

Significance
The specific cause of the long-term decrease in stable nitrogen isotope ratio (15N/14N) of ice core nitrate beginning ∼1850 is a subject of debate, hindering the efforts to understand changes in the global nitrogen cycle. Our high-resolution record of ice core 15N/14N combined with model calculations suggests that the decrease is mainly caused by equilibrium shift in gas−particle partitioning of atmospheric nitrate due to increasing atmospheric acidity resulting from anthropogenic emissions of nitrogen and sulfur oxides. Our high-resolution record also reveals a leveling off of 15N/14N ∼1970, synchronous with changes in acidity and sulfate and nitrate concentrations. This leveling off suggests a measurable reduction in air pollution following the implementation of the US Clean Air Act of 1970.
Keywords: fossil fuel emissions, proxy, industrial, acid deposition, clean air act
Abstract
Nitrogen stable isotope ratio (δ15N) in Greenland snow nitrate and in North American remote lake sediments has decreased gradually beginning as early as ∼1850 Christian Era. This decrease was attributed to increasing atmospheric deposition of anthropogenic nitrate, reflecting an anthropogenic impact on the global nitrogen cycle, and the impact was thought to be amplified ∼1970. However, our subannually resolved ice core records of δ15N and major ions (e.g.,  ,
, 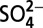 ) over the last ∼200 y show that the decrease in δ15N is not always associated with increasing
) over the last ∼200 y show that the decrease in δ15N is not always associated with increasing  concentrations, and the decreasing trend actually leveled off ∼1970. Correlation of δ15N with H+,
concentrations, and the decreasing trend actually leveled off ∼1970. Correlation of δ15N with H+,  , and HNO3 concentrations, combined with nitrogen isotope fractionation models, suggests that the δ15N decrease from ∼1850–1970 was mainly caused by an anthropogenic-driven increase in atmospheric acidity through alteration of the gas−particle partitioning of atmospheric nitrate. The concentrations of
, and HNO3 concentrations, combined with nitrogen isotope fractionation models, suggests that the δ15N decrease from ∼1850–1970 was mainly caused by an anthropogenic-driven increase in atmospheric acidity through alteration of the gas−particle partitioning of atmospheric nitrate. The concentrations of  and
and 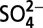 also leveled off ∼1970, reflecting the effect of air pollution mitigation strategies in North America on anthropogenic NOx and SO2 emissions. The consequent atmospheric acidity change, as reflected in the ice core record of H+ concentrations, is likely responsible for the leveling off of δ15N ∼1970, which, together with the leveling off of
also leveled off ∼1970, reflecting the effect of air pollution mitigation strategies in North America on anthropogenic NOx and SO2 emissions. The consequent atmospheric acidity change, as reflected in the ice core record of H+ concentrations, is likely responsible for the leveling off of δ15N ∼1970, which, together with the leveling off of  concentrations, suggests a regional mitigation of anthropogenic impact on the nitrogen cycle. Our results highlight the importance of atmospheric processes in controlling δ15N of nitrate and should be considered when using δ15N as a source indicator to study atmospheric flux of nitrate to land surface/ecosystems.
concentrations, suggests a regional mitigation of anthropogenic impact on the nitrogen cycle. Our results highlight the importance of atmospheric processes in controlling δ15N of nitrate and should be considered when using δ15N as a source indicator to study atmospheric flux of nitrate to land surface/ecosystems.
Since the beginning of the Industrial Revolution, human activities have profoundly changed the biogeochemical cycle of reactive nitrogen (Nr) through anthropogenic NOx (NOx = NO + NO2) emissions [∼90% from fossil fuel combustion, ∼10% from N-fertilized soil (1)]. Increased atmospheric NOx concentration has impacted the tropospheric oxidative capacity (2). In addition, input of anthropogenic Nr to the biosphere can increase the productivity of N-deficient ecosystems and alter ecosystems in relatively N-rich states inducing ecological changes that have impacts on carbon uptake rates (3).
The primary sink of NOx is formation of atmospheric nitrate that is distributed between gas phase (HNO3(g)) and/or aerosol phase (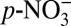 ). Nitrate is removed from the atmosphere via wet and dry deposition. Once deposited, nitrate can be preserved in polar ice sheets, or converted to and preserved as organic nitrogen in sediments. The stable nitrogen isotope ratio (δ15N) of nitrogen species (i.e., nitrate, total nitrogen) in natural archives has been tied to NOx source signatures and used to investigate climatic- (4) and/or anthropogenic-driven (5, 6) changes in NOx sources, providing information not available from concentration measurement alone. A thorough understanding of the records of nitrogen species and their δ15N signatures is important for assessing the history of NOx emissions and atmospheric Nr chemistry, and the deposition patterns of Nr. This understanding will further contribute to projections of future changes in atmospheric Nr chemistry and the potential impact on the global nitrogen cycle in response to continued anthropogenic NOx emissions.
). Nitrate is removed from the atmosphere via wet and dry deposition. Once deposited, nitrate can be preserved in polar ice sheets, or converted to and preserved as organic nitrogen in sediments. The stable nitrogen isotope ratio (δ15N) of nitrogen species (i.e., nitrate, total nitrogen) in natural archives has been tied to NOx source signatures and used to investigate climatic- (4) and/or anthropogenic-driven (5, 6) changes in NOx sources, providing information not available from concentration measurement alone. A thorough understanding of the records of nitrogen species and their δ15N signatures is important for assessing the history of NOx emissions and atmospheric Nr chemistry, and the deposition patterns of Nr. This understanding will further contribute to projections of future changes in atmospheric Nr chemistry and the potential impact on the global nitrogen cycle in response to continued anthropogenic NOx emissions.
