

Significance
Heme-based metalloproteins play a central role in respiration by transporting and storing oxygen, a function that is inhibited by carbon monoxide. Density-functional theory has been unable to provide a complete description of the binding of these ligands to heme's central iron atom, predicting an unrealistically high relative affinity for carbon monoxide. Here, we solve this problem using dynamical mean-field theory in combination with linear-scaling density-functional theory, thus allowing for a simultaneous description of crucial quantum entanglement and protein discrimination effects in the ground-state of the oxygen-heme complex. By simulating the binding process within a 1,000-atom quantum-mechanical model of the myoglobin metalloprotein, we obtain a significantly improved description of its spectroscopic and energetic observables.
Keywords: metalloprotein, strong correlation, optical absorption, quantum-mechanical simulation, natural bond orbitals
Abstract
We carry out a first-principles atomistic study of the electronic mechanisms of ligand binding and discrimination in the myoglobin protein. Electronic correlation effects are taken into account using one of the most advanced methods currently available, namely a linear-scaling density functional theory (DFT) approach wherein the treatment of localized iron 3d electrons is further refined using dynamical mean-field theory. This combination of methods explicitly accounts for dynamical and multireference quantum physics, such as valence and spin fluctuations, of the 3d electrons, while treating a significant proportion of the protein (more than 1,000 atoms) with DFT. The computed electronic structure of the myoglobin complexes and the nature of the Fe–O2 bonding are validated against experimental spectroscopic observables. We elucidate and solve a long-standing problem related to the quantum-mechanical description of the respiration process, namely that DFT calculations predict a strong imbalance between O2 and CO binding, favoring the latter to an unphysically large extent. We show that the explicit inclusion of the many-body effects induced by the Hund’s coupling mechanism results in the correct prediction of similar binding energies for oxy- and carbonmonoxymyoglobin.
The ability of metalloporphyrins to bind small ligands is of great interest in the field of biochemistry. One such example is the heme molecule, which reversibly binds diatomic ligands, such as oxygen (O2) and carbon monoxide (CO), and plays a crucial role in human respiration. Heme is used in myoglobin (Mb) and hemoglobin proteins to store and transport O2 in vertebrates. The heme group of Mb is packed within a predominantly α-helical secondary structure and is coordinated by a histidine residue (known as the proximal histidine) as the fifth ligand of the heme’s central Fe ion.
Despite intensive studies (1–6), the nature of the bonding of O2 to the iron-binding site of the heme molecule remains poorly understood, mainly due to the strong electronic correlation effects associated with its localized Fe 3d electrons. It is known that these electrons are energetically well-aligned with the 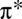 acceptor orbitals of CO and O2, and that the molecules’ bound conformations seek to maximize intermolecular orbital overlap (7–9). In the case of MbO2, the short Fe–O bond (1.81 Å) (9) implies that σ-bonding is supplemented, to some extent, by π-bonding (8). Indeed, calculations using the ab initio complete active space self-consistent method, in combination with a molecular mechanics force field to describe the protein (CASSCF/MM) (4), have identified a weak π-bonding mechanism in the Fe–O2 bond that gives rise to an antiferromagnetic (open-shell singlet) state.
acceptor orbitals of CO and O2, and that the molecules’ bound conformations seek to maximize intermolecular orbital overlap (7–9). In the case of MbO2, the short Fe–O bond (1.81 Å) (9) implies that σ-bonding is supplemented, to some extent, by π-bonding (8). Indeed, calculations using the ab initio complete active space self-consistent method, in combination with a molecular mechanics force field to describe the protein (CASSCF/MM) (4), have identified a weak π-bonding mechanism in the Fe–O2 bond that gives rise to an antiferromagnetic (open-shell singlet) state.
However, recent Fe L-edge X-ray absorption spectroscopy measurements on small biomimetic heme models lack the signature low-energy peak that is characteristic of the 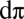 hole, formed by metal-to-ligand charge transfer into the ligand
hole, formed by metal-to-ligand charge transfer into the ligand 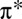 orbitals (10). Although these spectroscopic results are more consistent with a strong Fe–O π interaction, some uncertainty remains about whether the same bonding picture holds in MbO2 because the experiment was performed on a small model system [Fe(picket fence porphyrin)1-MeImO2], which, in particular, neglects the distal histidine (His-64) that hydrogen bonds directly with O2 in the protein.
orbitals (10). Although these spectroscopic results are more consistent with a strong Fe–O π interaction, some uncertainty remains about whether the same bonding picture holds in MbO2 because the experiment was performed on a small model system [Fe(picket fence porphyrin)1-MeImO2], which, in particular, neglects the distal histidine (His-64) that hydrogen bonds directly with O2 in the protein.
