling contribute to the sustained proliferation of cancer cells. However, many types of cancer cells show sustained proliferation without showing any mutations in these genes. The altered metabolism associated with most cancer cells, including upregulation of PKM2, may thus be a mechanism to sustain cell proliferation in those cells.
EXPERIMENTAL PROCEDURES
Protein kinase assays
The reaction solution [0.1–10 nM PKM2, 0–4 μM histone (NEB, typically 1 μM H3 monomer concentration), 150 μM PEP, 50 mM HEPES, pH 7.4, 100 mM potassium chloride, 6.2 mM magnesium acetate, and 5% glycerol] was incubated at 25°C for 1–120 minutes (typically 5 min). After the reaction, the solution was mixed with an equal volume of SDS-gel loading buffer (375 mM Tris-HCl, pH 8, 10% SDS, 50% glycerol, 1 mM DTT, 0.1% bromophenol blue) and incubated at 95°C for 10 minutes. All other protein kinase assays were performed as described, with a fixed concentration of PKM2 (10 nM), for 30 minutes, unless specifically noted otherwise. Solutions were subjected to SDS-PAGE, staining with ProQ Diamond (Invitrogen), or Western blot analysis. For the determination of catalytic efficiency (kcat), a reaction was carried out with higher concentration of PKM2 (10 nM) for 1 hour, and the resulting signal was assumed to represent the completed reaction.
Western Blot
Western blots were performed as described by AbCam (http://www.abcam.com/ps/pdf/protocols/WB-beginner.pdf). In brief, cells were lysed in ice-cold RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate) supplemented with 1 mM phenylmethylsulfonyl fluoride and 1 mM sodium fluoride, and total protein amount was measured using a Pierce 660 reagent (Thermo Scientific) with bovine serum albumin as a standard. Proteins were separated by SDS-PAGE, and transferred to nitrocellulose membranes. The membrane was blocked with phosphate-buffered saline (PBS) supplemented with 5% bovine serum albumin, and probed with the appropriate primary antibody (rabbit anti-phospho-p42/p44, rabbit anti-p42/p44, rabbit anti-pT11-Histone H3, and rabbit anti-Histone H3 from Cell Signaling; rabbit anti-GAPDH and mouse-anti-Myc from Sigma-Aldrich) overnight at 4°C. After primary antibody incubation, membranes were probed with horseradish peroxidase (HRP) conjugated goat anti-rabbit IgG or goat anti-mouse IgG secondary antibodies (Bio-Rad). HRP-conjugated secondary antibodies were then detected using commercial chemiluminescence substrates (Pierce). Chemiluminescence images were acquired using a FluorChem M FM0455 imager.
Protein microarray experiments
Protein microarray experiments were carried out using Invitrogen Human protein microarrays, recombinant PKM2, and PEP in the presence and in the absence of SAICAR. Phosphorylated proteins were detected using ProQ Diamond stain (Invitrogen). An extensive description of this experiment is provided in the Supplementary Information.
Phosphorylation of PKM2
Recombinant PKM2 (1 μM) and recombinant GST-Erk1 (10 nM) were incubated together in a buffer containing 1 mM ATP, 50 mM Tris-HCl, pH 7.0, 150 mM NaCl, 1 mM DTT, 1 mM MgCl2 for 30 minutes at 25°C. The reaction solution was passed through glutathione-agarose resin (Sigma-Aldrich) to deplete GST-Erk1. Unbound proteins were eluted with 50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT, 1 mM MgCl2 buffer. Fractions were then subjected to SDS-PAGE and either Coomassie or Pro-Q diamond staining (Invitrogen). Fractions containing phosphorylated PKM2 were identified using Pro-Q diamond staining. Concentration of phosphorylated PKM2 was determined by measuring A280 in denaturing conditions (Edelhoch, 1969) using a calculated extinction coefficient (28,910 M−1 cm−1).
Erk1/2 activity assay
A 10 μL reaction (25 nM rPKM2, 1 μM rErk1, 150 μM PEP, 50 mM HEPES pH 7.4, 100 mM potassium chloride, 10% glycerol, 6.2 mM magnesium chloride, and 1 mM DTT) was incubated for 30 minutes at room temperature. The Erk1 reaction was serially diluted using water, and 8 μL of diluted solution was added to 32 μL Omnia Kinase Assay solution (10 μM Omnia Peptide 17, 1 mM ATP, 0.2 mM DTT, and proprietary buffer components from the vendor; Invitrogen) in a black transparent bottom 96-well plate. The emission at 485 nm (excitation at 360 nm) was recorded every minute for 60 minutes at 30°C using a Tecan Infinity M2 fluorescence microplate reader.
Supplementary Material
HIGHLIGHTS.