A gradual decrease beginning as early as 1850 was observed in δ15N of nitrogen species in Greenland ice cores (5) and sediments of remote lakes in North America (6). This decrease has been attributed to direct input of atmospheric nitrate with low δ15N originating from fossil fuel combustion and/or fertilizer sources (5−7) and viewed as new evidence of the anthropogenic impact on the global nitrogen cycle. However, measurements of δ15N of NOx and atmospheric nitrate suggest that the δ15N from fossil fuel combustion is unlikely to be lower than that from natural sources (Table S1). A more recent study (7) attributed the decrease exclusively to NOx emissions from N-fertilized soil, assuming a significant (up to 40%) contribution of soil-emitted NOx to the atmospheric Nr budget. Greenland ice core records, however, indicate no increasing trend in  concentrations over the past 200 y (8), which should be expected as the precursors of
concentrations over the past 200 y (8), which should be expected as the precursors of  and
and 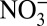 (NH3 and NOx, respectively) are produced simultaneously by soil biogenic activities.
(NH3 and NOx, respectively) are produced simultaneously by soil biogenic activities.
It is known that atmospheric processes, including NOx cycling (9, 10) and the partitioning of nitrate between the gas and aerosol phase (11, 12), affect δ15N of atmospheric Nr. In addition, after nitrate deposition to snow, postdepositional loss tends to increase δ15N remaining in snow (13, 14). Therefore, understanding the trend in δ15N observed in natural archives requires a comprehensive evaluation of these processes. To discern the exact cause of the decrease in δ15N in the industrial era, we measured the concentration and δ15N of nitrate in an ice core from Summit, Greenland [72°36’N, 38°30’W, 3200 meters above sea level (m.a.s.l.)] at subannual resolution over 235 y of snow accumulation (1772–2006). Profiles of soluble chemical impurities (Cl−, 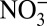 ,
, 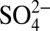 , Na+,
, Na+,  , K+, Mg2+ and Ca2+) spanning the same time period were obtained from a nearby parallel core. We examine the relationships among δ15N,
, K+, Mg2+ and Ca2+) spanning the same time period were obtained from a nearby parallel core. We examine the relationships among δ15N, 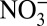 and
and 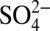 concentrations, and the calculated H+ and acid-form nitrate (HNO3) concentrations (SI Text), and use nitrogen isotope fractionation models to explore mechanisms leading to the observed δ15N trend since ∼1850.
concentrations, and the calculated H+ and acid-form nitrate (HNO3) concentrations (SI Text), and use nitrogen isotope fractionation models to explore mechanisms leading to the observed δ15N trend since ∼1850.
The subannual nitrate concentration and δ15N data from the Summit ice core are shown in Fig. 1. Consistent with previously published results (5), as early as 1850, δ15N began to decrease gradually but steadily, with a more rapid decrease from 1960 to 1970. From ∼1970 to 2006, the values of δ15N are relatively constant with a mean of −1.0 ± 4.6 ‰ (1σ), significantly lower than the pre-1850 mean of 13.4 ± 4.5 ‰. The leveling off of δ15N after ∼1970 was not observed in the previously reported records from a Greenland ice core (5) and in lake sediment cores (6), perhaps because of the relatively low temporal resolution of those records. The nitrate concentration record appears to be negatively correlated with the δ15N record, with an apparently rapid increase around 1950 and relatively little change after ∼1970. However, our high-resolution record shows that although nitrate concentrations from 1890 to 1950 are higher than those before 1890, the monotonically increasing trend in nitrate concentrations did not commence until ∼1950. In contrast, the decreasing trend in δ15N is clearly evident from 1890 through 1970. Due to the dominance of North American pollution at Summit (15, 16), the leveling off of nitrate concentrations after the 1970s is likely owing to stabilized or slightly reduced NOx emissions resulting from air pollution mitigation strategies in the United States.
Fig. 1.

Subannually resolved δ15N( ) (top) and nitrate concentration data (bottom) in the Summit, Greenland ice core. Heavy curves are five-point (∼2 y) running averages.
) (top) and nitrate concentration data (bottom) in the Summit, Greenland ice core. Heavy curves are five-point (∼2 y) running averages.
Mass balance calculations show that if the δ15N trend is assumed to result exclusively from changes in fossil fuel sourced NOx, nitrate from fossil fuel combustion must possess δ15N ∼23 ‰ lower than that from natural NOx sources to account for the observed 14.4 ‰ decrease (see SI Text for detailed calculations). This is highly unlikely, because available measurements of δ15N(NOx) from fossil fuel combustion sources generally show higher values than that from natural sources (Table S1). In addition, nitrate sampled in locations in close proximity to local fossil fuel combustion sources often have higher δ15N than that in relatively pristine areas (e.g., refs. 17 and 18). Although δ15N(NOx) from N-fertilized soil is lower than that from natural sources (Table S1), the quantity of NOx produced by N-fertilized soil must be as much as 70–360% of that produced by fossil fuel combustion to explain the observed 14.4 ‰ decrease (SI Text). For comparison, a global chemical transport model constrained by satellite observations suggests that NOx emitted from N-fertilized soil is only 10–17% of that from fuel combustion (1). Together, these observations suggest that NOx source changes since the Industrial Revolution are unlikely to have caused the observed decrease in δ15N in the ice core records.
In Fig. 2, the annual concentrations of 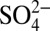 ,
, 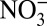 , and δ15N, and annual H+ and HNO3 concentrations calculated from ionic balance (SI Text) are shown. The concentrations of H+,
, and δ15N, and annual H+ and HNO3 concentrations calculated from ionic balance (SI Text) are shown. The concentrations of H+, 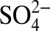 , and HNO3 started increasing at the same time (∼1850) as δ15N began to decrease, preceding the onset of the
, and HNO3 started increasing at the same time (∼1850) as δ15N began to decrease, preceding the onset of the 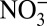 concentration increase by about 100 y. The increase in H+ and
concentration increase by about 100 y. The increase in H+ and 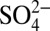 concentrations after 1850 is mainly due to atmospheric sulfate production arising from growing anthropogenic SO2 emissions (19), whereas their decrease after 1970 almost certainly reflects reduced SO2 emissions following the US Clean Air Act. As shown in Fig. 2B, by the year ∼2000, sulfate concentrations decreased to levels similar to that before 1850, which is consistent with observations from another Greenland ice core (20). However, SO2 emissions in North America in 2000 are still significantly higher than those before 1850 (21). Therefore, it appears that Greenland ice core records of sulfate after ∼1970 show a faster rate of decrease than that of North American anthropogenic SO2 emissions. Lamarque et al. (22) also found that global chemical transport models tend to overestimate sulfate deposition over Greenland after 1980, for reasons that remain unknown.