Furthermore, although the diamagnetic nature of MbO2 is well established, there is little experimental evidence that directly addresses the extent of the charge transfer from Fe to O2. Both CASSCF/MM (4) and L-edge X-ray absorption spectroscopy (10) suggest strong σ-donation from O2 into the 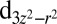 orbital of iron (ligand-to-metal back charge transfer), which is hypothesized to limit the charge on the O2 molecule to around
orbital of iron (ligand-to-metal back charge transfer), which is hypothesized to limit the charge on the O2 molecule to around 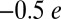 (4). However, the stretching frequency of the O–O bond in MbO2 has been shown to be close to that of the free
(4). However, the stretching frequency of the O–O bond in MbO2 has been shown to be close to that of the free 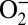 ion (8, 11), which motivates further study.
ion (8, 11), which motivates further study.
The energetics of diatomic ligand binding to the Mb protein are expected to depend strongly on the electronic structure of the heme site, and in particular on its orbital polarization. Specifically, Mb reduces the heme group’s natural preference for CO binding: Based on experimental equilibrium association constants, the binding free energy of CO, relative to O2, is reduced from around 5.9 kcal/mol in a nonpolar solvent to 1.9 kcal/mol in the protein environment (12). The combination of the strong electronic correlation centered at the Fe-binding site and long-ranged interactions between the protein and the charged O2 molecule make computational modeling of the energetics of these complexes extremely challenging.
Typically, such studies calculate the protein effect (13–15), or relative spin state energies (16), or focus on small model systems (6). However, most approaches applied to large system sizes did not include a proper treatment of electronic correlations. The effect of electronic correlations in the iron 3d states was investigated by some of us (13), where it was included, to an extent, in ab initio simulations of ligand discrimination in Mb via a density functional theory (DFT) + U treatment. DFT + U has been shown to be an efficient method for correcting self-interaction errors in the approximate DFT description of transition-metal chemistry (17), and when combined with linear-scaling approaches (18, 19), it allows us to tackle such systems comprising thousands of atoms. It was found, in the case of Mb (13), that the protein discrimination effect is dominated by polar interactions between O2 and the distal protein residue His-64. However, a problem in the DFT + U calculations is that a strong residual energetic imbalance that favors CO over O2 binding was observed (13), suggesting that approaches beyond static DFT + U are called for to obtain a proper description of Mb.
Recent progress has been made in the study of strongly correlated electrons by means of dynamical mean-field theory (DMFT) (20), a sophisticated method which includes quantum dynamical effects, and takes into account both valence and spin fluctuations. DMFT is routinely used to describe materials, and recently has also been extended to nanoscopic systems (21, 22). DMFT also explicitly includes the Hund’s exchange coupling typically, although not always, neglected in DFT and DFT + U studies. DMFT was recently combined with linear-scaling DFT (23) to produce a linear-scaling DFT + DMFT approach (24). By means of the latter, we have pointed out that strong correlation effects in heme are controlled by the Hund’s coupling J, and not the Hubbard repulsion U alone (25), suggesting that subtle quantum many-body effects are missing in the DFT + U treatment of Mb (13). However, the computational model included just the heme group and diatomic ligands, neglecting entirely the protein environment and proximal histidine ligand, thereby strongly overestimating the binding energy of CO relative to O2.
In the present work, we bridge state-of-the-art DMFT many-body calculations with large-scale DFT calculations. We perform simulations of realistic models of the MbO2 and MbCO complexes, comprising 1,007 atoms, using linear-scaling DFT + DMFT. We thereby treat the electrostatic, steric, and hydrogen-bonding effects due to protein materials together with the multireference, finite-temperature, and explicit Hund’s exchange coupling effects associated with the iron 3d-binding site in single, self-consistent calculations for, to our knowledge, the first time. We systematically investigate how the Hund’s coupling alters the electronic structure at the heme site and, at the same time, corrects the long-simulated, unphysical imbalance between CO- and O2-binding affinities.