The binding of SAICAR induces protein kinase activity of PKM2
The PKM2-SAICAR complex phosphorylates >100 human proteins.
The PKM2-SAICAR complex activates Erk1 in vitro and in cells.
PKM2-SAICAR is necessary for sustained proliferative signaling.
Acknowledgments
We thank B. Wendland, G. Bowman, H. Zhao, and members of our department for reagents and for allowing access to their instruments, E. Pryce and J. M. McCaffery for technical assistance with fluorescence microscopy, T. Coupet for preparation of reagents, and W. An (U. Southern California) for plasmids encoding HA-tagged H3.1 and H3.3. We also thank to R. Kuruvilla, X. Chen, H. Zhao, G. Bowman and members of our department for their comments on this manuscript. K.E.K. was supported by an NIH training grant (T32GM007231) to the Department. This work was supported in part by an NIH grant (R01CA168658), the Sidney Kimmel Foundation for Cancer Research Kimmel Scholar’s Award (SKF-13-082), and the Johns Hopkins University Krieger School of Arts and Sciences startup package to Y-S.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Carpenter G. Receptors for epidermal growth-factor and other polypeptide mitogens. Annu Rev Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- Chang LF, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30:4297–4306. doi: 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumor growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP. Glycolytic network restructuring integral to energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol. 2010;48:725–734. doi: 10.1016/j.yjmcc.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews CM, Erickson RL. Extracellular signals and reversible protein phosphorylation – What to MEK of it all. Cell. 1993;74:215–217. doi: 10.1016/0092-8674(93)90411-i. [DOI] [PubMed] [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1969;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Bamezai RN. Human pyruvate kinase M2: a multifunctional protein. Protein Sci. 2013;19:2031–2044. doi: 10.1002/pro.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc Natl Acad Sci USA. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Haystead TAJ, Dent P, Wu J, Haystead CMM, Sturgill TW. Ordered phosphorylation of p42MAPK by MAP kinase kinase. FEBS Lett. 1992;306:17–22. doi: 10.1016/0014-5793(92)80828-5. [DOI] [PubMed] [Google Scholar]
- Jeyasingham MD, Carlson GM. Evaluation of phosphoenolpyruvate as a phosphoryl group donor for phosphoproteins in skeletal muscle. Arch Biochem Biophys. 1998;357:285–292. doi: 10.1006/abbi.1998.0795. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Li X, Yang W, Hawke DH, Xia Y, Aldape K, Wei C, Guo F, Chen Y, Lu Z. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Molecular Cell. 2014 doi: 10.1016/j.molcel.2013.11.001. pii: S1097-2765(13)00826-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal RL, Mattoo RL, Waygood EB. Phosphoenolpyruvate-dependent protein kinase activity in rat skeletal muscle. FEBS Lett. 1983;162:127–132. doi: 10.1016/0014-5793(83)81063-1. [DOI] [PubMed] [Google Scholar]
- Keller KE, Tan IS, Lee YS. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science. 2012;338:1069–1072. doi: 10.1126/science.1224409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Heo K, Choi J, Kim K, An W. Histone variant H3.3 stimulates HSP70 transcription through cooperation with HP1 γ. Nucleic Acids Res. 2011;39:8329–8341. doi: 10.1093/nar/gkr529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim HK, Han YM, Kim J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int J Biochem Cell Biol. 2008;40:1043–1054. doi: 10.1016/j.biocel.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab. 2012;23:560–566. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E. Pyruvate kinase type M2: a crossroad in the tumor metabolome. Br J Nutr. 2002;S1:S23–S29. [PubMed] [Google Scholar]
- Mazurek S. Pyurvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Mitrophanov AY, Groisman EA. Positive feedback in cellular control systems. Bioessays. 2008;30:542–555. doi: 10.1002/bies.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I, Hoffmann I. Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res. 2000;4:107–114. doi: 10.1007/978-1-4615-4253-7_10. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Gibson TB, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Postma PW, Lengeler JW, Jacobson GR. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Mol Biol Rev. 1993;57:543–594. doi: 10.1128/mr.57.3.543-594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr Erk1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Markou T, Fuller SJ, Tham EL, Molkentin JD, Paterson HF, Clerk A. Monophosphothreonyl extracellular signal-regulated kinases 1 and 2 (ERK1/2) are formed endogenously in intact cardiac myocytes and are enzymically active. Cell Signal. 2011;23:468–477. doi: 10.1016/j.cellsig.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG. Exploiting tumor metabolism: challenges for clinical translation. J Clin Invest. 2013;123:3648–3651. doi: 10.1172/JCI72391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012a;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. Erk1/2-dependent phosphorylation and nuclear translocalization of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012b;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.