concentrations after 1850 is mainly due to atmospheric sulfate production arising from growing anthropogenic SO2 emissions (19), whereas their decrease after 1970 almost certainly reflects reduced SO2 emissions following the US Clean Air Act. As shown in Fig. 2B, by the year ∼2000, sulfate concentrations decreased to levels similar to that before 1850, which is consistent with observations from another Greenland ice core (20). However, SO2 emissions in North America in 2000 are still significantly higher than those before 1850 (21). Therefore, it appears that Greenland ice core records of sulfate after ∼1970 show a faster rate of decrease than that of North American anthropogenic SO2 emissions. Lamarque et al. (22) also found that global chemical transport models tend to overestimate sulfate deposition over Greenland after 1980, for reasons that remain unknown.
Fig. 2.

Annual concentration of H+ (A), 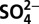 (B),
(B),  (C), and δ15N(
(C), and δ15N( ) (D) from 1772 to 2006. Large H+ and
) (D) from 1772 to 2006. Large H+ and 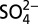 peaks in A and B indicate volcanic eruptions. The blue curve in C represents the calculated acid form nitrate (HNO3) concentrations; the blue and red curves in D are the modeled δ15N(
peaks in A and B indicate volcanic eruptions. The blue curve in C represents the calculated acid form nitrate (HNO3) concentrations; the blue and red curves in D are the modeled δ15N( ) trends throughout the record calculating with ε = −8.5 ‰ and −21 ‰, respectively, assuming ideal conditions. Vertical dashed lines mark the years of interest.
) trends throughout the record calculating with ε = −8.5 ‰ and −21 ‰, respectively, assuming ideal conditions. Vertical dashed lines mark the years of interest.
To assess possible causal relationships, we calculated correlation coefficients between the annual averages as well as the detrended annual averages of δ15N with those of H+, 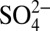 , HNO3, and
, HNO3, and 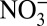 (Table 1). High correlations between annual δ15N and H+,
(Table 1). High correlations between annual δ15N and H+, 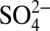 , HNO3, and
, HNO3, and 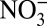 concentrations are apparently driven by their similar long-term trends, as no significant correlations exist among the detrended data. Thus, in the following discussion, we only analyze the correlations among the nondetrended annual data. Although correlations are found between δ15N and H+,
concentrations are apparently driven by their similar long-term trends, as no significant correlations exist among the detrended data. Thus, in the following discussion, we only analyze the correlations among the nondetrended annual data. Although correlations are found between δ15N and H+, 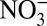 , and HNO3 over the entire 235-y record, the correlation between δ15N and H+ during the 1850–1970 δ15N decrease is stronger than that with
, and HNO3 over the entire 235-y record, the correlation between δ15N and H+ during the 1850–1970 δ15N decrease is stronger than that with 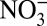 and HNO3 (Table 1). For the period of 1850–1950, before the dramatic decrease in δ15N and increase in
and HNO3 (Table 1). For the period of 1850–1950, before the dramatic decrease in δ15N and increase in 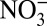 concentration (Fig. 2), the significant correlations between δ15N and H+,
concentration (Fig. 2), the significant correlations between δ15N and H+, 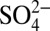 and HNO3 persist, whereas significant correlation between δ15N and
and HNO3 persist, whereas significant correlation between δ15N and 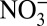 disappears. These results suggest that the general correlation between δ15N and
disappears. These results suggest that the general correlation between δ15N and 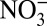 over 1772–2006 is likely driven by independent processes linked to anthropogenic activities, rather than a simple causal effect of NOx source change.
over 1772–2006 is likely driven by independent processes linked to anthropogenic activities, rather than a simple causal effect of NOx source change.
Table 1.
Correlations between annual data of δ15N and H+, 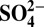 , HNO3, and
, HNO3, and 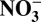 over the entire record (1772–2006), and in the periods of 1850–1970, 1850–1950
over the entire record (1772–2006), and in the periods of 1850–1970, 1850–1950
| Period | Data type | [H+], μeq L−1 |
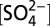 , μeq L−1 , μeq L−1
|
[HNO3], μeq L−1 |
 , μeq L−1 , μeq L−1
|
|
r (P1; P2)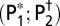
|
r (P1; P2) | r (P1; P2) | r (P1; P2) | |||
| δ15N (‰) | 1772∼2006 | annual data | −0.78 (<0.01; <0.01) | −0.60 (<0.01; <0.01) | −0.78 (<0.01; <0.01) | −0.79 (<0.01; <0.01) |
| detrended annual data | −0.09 (>0.05; >0.05) | 0.04 (>0.05; >0.05) | −0.08 (>0.05; >0.05) | −0.14 (<0.05; <0.05) | ||
| δ15N (‰) | 1850∼1970 | annual data | −0.72 (<0.01; <0.01) | −0.63 (<0.01; <0.01) | −0.58 (<0.01; <0.01) | −0.59 (<0.01; <0.01) |
| detrended annual data | −0.11 (>0.05; >0.05) | −0.01 (>0.05; >0.05) | −0.07 (>0.05; >0.05) | −0.03 (>0.05; >0.05) | ||
| δ15N (‰) | 1850∼1950 | annual data | −0.42 (<0.01; >0.05) | −0.37 (<0.01; >0.05) | −0.21 (<0.05; >0.05) | −0.18 (>0.05; >0.05) |
| detrended annual data | −0.01 (>0.05; >0.05) | 0.03 (>0.05; >0.05) | −0.04 (>0.05; >0.05) | 0.01 (>0.05; >0.05) |
Significant correlations are in bold. P1, P value; P2, adjusted P value taking into account the effective degree of freedom (autocorrelation).