Results
We first discuss our results for the porphyrin-plane component of the optical absorption spectra of ligated Mb, computed using DFT + DMFT (Fig. 1), where a realistic value of the Hund’s coupling 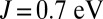 is considered (results obtained for
is considered (results obtained for 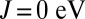 are shown for comparison). The absorption spectrum of this protein has been reported experimentally to be qualitatively dependent on its ligation state, in that a peak is present in the infrared region in the MbO2 case but not for MbCO (26). Our theoretical absorption spectrum is in good agreement with the experimental data obtained from sperm whale MbO2 single crystals (27), and reproduces an MbO2 infrared absorption band at
are shown for comparison). The absorption spectrum of this protein has been reported experimentally to be qualitatively dependent on its ligation state, in that a peak is present in the infrared region in the MbO2 case but not for MbCO (26). Our theoretical absorption spectrum is in good agreement with the experimental data obtained from sperm whale MbO2 single crystals (27), and reproduces an MbO2 infrared absorption band at 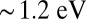 , observed experimentally at 1.3 eV (28). Our calculations associate this feature with a charge-transfer band generated by the hybridization of the Fe atom and the O2 molecule, in particular transitions of occupied porphyrin π and iron d orbital states into empty O2
, observed experimentally at 1.3 eV (28). Our calculations associate this feature with a charge-transfer band generated by the hybridization of the Fe atom and the O2 molecule, in particular transitions of occupied porphyrin π and iron d orbital states into empty O2
 orbitals. In MbCO, due to the strong covalent bond, the porphyrin π and d hybridized orbitals are at a lower energy and, hence, there is no contribution to the infrared spectrum. The double-peak structure in the optical transition obtained at
orbitals. In MbCO, due to the strong covalent bond, the porphyrin π and d hybridized orbitals are at a lower energy and, hence, there is no contribution to the infrared spectrum. The double-peak structure in the optical transition obtained at 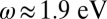 and 2.2 eV is also very close to experiments, where they are obtained respectively at 2.1 and 2.3 eV (27, 28). We attribute this feature to the porphyrin Q band (π to
and 2.2 eV is also very close to experiments, where they are obtained respectively at 2.1 and 2.3 eV (27, 28). We attribute this feature to the porphyrin Q band (π to  absorptions) and to corresponding charge transfer excitations. We find a broad Soret band centered theoretically at
absorptions) and to corresponding charge transfer excitations. We find a broad Soret band centered theoretically at 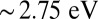 , close to the experimental peak obtained at 2.95 eV (27, 28). For MbO2, the spectrum at
, close to the experimental peak obtained at 2.95 eV (27, 28). For MbO2, the spectrum at 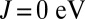 is qualitatively similar. However, Fig. 1 also reveals that a nonzero J is required to recover the experimentally observed double-peak structure of the MbCO Q band (27). Analysis of the spectral weight below the Fermi level in MbCO reveals the source of this splitting. For
is qualitatively similar. However, Fig. 1 also reveals that a nonzero J is required to recover the experimentally observed double-peak structure of the MbCO Q band (27). Analysis of the spectral weight below the Fermi level in MbCO reveals the source of this splitting. For  , the orbital character of the HOMO is almost degenerate between the three
, the orbital character of the HOMO is almost degenerate between the three 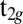 orbitals. However, for J = 0.7 eV, we observe a splitting of the spectral weight of the dxy and the dxz,yz orbitals of
orbitals. However, for J = 0.7 eV, we observe a splitting of the spectral weight of the dxy and the dxz,yz orbitals of 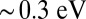 , thus recovering the expected splitting of the charge transfer Q band in the optical absorption spectrum.
, thus recovering the expected splitting of the charge transfer Q band in the optical absorption spectrum.
Fig. 1.

Optics. Porphyrin-plane component of the optical absorption spectrum calculated using DFT + DMFT at 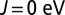 (dashed curves) and
(dashed curves) and 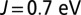 (continuous curves) for MbCO (blue) and MbO2 (red).
(continuous curves) for MbCO (blue) and MbO2 (red).
To further understand the nature of the bonding in the MbO2 complex, we have found it instructive to transform the atomic basis functions, used to expand the DFT + DMFT density matrix (that is, the frequency-integrated Green’s function), into a set of natural bond orbitals (NBOs) (29–31). The transformation is constructed such that the resulting orbitals may be categorized into localized Lewis-type bonding and lone pair orbitals, as well as their antibonding and Rydberg counterparts, thus allowing a chemically intuitive population analysis to be applied to the DFT + DMFT many-body wave-function. Fig. 2 shows the σ- and π-bonding many-body NBOs of the MbO2 complex. We find, in particular, that an O2
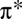 NBO (Fig. 2A), which has an occupancy of 1.5 e, and an antibonding NBO formed between Fe and the proximal histidine, with occupancy 0.5 e and a strong
NBO (Fig. 2A), which has an occupancy of 1.5 e, and an antibonding NBO formed between Fe and the proximal histidine, with occupancy 0.5 e and a strong 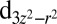 character, interact strongly via the DFT Hamiltonian. We note that the 0.5 e occupancy of the antibonding orbital on Fe is consistent with the ligand–metal back charge transfer process between O2 and the Fe
character, interact strongly via the DFT Hamiltonian. We note that the 0.5 e occupancy of the antibonding orbital on Fe is consistent with the ligand–metal back charge transfer process between O2 and the Fe 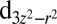 orbital, which is observed both in CASSCF/MM (4) and L-edge X-ray absorption spectroscopy (10). Ligand–metal back charge transfer is also present at
orbital, which is observed both in CASSCF/MM (4) and L-edge X-ray absorption spectroscopy (10). Ligand–metal back charge transfer is also present at 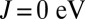 , albeit with a smaller magnitude (0.34 e). Thus, electronic delocalization is expected to provide a greater energetic stabilization in the MbO2 complex at
, albeit with a smaller magnitude (0.34 e). Thus, electronic delocalization is expected to provide a greater energetic stabilization in the MbO2 complex at 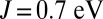 .