The significant correlation between δ15N and H+ (acidity), and that between δ15N and the acidic species (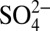 , HNO3), over the three time periods we examined suggests that the decreasing δ15N from 1850 to 1970 is likely driven by the concurrent increase in atmospheric acidity. We hypothesize that the link between δ15N and H+ is the effect of acidity on the thermodynamic equilibrium partitioning of atmospheric nitrate between HNO3(g) and
, HNO3), over the three time periods we examined suggests that the decreasing δ15N from 1850 to 1970 is likely driven by the concurrent increase in atmospheric acidity. We hypothesize that the link between δ15N and H+ is the effect of acidity on the thermodynamic equilibrium partitioning of atmospheric nitrate between HNO3(g) and 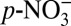 . In the atmosphere, the equilibrium partitioning of nitrate is achieved quickly (minutes to several hours) between the gas phase and the fine-mode aerosol phase (< 1 μm) (23). A simplified process describing the equilibrium between
. In the atmosphere, the equilibrium partitioning of nitrate is achieved quickly (minutes to several hours) between the gas phase and the fine-mode aerosol phase (< 1 μm) (23). A simplified process describing the equilibrium between 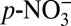 and HNO3(g) (24) is:
and HNO3(g) (24) is:
where aq represents the aqueous phase on an aerosol surface, and 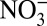 (aq) + HNO3(aq) together represent
(aq) + HNO3(aq) together represent 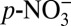 . Experiments have shown that HNO3(g) is depleted in 15N relative to
. Experiments have shown that HNO3(g) is depleted in 15N relative to 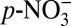 due to nitrogen isotope fractionation in conversion between
due to nitrogen isotope fractionation in conversion between 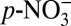 and HNO3(g) (11, 12). Eq. 1 suggests that an increase in atmospheric acidity elevates the fraction of nitrate as HNO3(g), and therefore lowers δ15N(HNO3(g)) according to a Rayleigh type fractionation (25). The atmospheric acidity increase from ∼1850–1970, as reflected by the H+ concentration record (Fig. 2A), should lead to decreasing δ15N(HNO3(g)) over this time period.
and HNO3(g) (11, 12). Eq. 1 suggests that an increase in atmospheric acidity elevates the fraction of nitrate as HNO3(g), and therefore lowers δ15N(HNO3(g)) according to a Rayleigh type fractionation (25). The atmospheric acidity increase from ∼1850–1970, as reflected by the H+ concentration record (Fig. 2A), should lead to decreasing δ15N(HNO3(g)) over this time period.
For the decrease in ice core δ15N to be explained by the impact of increases in atmospheric acidity, HNO3(g) must be preferentially transported to Greenland from the NOx source region(s) relative to 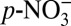 . En route to Greenland, there is further partitioning of nitrate between the gas and aerosol phases, and thus further fractionation of nitrogen isotopes. However, as the air mass approaches Greenland, the effect of gas−particle partitioning within the air mass on ice core δ15N decreases because HNO3(g) and
. En route to Greenland, there is further partitioning of nitrate between the gas and aerosol phases, and thus further fractionation of nitrogen isotopes. However, as the air mass approaches Greenland, the effect of gas−particle partitioning within the air mass on ice core δ15N decreases because HNO3(g) and 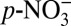 are both locally deposited to the surface snow. In fact, field studies (26, 27) have suggested that nitrate primarily exists as HNO3(g) at Summit, Greenland. Global model calculations (2) also show that HNO3(g) is the majority (> 90%) of atmospheric nitrate in central Greenland for both the preindustrial period and the present day. For the entire Arctic region, the global model (2) predicts that the fraction of atmospheric nitrate in the gas phase has increased from the preindustrial period (50–70%) to the present day (>90%). This prediction is consistent with our calculations showing that the fraction of acid-form nitrate (HNO3) in ice [representing the contribution of HNO3(g) deposition to total ice core nitrate concentration] has increased from ∼50% before 1850 to ∼90% after 1950 (Fig. 2C). In contrast, the fraction of HNO3(g) in the lower-latitude NOx source regions is ∼20% (28). Therefore, the observed decrease in δ15N can be qualitatively explained by the impact of the atmospheric acidity increase on gas−aerosol partitioning of nitrate, given the preferential transport of HNO3(g). The preferential transport of HNO3(g) also explains why calculated HNO3 concentrations increased earlier than
are both locally deposited to the surface snow. In fact, field studies (26, 27) have suggested that nitrate primarily exists as HNO3(g) at Summit, Greenland. Global model calculations (2) also show that HNO3(g) is the majority (> 90%) of atmospheric nitrate in central Greenland for both the preindustrial period and the present day. For the entire Arctic region, the global model (2) predicts that the fraction of atmospheric nitrate in the gas phase has increased from the preindustrial period (50–70%) to the present day (>90%). This prediction is consistent with our calculations showing that the fraction of acid-form nitrate (HNO3) in ice [representing the contribution of HNO3(g) deposition to total ice core nitrate concentration] has increased from ∼50% before 1850 to ∼90% after 1950 (Fig. 2C). In contrast, the fraction of HNO3(g) in the lower-latitude NOx source regions is ∼20% (28). Therefore, the observed decrease in δ15N can be qualitatively explained by the impact of the atmospheric acidity increase on gas−aerosol partitioning of nitrate, given the preferential transport of HNO3(g). The preferential transport of HNO3(g) also explains why calculated HNO3 concentrations increased earlier than 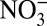 concentrations (Fig. 2C): Increased acidity elevated the fraction of HNO3(g) so that more HNO3(g) was transported to Greenland despite no increase in total atmospheric nitrate. The total nitrate concentration did not begin to increase until HNO3 became the dominant fraction of ice core nitrate (Fig. 2C).
concentrations (Fig. 2C): Increased acidity elevated the fraction of HNO3(g) so that more HNO3(g) was transported to Greenland despite no increase in total atmospheric nitrate. The total nitrate concentration did not begin to increase until HNO3 became the dominant fraction of ice core nitrate (Fig. 2C).
We also used a Rayleigh type fractionation model (13, 25) to quantify the impact of atmospheric acidity change on δ15N assuming ideal conditions (see Materials and Methods and SI Text for model details; data are available in Dataset S1). The model requires a known isotopic fractionation constant (ε) between HNO3(g) and 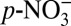 , which has not been determined to date. To estimate the fractionation, we applied ε = −8.5 ‰ determined for the fractionation between HNO3(g) and snow nitrate (25) in one set of model calculations, and ε = −21 ‰ determined in the laboratory for the fractionation between HNO3(g) and NH4NO3 particles (12) in another. As shown in Fig. 2D, the model is able to predict the long-term decreasing trend in δ15N from 1850 to 1970 by only including the effect of acidity change on the phase partitioning of atmospheric nitrate. Using ε = −8.5 ‰ underestimates the magnitude of the decrease by 5.9 ‰. The model with ε = −21 ‰ predicts a 15.6 ‰ decrease from 1850 to 1970, comparable to the observed 14.4 ‰ decrease, and captures many features of the observed δ15N variations throughout the entire 235-y record. Discrepancies between the model and the observations are expected, as the model only considers the effect of changes in atmospheric acidity on δ15N and ignores the role of changes in NOx source strengths and postdepositional processing. In particular, the model predicts a faster rate of increase in δ15N than the observations after ∼1990 (Fig. 2D). This discrepancy is likely due to the faster rate of decrease in ice core sulfate concentrations compared with that in anthropogenic SO2 emissions in North America during this time period. Because of this, the observed decreases in snow acidity may overrepresent decreases in atmospheric acidity in SO2 source regions after ∼1990. To assess the effect of the potential difference in the trends in SO2 emissions and ice core sulfate concentrations after ∼1990, we scale the post-1970 ice core annual sulfate concentrations according to annual SO2 emission data (21): first, the ratio of sulfate concentration and SO2 emission in 1970 is calculated as the scaling factor; subsequently, sulfate concentrations in each year after 1970 are calculated by multiplying annual SO2 emission data by this scaling factor. We then replace the measured sulfate concentrations after 1970 with these scaled concentrations to calculate the concentration of H+ based on ionic balance. Using these acidity calculations, our model predicts an almost identical trend in δ15N after ∼1990 as the observations (Fig. S1).
, which has not been determined to date. To estimate the fractionation, we applied ε = −8.5 ‰ determined for the fractionation between HNO3(g) and snow nitrate (25) in one set of model calculations, and ε = −21 ‰ determined in the laboratory for the fractionation between HNO3(g) and NH4NO3 particles (12) in another. As shown in Fig. 2D, the model is able to predict the long-term decreasing trend in δ15N from 1850 to 1970 by only including the effect of acidity change on the phase partitioning of atmospheric nitrate. Using ε = −8.5 ‰ underestimates the magnitude of the decrease by 5.9 ‰. The model with ε = −21 ‰ predicts a 15.6 ‰ decrease from 1850 to 1970, comparable to the observed 14.4 ‰ decrease, and captures many features of the observed δ15N variations throughout the entire 235-y record. Discrepancies between the model and the observations are expected, as the model only considers the effect of changes in atmospheric acidity on δ15N and ignores the role of changes in NOx source strengths and postdepositional processing. In particular, the model predicts a faster rate of increase in δ15N than the observations after ∼1990 (Fig. 2D). This discrepancy is likely due to the faster rate of decrease in ice core sulfate concentrations compared with that in anthropogenic SO2 emissions in North America during this time period. Because of this, the observed decreases in snow acidity may overrepresent decreases in atmospheric acidity in SO2 source regions after ∼1990. To assess the effect of the potential difference in the trends in SO2 emissions and ice core sulfate concentrations after ∼1990, we scale the post-1970 ice core annual sulfate concentrations according to annual SO2 emission data (21): first, the ratio of sulfate concentration and SO2 emission in 1970 is calculated as the scaling factor; subsequently, sulfate concentrations in each year after 1970 are calculated by multiplying annual SO2 emission data by this scaling factor. We then replace the measured sulfate concentrations after 1970 with these scaled concentrations to calculate the concentration of H+ based on ionic balance. Using these acidity calculations, our model predicts an almost identical trend in δ15N after ∼1990 as the observations (Fig. S1).
Variations in O3 concentration have also been suggested to impact δ15N by altering the ratio of atmospheric NO and NO2 (10, 11). Using a box model (10), we estimate that the ∼40% increase in tropospheric O3 concentration from the preindustrial period to the present day (2) would lower δ15N of ice core nitrate by only 2.2 ‰ (SI Text). Increased O3 could thus account for no more than 15% of the observed decrease in δ15N from 1850 to 1970.
Postdepositional processing via evaporation and photolysis of snow nitrate also affects ice core δ15N (14, 25). The observed positive δ15N values (∼13.4 ‰) before 1850, in comparison with negative or near-zero δ15N of natural NOx (Table S1), is likely the result of postdepositional processing. However, factors influencing postdepositional processing at Summit, such as snow accumulation rate and concentrations of light-absorbing impurities, do not show any long-term trends that parallel the decrease in δ15N from 1850 to 1970 (SI Text and Fig. S2). Although postdepositional processing isotopically enriches nitrate remaining in snow, the photolysis of snow nitrate leads to a regional source of NOx (29) with depleted δ15N throughout the snow-covered Arctic. Snow nitrate photolysis is promoted by acidic conditions (30); thus the snow-sourced NOx is likely enhanced since ∼1850 due to the snow acidity increase (Fig. 2A). In addition, the strength of this snow-sourced NOx is amplified by increasing snow nitrate concentrations originating from transport and deposition of anthropogenic nitrate from lower latitudes (5, 19). The net isotopic effect of this process on ice core δ15N depends on the net loss or gain of snow nitrate resulting in enrichment and depletion in δ15N, respectively, the magnitude of which is unknown. Additional studies are necessary to quantify the impact of acidity on the photolysis of snow nitrate and its implications for the nitrogen budget in the Arctic.