.
Fig. 2.

NBO analysis of the DFT + DMFT Green’s function for MbO2. (A) An O2
 type many-body NBO (cyan/magenta) of occupancy 1.5 e, which strongly interacts (via the DFT Hamiltonian) with an antibonding Fe-based many-body NBO (red/blue) of occupancy 0.5 e. (B and C) Many-body NBOs showing π-bonding between Fe and O2; the NBO occupancies are 1.4 and 1.8 e, respectively.
type many-body NBO (cyan/magenta) of occupancy 1.5 e, which strongly interacts (via the DFT Hamiltonian) with an antibonding Fe-based many-body NBO (red/blue) of occupancy 0.5 e. (B and C) Many-body NBOs showing π-bonding between Fe and O2; the NBO occupancies are 1.4 and 1.8 e, respectively.
The net charge on the O2 molecule, from natural population analysis, is 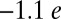 , which is consistent with the Weiss picture of bonding in MbO2 (2). It is worth noting that state-of-the-art CASSCF/MM calculations point toward a smaller O2 charge of
, which is consistent with the Weiss picture of bonding in MbO2 (2). It is worth noting that state-of-the-art CASSCF/MM calculations point toward a smaller O2 charge of 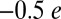 (4). Metal-to-ligand charge transfer is expected to occur via π-bonding interactions between Fe d orbitals and O2
(4). Metal-to-ligand charge transfer is expected to occur via π-bonding interactions between Fe d orbitals and O2
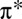 (4, 10). Indeed, Fig. 2 B and C are characteristic of the multiconfigurational CASSCF orbitals that make up the proposed π-type bonding in a previous study (4). A notable difference between these calculations and the CASSCF/MM study is that the π-bonding is much stronger than previously reported. Here, 3.25 e are involved in π-bonding, as opposed to ∼2 e in CASSCF/MM. Our calculations yield a
(4, 10). Indeed, Fig. 2 B and C are characteristic of the multiconfigurational CASSCF orbitals that make up the proposed π-type bonding in a previous study (4). A notable difference between these calculations and the CASSCF/MM study is that the π-bonding is much stronger than previously reported. Here, 3.25 e are involved in π-bonding, as opposed to ∼2 e in CASSCF/MM. Our calculations yield a 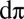 hole character of 19%. This compares extremely favorably with recent Fe L-edge X-ray absorption spectroscopy measurements of a small biomimetic heme model, which estimates the
hole character of 19%. This compares extremely favorably with recent Fe L-edge X-ray absorption spectroscopy measurements of a small biomimetic heme model, which estimates the 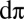 hole character to be
hole character to be 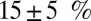 (10). We therefore find that the π-bonding character in MbO2 is similar to that in isolated porphyrins. We note that the stronger π-bonding interaction between the iron and O2 also suggests that spin polarization of the π electrons is less likely (10), suggesting that a broken spin symmetry description of MbO2 might not be entirely reliable.
(10). We therefore find that the π-bonding character in MbO2 is similar to that in isolated porphyrins. We note that the stronger π-bonding interaction between the iron and O2 also suggests that spin polarization of the π electrons is less likely (10), suggesting that a broken spin symmetry description of MbO2 might not be entirely reliable.