Although multiple factors influence δ15N, only changes in atmospheric acidity and tropospheric ozone appear to be able to lead to a decreasing trend in δ15N from 1850 to 1970. Model calculations of the impact of atmospheric acidity on δ15N further demonstrate that the acidity change alone is able to account for the magnitude of the observed decrease from 1850 to 1970, and explain many features of the observed variability in δ15N over the entire 235-y record (Fig. 2D). For short-term (annual and subannual) variations in ice core δ15N, atmospheric acidity does not appear to be important, as suggested by the fact that there is no significant correlation between the detrended data of acidity and δ15N (Table 1). The preindustrial-to-industrial increase in ozone concentrations contributes up to 15% to the δ15N decrease. This, combined with the result of the correlation analysis (Table 1), leads us to conclude that the observed decrease in Greenland ice core δ15N from 1850 to 1970 is likely dominated by the increase in atmospheric acidity originating mainly from anthropogenic SO2 emissions. We note that there are periods when volcanic eruptions (e.g., 1991 Pinatubo) could lead to episodically high atmospheric acidity similar to (or even higher than) that in the 1970s (Fig. 2A). However, decreases in δ15N are not observed in response to these eruptions because the atmospheric acidity in the source regions of nitrate (i.e., North America) is not perturbed by such events (see detailed discussion in SI Text).
Our results highlight the importance of atmospheric processes in regulating δ15N of Nr species, and that these processes must be considered in efforts to use δ15N to investigate changes in the global nitrogen cycle. In addition, in contrast with previous work (5, 6), our results indicate that δ15N in ice cores or sediments is unlikely to be an indicator solely of changes in NOx sources, as multiple processes, particularly changes in atmospheric acidity since the Industrial Revolution, must be considered. Based on the results reported here, the interpretation of the observed glacial−interglacial change in Greenland ice core δ15N (4), and that of the seasonal variations in δ15N of Greenland snow nitrate (31), as an indicator of changes in NOx sources, should be reevaluated to include the impacts of atmospheric and/or postdepositional processing. The 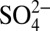 and
and 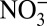 concentrations in Greenland snow stopped increasing around 1970, resulting from the mitigation of air pollution (acidic aerosols) under regulation strategies in North America. Input of anthropogenic Nr to ecosystems may have accelerated after 1970 (6, 32) in agricultural areas due to increased N-fertilizer use; however, in remote areas of North America (e.g., high-elevation lakes) and in Greenland, where nitrogen inputs mainly depend on atmospheric transport of Nr from North America (15, 16), anthropogenic nitrogen deposition likely stopped increasing after 1970 due to regulations on NOx emissions in the United States.
concentrations in Greenland snow stopped increasing around 1970, resulting from the mitigation of air pollution (acidic aerosols) under regulation strategies in North America. Input of anthropogenic Nr to ecosystems may have accelerated after 1970 (6, 32) in agricultural areas due to increased N-fertilizer use; however, in remote areas of North America (e.g., high-elevation lakes) and in Greenland, where nitrogen inputs mainly depend on atmospheric transport of Nr from North America (15, 16), anthropogenic nitrogen deposition likely stopped increasing after 1970 due to regulations on NOx emissions in the United States.
Materials and Methods
Samples and Analysis.
This study is based on two parallel ice cores (Core 1 and Core 3, 79.86 m and 79.985 m long, respectively) drilled at Summit, Greenland (72°36’N, 38°30’W, 3200 m.a.s.l.) in June 2007. Each core was drilled as 1-m-long tubes with a diameter of 10 cm. The tubes were stored in clean plastic sleeves and transported frozen to South Dakota State University for processing and analyses. In the Ice Core and Environmental Chemistry Lab at South Dakota State University, one core (Core 1) was analyzed for concentrations of major ion species (Cl−,  ,
, 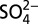 , Na+,
, Na+, 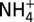 , K+, Mg2+, and Ca2+) using the technique of continuous flow analysis with ion chromatography detection (33). The concentration profile of nitrate obtained from this core was used to guide the sampling for isotopic measurements from the other core (Core 3), as a minimum of 200 nmol nitrate in each sample is required for measurements of nitrate isotopic composition. In total, 538 samples with depth intervals varying from 10 cm to 30 cm were obtained from the other core. There were several tubes in this core that had been consumed for other purposes. Thus, the isotopic data were not continuous below the depth of 66 m. This makes the data of δ15N
, K+, Mg2+, and Ca2+) using the technique of continuous flow analysis with ion chromatography detection (33). The concentration profile of nitrate obtained from this core was used to guide the sampling for isotopic measurements from the other core (Core 3), as a minimum of 200 nmol nitrate in each sample is required for measurements of nitrate isotopic composition. In total, 538 samples with depth intervals varying from 10 cm to 30 cm were obtained from the other core. There were several tubes in this core that had been consumed for other purposes. Thus, the isotopic data were not continuous below the depth of 66 m. This makes the data of δ15N discontinuous before ∼1830, so it is difficult to determine whether there is a trend in δ15N
discontinuous before ∼1830, so it is difficult to determine whether there is a trend in δ15N before 1850. However, according to the previously published data, there is no trend before 1850 (5).
before 1850. However, according to the previously published data, there is no trend before 1850 (5).
The comprehensive isotopic composition of nitrate [i.e., δ17O, δ18O, and δ15N; only δ15N was reported in this study; δ15N ‰ = (Rs/Rair − 1) × 1000, where R is the ratio of 15N to 14N in sample (Rs) or in air (Rair)] was measured using the denitrifier method (34) in the IsoLab at the University of Washington, after the samples were preprocessed at South Dakota State University. Briefly, nitrate in each sample is converted to N2O by the bacteria, Pseudomonas aureofaciens. N2O is then thermally decomposed into N2 and O2 in a gold tube at 800 °C. The isotopic ratios of each gas are then measured by a Finnigan Delta-Plus Advantage IR-MS after being separated by a gas chromatograph. International nitrate reference materials, USGS-34 and IAEA-NO3, were used for calibration. The uncertainty (1σ) of δ15N measurement is 0.7 ‰ based on replicate measurements of the international reference materials.
The concentration profiles of Ca2+ and Mg2+ were used to build the depth−age scale of the cores. As a result, the two cores were dated from 1771 to 2007. However, as the samples in the years of 1771 an 2007 do not cover the entire year, we only reported the annual averages from 1772 to 2006. The dating of Core 3 samples was completed by applying the depth−age scale of Core 1, as Core 3 and Core 1 were drilled almost simultaneously and in close proximity (within 10 m). The error of Core 3 sample dating with this method is estimated to be less than a half year by comparing the depths of well-known volcanic eruption events in Core 3 to Core 1 (∼10 cm different in depth).
Model Description.
We assume a Rayleigh type fractionation process between HNO3(g) and  , and the δ15N value of HNO3(g) and
, and the δ15N value of HNO3(g) and  can be calculated with the following equations (13, 25):
can be calculated with the following equations (13, 25):
δ15Nrem. represents δ15N of  (the portion of nitrate remaining in aerosols); δ15Neva. represents δ15N of HNO3(g) (the portion of nitrate evaporating from aerosols); and δ15N0 is the δ15N of total nitrate and is assumed to be zero (could be any value as we focused on the relative changes in δ15N of HNO3(g) in this study). The ε is the equilibrium fractionation constant between HNO3(g) and
(the portion of nitrate remaining in aerosols); δ15Neva. represents δ15N of HNO3(g) (the portion of nitrate evaporating from aerosols); and δ15N0 is the δ15N of total nitrate and is assumed to be zero (could be any value as we focused on the relative changes in δ15N of HNO3(g) in this study). The ε is the equilibrium fractionation constant between HNO3(g) and  , and f in the equation is the fraction of
, and f in the equation is the fraction of  in total atmospheric nitrate, which is calculated using effective Henry's law constant, assuming ideal conditions (the implications of this assumption for our results are discussed in SI Text):
in total atmospheric nitrate, which is calculated using effective Henry's law constant, assuming ideal conditions (the implications of this assumption for our results are discussed in SI Text):
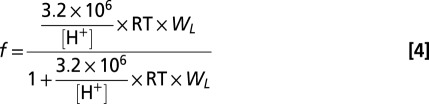 |
WL is the liquid water content of aerosols (grams per liter of air), R is the ideal gas constant, T is the thermodynamic temperature measured in kelvin, and (3.2 × 106)/[H+] is the effective Henry's law constant.
For model details, and sensitive discussions, please refer to SI Text.
Supplementary Material
Acknowledgments
L.G. thanks A. Lanciki for concentration analysis of Core 1, and M.C. Zatko for discussions and help on quantitatively estimating the effect of postdepositional loss of snow nitrate on the δ15N trend. We thank two anonymous reviewers for helpful suggestions. We acknowledge financial support from National Science Foundation (Awards 0612461 and 0839066 to J.C.-D. and PLR1244817 and PLR 1106317 to B.A.) and logistical support from Ice Drilling Design and Operations. J.S. has been partially supported by a grant from Labex OSUG@2020 (Investissements d’avenir – ANR10 LABX56), the Institut Polaire Français IPEV, and LEFE-IMAGO, a scientific program of the Institut National des Sciences de l’Univers (Centre National de la Recherche Scientifique).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319441111/-/DCSupplemental.
References
- 1.Jaegle L, Steinberger L, Martin RV, Chance K. Global partitioning of NOx sources using satellite observations: Relative roles of fossil fuel combustion, biomass burning and soil emissions. Faraday Discuss. 2005;130:407–423. doi: 10.1039/b502128f. [DOI] [PubMed] [Google Scholar]
-
2.Sofen ED, Alexander B, Kunasek SA. The impact of anthropogenic emissions on atmospheric sulfate production pathways, oxidants, and ice core Δ17O
 . Atmos Chem Phys. 2011;11(7):3565–3578. [Google Scholar]
. Atmos Chem Phys. 2011;11(7):3565–3578. [Google Scholar] - 3.Galloway JN, et al. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science. 2008;320(5878):889–892. doi: 10.1126/science.1136674. [DOI] [PubMed] [Google Scholar]
- 4.Hastings MG, Sigman DM, Steig EJ. Glacial/interglacial changes in the isotopes of nitrate from the Greenland Ice Sheet Project 2 (GISP2) ice core. Global Biogeochem Cycles. 2005;19:GB4024. [Google Scholar]
- 5.Hastings MG, Jarvis JC, Steig EJ. Anthropogenic impacts on nitrogen isotopes of ice-core nitrate. Science. 2009;324(5932):1288. doi: 10.1126/science.1170510. [DOI] [PubMed] [Google Scholar]
- 6.Holtgrieve GW, et al. A coherent signature of anthropogenic nitrogen deposition to remote watersheds of the Northern Hemisphere. Science. 2011;334(6062):1545–1548. doi: 10.1126/science.1212267. [DOI] [PubMed] [Google Scholar]
-
7.Felix JD, Elliott EM. The agricultural history of human-nitrogen interactions as recorded in ice core
 . Geophys Res Lett. 2013;40(8):1642–1646. [Google Scholar]
. Geophys Res Lett. 2013;40(8):1642–1646. [Google Scholar] - 8.Savarino J, Legrand M. High northern latitude forest fires and vegetation emissions over the last millennium inferred from the chemistry of a central Greenland ice core. J Geophys Res. 1998;103(D7):8267–8279. [Google Scholar]