Next we show in Table 1 a comparison of the computed Fe orbital density with and without the explicit inclusion of Hund’s coupling J in the Hamiltonian. We find that the effect of J in MbO2 is to bring the 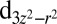 , dxy, and
, dxy, and  orbitals closer to single-electron occupation, so that the Hund’s coupling enhances the spin magnetic moment on the Fe atom. Indeed, we find, in our calculations, a build up of a magnetic moment in the
orbitals closer to single-electron occupation, so that the Hund’s coupling enhances the spin magnetic moment on the Fe atom. Indeed, we find, in our calculations, a build up of a magnetic moment in the  ,
, 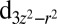 , and dxy orbitals, with a concomitant electron occupation of
, and dxy orbitals, with a concomitant electron occupation of  absent from our
absent from our 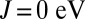 calculation and from the CASSCF/MM approach. The latter discrepancy may be due to the fact that CASSCF does not include dynamical correlation effects and may also be dependent on the chosen active space. In MbCO, unlike MbO2, we observe that the doublet on the dxy orbital is not emptied as the Hund’s coupling is increased. In Fig. 3A we show the dependence of the Fe charge, computed using Mulliken analysis in the nonorthogonal generalized Wannier function (NGWF) (32) basis, on the Hund’s coupling J for MbO2 and MbCO, respectively. For MbO2, we find that the charge of the Fe is transferred to the porphyrin ring and protein as the Hund’s coupling is increased. In contrast, for MbCO we find a very weak dependence of the Fe charge on the Hund’s coupling parameter (Table 1). In our view, the latter indicates a very strong Fe–CO covalent bond, which remains stable against the Hund’s coupling. We find that the charge transferred from the Fe to O2 is 1.1 e at the physical value of the Hund’s coupling
calculation and from the CASSCF/MM approach. The latter discrepancy may be due to the fact that CASSCF does not include dynamical correlation effects and may also be dependent on the chosen active space. In MbCO, unlike MbO2, we observe that the doublet on the dxy orbital is not emptied as the Hund’s coupling is increased. In Fig. 3A we show the dependence of the Fe charge, computed using Mulliken analysis in the nonorthogonal generalized Wannier function (NGWF) (32) basis, on the Hund’s coupling J for MbO2 and MbCO, respectively. For MbO2, we find that the charge of the Fe is transferred to the porphyrin ring and protein as the Hund’s coupling is increased. In contrast, for MbCO we find a very weak dependence of the Fe charge on the Hund’s coupling parameter (Table 1). In our view, the latter indicates a very strong Fe–CO covalent bond, which remains stable against the Hund’s coupling. We find that the charge transferred from the Fe to O2 is 1.1 e at the physical value of the Hund’s coupling 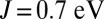 (Fig. 3B), confirming the estimation obtained using natural population analysis.
(Fig. 3B), confirming the estimation obtained using natural population analysis.
Table 1.
3d orbital occupations of the Fe atom in ligated Mb
| Protein | 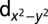 |
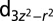 |
dxy | dxz | dyz |
MbO2
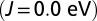
|
0.82 | 0.38 | 1.99 | 1.90 | 1.92 |
MbO2
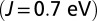
|
0.96 | 0.96 | 1.10 | 1.87 | 1.96 |
MbCO 
|
0.95 | 1.13 | 1.99 | 1.88 | 1.89 |
MbCO 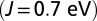
|
1.00 | 1.15 | 1.99 | 1.84 | 1.84 |
| MbO2 (CASSCF/MM) | 0.44 | 0.59 | 1.93 | 1.86 | 1.13 |
The CASSCF/MM values are shown for comparison (4).
Fig. 3.

Polarization. (A) The dependence on J of the DFT + DMFT Mulliken charge of the Fe atom in MbO2 and MbCO. (B) Mulliken charges in MbO2 of the two O atoms of O2 (O-1 is bonded to Fe and O-2 forms a hydrogen bond with His-64). Also shown are the corresponding charges of the in-plane N atoms of the heme group and His93 (the fifth ligand of Fe).
Fig. 4 depicts the computed spin fluctuations in MbO2 and MbCO, specifically, a histogram of the spin quantum number distribution obtained by looking at the 16 dominant states in the reduced (3d subspace) density matrix, obtained by tracing the atomic DMFT problem over the bath degrees of freedom. This gives an effective representation of the quantum states of the Fe atom. The ground-state wave function is not a pure state with a single allowed value for the magnetic moment (singlet, doublet, triplet, etc.), yet we can describe the fluctuating magnetic moment of the Fe atom by analyzing the distribution of the magnetic moments obtained from the dominant configurations. In particular, we find that the reduced density matrix of MbCO has states with dominant 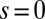 configurations, and MbO2 has dominant contributions from
configurations, and MbO2 has dominant contributions from 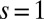 , with higher spin contributions at
, with higher spin contributions at 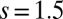 . This is consistent with our general observation that MbO2 has larger valence fluctuations (entanglement in the ground-state) than MbCO.
. This is consistent with our general observation that MbO2 has larger valence fluctuations (entanglement in the ground-state) than MbCO.
Fig. 4.

Spin fluctuations. Spin-state distribution obtained by analyzing the 16 dominant states in the DFT + DMFT reduced density matrix (see Results) at 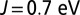 . The reduced density matrix of MbCO has states with dominant
. The reduced density matrix of MbCO has states with dominant 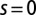 configurations, and MbO2 has dominant contributions from
configurations, and MbO2 has dominant contributions from 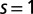 , with higher spin contributions at
, with higher spin contributions at 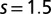 .
.