- 9.Freyer HD, Kley D, Volzthomas A, Kobel K. On the interaction of isotopic exchange processes with photochemical reactions in atmospheric oxides of nitrogen. J Geophys Res. 1993;98(D8):14791–14796. [Google Scholar]
- 10.Jarvis JC, Steig EJ, Hastings MG, Kunasek SA. Influence of local photochemistry on isotopes of nitrate in Greenland snow. Geophys Res Lett. 2008;35:L21804. [Google Scholar]
- 11.Freyer HD. Seasonal variation of 15N/14N ratios in atmospheric nitrate species. Tellus Ser B. 1991;43(1):30–44. [Google Scholar]
- 12.Heaton THE, Spiro B, Madeline S, Robertson C. Potential canopy influences on the isotopic composition of nitrogen and sulphur in atmospheric deposition. Oecologia. 1997;109(4):600–607. doi: 10.1007/s004420050122. [DOI] [PubMed] [Google Scholar]
- 13.Blunier T, Floch GL, Jacobi HW, Quansah E. Isotopic view on nitrate loss in Antarctic surface snow. Geophys Res Lett. 2005;32:L13501. [Google Scholar]
- 14.Frey MM, Savarino J, Morin S, Erbland J, Martins JMF. Photolysis imprint in the nitrate stable isotope signal in snow and atmosphere of East Antarctica and implications for reactive nitrogen cycling. Atmos Chem Phys. 2009;9(22):8681–8696. [Google Scholar]
- 15.Hirdman D, et al. Source identification of short-lived air pollutants in the Arctic using statistical analysis of measurement data and particle dispersion model output. Atmos Chem Phys. 2010;10(2):669–693. [Google Scholar]
- 16.Kahl JDW, et al. Air mass trajectories to Summit, Greenland: A 44-year climatology and some episodic events. J Geophys Res. 1997;102(C12):26861–26875. [Google Scholar]
- 17.Elliott EM, et al. Nitrogen isotopes as indicators of NOx source contributions to atmospheric nitrate deposition across the Midwestern and northeastern United States. Environ Sci Technol. 2007;41(22):7661–7667. doi: 10.1021/es070898t. [DOI] [PubMed] [Google Scholar]
- 18.Morin S, et al. Comprehensive isotopic composition of atmospheric nitrate in the Atlantic Ocean boundary layer from 65°S to 79°N. J Geophys Res. 2009;114:D05303. [Google Scholar]
- 19.Mayewski PA, et al. An ice-core record of atmospheric response to anthropogenic sulphate and nitrate. Nature. 1990;346(6284):554–556. [Google Scholar]
- 20.McConnell JR, et al. 20th-century industrial black carbon emissions altered Arctic climate forcing. Science. 2007;317(5843):1381–1384. doi: 10.1126/science.1144856. [DOI] [PubMed] [Google Scholar]
- 21.Smith SJ, et al. Anthropogenic sulfur dioxide emissions: 1850−2005. Atmos Chem Phys. 2011;11(3):1101–1116. [Google Scholar]
- 22.Lamarque JF, et al. Multi-model mean nitrogen and sulfur deposition from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP): Evaluation of historical and projected future changes. Atmos Chem Phys. 2013;13(16):7997–8018. [Google Scholar]
- 23.Meng ZY, Seinfeld JH. Time scales to achieve atmospheric gas-aerosol equilibrium for volatile species. Atmos Environ. 1996;30(16):2889–2900. [Google Scholar]
-
24.Fountoukis C, Nenes A. ISORROPIA II: A computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-
 -Na+-
-Na+- -
-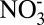 -Cl--H2O aerosols. Atmos Chem Phys. 2007;7(17):4639–4659. [Google Scholar]
-Cl--H2O aerosols. Atmos Chem Phys. 2007;7(17):4639–4659. [Google Scholar] - 25.Erbland J, et al. Air−snow transfer of nitrate on the East Antarctic Plateau - Part 1: Isotopic evidence for a photolytically driven dynamic equilibrium. Atmos Chem Phys. 2013;13:6403–6419. [Google Scholar]
- 26.Bergin MH, et al. The contributions of snow, fog, and dry deposition to the summer flux of anions and cations at Summit, Greenland. J Geophys Res. 1995;100(D8):16275–16288. [Google Scholar]
- 27.Dibb JE, Talbot RW, Bergin MH. Soluble acidic species in air and snow at Summit, Greenland. Geophys Res Lett. 1994;21(15):1627–1630. [Google Scholar]
- 28.Song CH, Carmichael GR. Gas-particle partitioning of nitric acid modulated by alkaline aerosol. J Atmos Chem. 2001;40(1):1–22. [Google Scholar]
- 29.Zatko MC, et al. The influence of snow grain size and impurities on the vertical profiles of actinic flux and associated NOx emissions on the Antarctic and Greenland ice sheets. Atmos Chem Phys. 2013;13(7):3547–3567. [Google Scholar]
- 30.Abida O, Osthoff HD. On the pH dependence of photo-induced volatilization of nitrogen oxides from frozen solutions containing nitrate. Geophys Res Lett. 2011;38:L16808. [Google Scholar]
- 31.Hastings MG, Steig EJ, Sigman DM. Seasonal variations in N and O isotopes of nitrate in snow at Summit, Greenland: Implications for the study of nitrate in snow and ice cores. J Geophys Res. 2004;109:D20306. [Google Scholar]
- 32.Elser JJ. A world awash with nitrogen. Science. 2011;334(6062):1504–1505. doi: 10.1126/science.1215567. [DOI] [PubMed] [Google Scholar]
- 33.Cole-Dai JH, Budner DM, Ferris DG. High speed, high resolution, and continuous chemical analysis of ice cores using a melter and ion chromatography. Environ Sci Technol. 2006;40(21):6764–6769. doi: 10.1021/es061188a. [DOI] [PubMed] [Google Scholar]
- 34.Kaiser J, Hastings MG, Houlton BZ, Rockmann T, Sigman DM. Triple oxygen isotope analysis of nitrate using the denitrifier method and thermal decomposition of N2O. Anal Chem. 2007;79(2):599–607. doi: 10.1021/ac061022s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.