Having shown how the Hund’s coupling affects the orbital occupancy of the Fe site in Mb, and the associated charge transfer to the O2 molecule, we investigate in what follows how the energetics of ligand binding to Mb are determined by these effects, and how the protein uses quantum fluctuations to discriminate between O2 and CO. Fig. 5 shows the binding energies of O2 and CO to Mb (to within a constant shift), calculated using DFT + DMFT, as a function of the Hund’s exchange coupling J. At 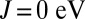 , the binding energy of MbCO is ∼1 eV more favorable than the binding energy of MbO2, yielding an unphysical energetic imbalance. For
, the binding energy of MbCO is ∼1 eV more favorable than the binding energy of MbO2, yielding an unphysical energetic imbalance. For 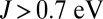 , we find on the contrary that the binding energy of MbO2 is dramatically reduced. We attribute this to the enhancement of the spin magnetic moment in the Fe atom. In the intermediate regime, close to
, we find on the contrary that the binding energy of MbO2 is dramatically reduced. We attribute this to the enhancement of the spin magnetic moment in the Fe atom. In the intermediate regime, close to 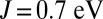 , we find that the imbalance between MbO2 and MbCO is thereby also dramatically reduced. In fact, the experimental binding free-energy difference between the two ligands of 1.9 kcal/mol is recovered from our calculations when J is near 0.7 eV, a typical value used for iron-based materials (33). In this case, the effect of J on the binding energy of O2 may be regarded as a balance between two competing effects. The charge analysis (Fig. 3) reveals that metal-to-ligand charge transfer is higher for
, we find that the imbalance between MbO2 and MbCO is thereby also dramatically reduced. In fact, the experimental binding free-energy difference between the two ligands of 1.9 kcal/mol is recovered from our calculations when J is near 0.7 eV, a typical value used for iron-based materials (33). In this case, the effect of J on the binding energy of O2 may be regarded as a balance between two competing effects. The charge analysis (Fig. 3) reveals that metal-to-ligand charge transfer is higher for 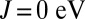 , which is expected to enhance ligand–protein interactions for small values of J. However, NBO analysis reveals a larger ligand-to-metal back charge transfer for
, which is expected to enhance ligand–protein interactions for small values of J. However, NBO analysis reveals a larger ligand-to-metal back charge transfer for  , which is consistent with the increased occupancy of the Fe
, which is consistent with the increased occupancy of the Fe 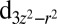 orbital (Table 1), and is expected to cause variational energetic lowering at higher values of J due to electronic delocalization.
orbital (Table 1), and is expected to cause variational energetic lowering at higher values of J due to electronic delocalization.
Fig. 5.

Binding energetics. Hund’s exchange coupling J dependence of the DFT + DMFT total energy of MbCO (blue) and MbO2 (red), where the DFT energy of the diatomic ligand CO or O2 has been subtracted to give 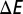 . A global arbitrary constant shift of the energy has been applied for clarity. The difference of binding energies
. A global arbitrary constant shift of the energy has been applied for clarity. The difference of binding energies  is obtained by
is obtained by 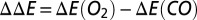 . The physical regime, where
. The physical regime, where 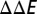 is small, is shaded.
is small, is shaded.
Compared with previously reported DFT studies, including our own DFT + U study of the same system (13), the present study predicts a significantly larger charge on the O2 molecule (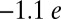 versus
versus 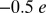 ). This charge is expected to stabilize the O2 molecule in the Mb protein via hydrogen bond interactions with His-64. Hence, we propose that both dynamical and multireference quantum effects, and large system sizes, must be accounted in order to correctly determine the energetics of ligand binding in proteins with strongly correlated subspaces.
). This charge is expected to stabilize the O2 molecule in the Mb protein via hydrogen bond interactions with His-64. Hence, we propose that both dynamical and multireference quantum effects, and large system sizes, must be accounted in order to correctly determine the energetics of ligand binding in proteins with strongly correlated subspaces.
Conclusions
We have presented the application of a recently developed methodology, designed to treat strong electronic interaction and multireference effects in systems of relatively very large numbers of atoms, to a molecule of important biological function. In particular, we have found that the Hund’s coupling J is the crucial ingredient necessary to increase the multireference high-spin character of the ground state, and so to bring binding energetics into qualitative agreement with experiment. This provides a route to the solution of a long-standing problem in the density-functional theory-based simulation of heme proteins, which often underestimates the Hund’s coupling and incorrectly describes multireference effects, namely an unphysically large imbalance of CO- and O2-binding energies. Our many-body description of the ligated Mb ground state is further supported by quantitative agreement with experimental findings on both the ligand dependence of the optical absorption spectra and the nature of the π-bonding in Fe–O2. Our approach, optimized to describe molecules and nanoparticles involving transition metal ions, supports a large range of applications, e.g., to strongly correlated oxide nanoparticles (34) or to enzymes (35).
Methodology
In this work we have carried out a detailed theoretical study of the electronic structure of the Mb molecule by means of a combination (19, 24) of linear-scaling DFT and DMFT (20, 36), a model which treats subspace local dynamical, finite temperature and multideterminantal effects, for given Hamiltonian parameters. The Order-N Electronic Total Energy Package (ONETEP) linear-scaling DFT code (19, 23, 37) was used to obtain the DFT ground state. The ONETEP method is particularly advanced in terms of its accuracy, equivalent to that of a plane-wave method, which is arrived at by means of an in situ variational optimization of the expansion coefficients of a minimal set of spatially truncated NGWFs (32), and is based on direct minimization of the total energy with respect to the single-particle density matrix. The use of a minimal, optimized Wannier function representation of the density matrix allows for the DFT ground state to be solved with relative ease in large systems, particularly in molecules where their explicit truncation implies that the addition of vacuum does not increase the computational cost. Preparation of the structures for DFT + DMFT analysis have been described in detail elsewhere (13). Briefly, the computational models are based on the X-ray crystal structures of sperm whale Mb in oxygenated and carbonmonoxygenated ligation states (Protein Data Bank ID codes 1A6M and 1A6G) (9). The heme group, ligand, and 53 closest residues (1,007 atoms in total) were extracted from the MbO2 crystal structure and optimized using spin-polarized DFT, with the Perdew–Burke–Ernzerhof (38) gradient-corrected exchange-correlation functional augmented by damped London potentials to describe van der Waals interactions (39). Following optimization, the heme group and three closest residues were replaced by their positions in the MbCO crystal structure and reoptimized. This scheme ensures that energy differences are directly attributable to local changes in the binding site, while accounting for long-ranged polarization and constraints of the protein scaffold. The DFT binding energy was converged to better than 0.02 eV with respect to changes in the plane-wave energy cutoff and NGWF cutoff radii, and no additional restrictions on the variational freedom, such as the density kernel truncation, were invoked.
We refined our DFT calculations using the DFT + DMFT method (20, 36) to obtain a more accurate treatment of strong electronic correlation effects. In particular, DMFT introduces both quantum and thermal fluctuations, which are multireference effects not captured at the level of the Kohn–Sham DFT. In this, the Mb molecule was mapped within DMFT to an Anderson impurity model Hamiltonian (40), and we used a recently developed extended Lanczos solver (41) to obtain the DMFT self-energy. Because only a single impurity site (3d orbital subspace) is present, the system becomes crystal momentum-independent in the molecular limit, and because the Kohn–Sham Green’s function is computed in full by inversion before projection onto the impurity subspace, the Anderson impurity mapping is effectively exact and the necessity of invoking the DMFT self-consistency is not required. However, in DFT + DMFT there is also a charge self-consistency cycle, albeit not routinely invoked at present due to computational cost, where the DFT + DMFT density kernel is used to generate a new Kohn–Sham Hamiltonian, which in turn provides a new input to the DMFT; the procedure being repeated until convergence is achieved. In this work our data are obtained in the absence of charge self-consistency, however we checked that the corrections are small. Indeed, for MbO2 at 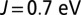 , the changes obtained by converging the charge self-consistent DFT + DMFT induce a change in the energy of
, the changes obtained by converging the charge self-consistent DFT + DMFT induce a change in the energy of 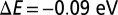 , which corresponds to the energy of MbO2 at
, which corresponds to the energy of MbO2 at 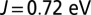 when the charge self-consistency is absent. Other changes are also small, for example, the chemical potential changes by
when the charge self-consistency is absent. Other changes are also small, for example, the chemical potential changes by 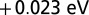 , and we find a change in the Fe charge of
, and we find a change in the Fe charge of 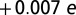 . All these variations are consistent with a renormalized J (J increased by 3% at
. All these variations are consistent with a renormalized J (J increased by 3% at 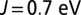 ).
).
To obtain the Kohn–Sham Green’s function, we performed the matrix inversion, as well as all matrix multiplications involved in the DMFT algorithm, on graphical computational units (GPUs) using a tailor-made parallel implementation of the Cholesky decomposition written in the CUDA programming language.
Electronic correlation effects are described within the localized subspace by the Slater–Kanamori form of the Anderson impurity Hamiltonian (42, 43), specifically
 |
where 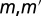 are orbital indices,
are orbital indices, 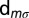
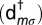 annihilates (creates) an electron with spin σ in the orbital m,
annihilates (creates) an electron with spin σ in the orbital m, 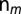 is the orbital occupation operator. The first term describes the effect of intraorbital Coulomb repulsion, parametrized by U, and the second term describes the interorbital repulsion, proportional to
is the orbital occupation operator. The first term describes the effect of intraorbital Coulomb repulsion, parametrized by U, and the second term describes the interorbital repulsion, proportional to 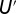 , which is renormalized by the Hund’s exchange coupling parameter J to ensure a fully rotationally invariant Hamiltonian (for further information on this topic, we refer the reader to ref. 44). The third term is the Hund’s rule exchange coupling, described by a spin exchange coupling of amplitude J.
, which is renormalized by the Hund’s exchange coupling parameter J to ensure a fully rotationally invariant Hamiltonian (for further information on this topic, we refer the reader to ref. 44). The third term is the Hund’s rule exchange coupling, described by a spin exchange coupling of amplitude J. 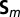 denotes the spin corresponding to orbital m, so that
denotes the spin corresponding to orbital m, so that 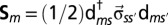 , where
, where 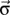 is the vector of Pauli matrices indexed by s and
is the vector of Pauli matrices indexed by s and  . In this work, we used
. In this work, we used  for the screened Coulomb interaction (16) and we explored the dependence of several observables on the Hund’s coupling (in the range
for the screened Coulomb interaction (16) and we explored the dependence of several observables on the Hund’s coupling (in the range 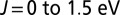 ). Our DMFT calculations were carried out at room temperature,
). Our DMFT calculations were carried out at room temperature, 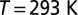 . In this work, we used the canonical form of the double-counting potential
. In this work, we used the canonical form of the double-counting potential 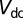 , given by
, given by 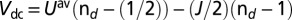 , assuming paramagnetic occupancy
, assuming paramagnetic occupancy  of the d orbitals. Here, the parameter
of the d orbitals. Here, the parameter 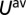 is the intra- and interorbital averaged repulsion (45). In our calculations we found that the DMFT solution remains paramagnetic, although the possibility of spontaneous formation of a magnetic moment (spin symmetry broken state) was allowed for. However, the low energy states are in a quantum superposition of polarized states, giving a fluctuating magnetic moment at the iron site. The theoretical optical absorption was obtained in DFT + DMFT within the linear-response regime (Kubo formalism), in the no-vertex-corrections approximation (46), where it is given by
is the intra- and interorbital averaged repulsion (45). In our calculations we found that the DMFT solution remains paramagnetic, although the possibility of spontaneous formation of a magnetic moment (spin symmetry broken state) was allowed for. However, the low energy states are in a quantum superposition of polarized states, giving a fluctuating magnetic moment at the iron site. The theoretical optical absorption was obtained in DFT + DMFT within the linear-response regime (Kubo formalism), in the no-vertex-corrections approximation (46), where it is given by
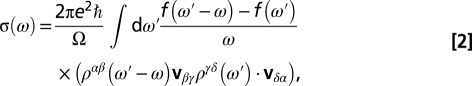 |
and the factor of two accounts for spin degeneracy, Ω is the simulation-cell volume, e is the electron charge, ћ is the reduced Planck constant, 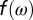 is the Fermi–Dirac distribution, and
is the Fermi–Dirac distribution, and 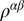 is the density matrix given by the frequency integral of the interacting DFT + DMFT Green’s function. The matrix elements of the velocity operator,
is the density matrix given by the frequency integral of the interacting DFT + DMFT Green’s function. The matrix elements of the velocity operator,  , noting that we do not invoke the Peierls substitution (46), are given by
, noting that we do not invoke the Peierls substitution (46), are given by
This expression is general to the NGWF representation (47), used in this work, where the contribution to the noninteracting Hamiltonian due to the nonlocal part of the norm-conserving pseudopotentials (48, 49), represented by 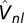 , is included. Once the self-energy matrix is obtained, it can be used to correct the DFT total energy with the DMFT correction (50, 51):
, is included. Once the self-energy matrix is obtained, it can be used to correct the DFT total energy with the DMFT correction (50, 51):
where 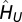 indicates the many-body interaction vertex of the DMFT, and the primed sum is over the occupied states. Tr indicates the one-electron trace for a generic representation and the sum over the Matsubara frequencies
indicates the many-body interaction vertex of the DMFT, and the primed sum is over the occupied states. Tr indicates the one-electron trace for a generic representation and the sum over the Matsubara frequencies 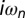 of the finite-temperature many-body formalism. The interaction term
of the finite-temperature many-body formalism. The interaction term  is obtained with the Galitskii–Migdal equation (52):
is obtained with the Galitskii–Migdal equation (52): 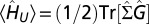 ,
, 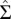
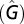 is the self-energy (Green’s function) matrix in the NGWF representation. We note that both the self-energy and the Green’s function are slowly decaying functions, hence the trace over Matsubara frequencies has to be done with care (50, 51). Finally, the double-counting correction
is the self-energy (Green’s function) matrix in the NGWF representation. We note that both the self-energy and the Green’s function are slowly decaying functions, hence the trace over Matsubara frequencies has to be done with care (50, 51). Finally, the double-counting correction 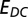 must be introduced because the contribution of interactions between the correlated orbitals to the total energy is already partially included in the exchange-correlation potential derived from DFT. The most commonly used form of the double-counting term is
must be introduced because the contribution of interactions between the correlated orbitals to the total energy is already partially included in the exchange-correlation potential derived from DFT. The most commonly used form of the double-counting term is 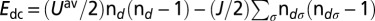 (45).
(45).
An important approach used in this work is the generation of NBOs based on the many-body Green’s function provided by DFT + DMFT, to obtain greater chemical insight into the ligand binding process. NBOs (29) are postprocessed linear combinations of the basis functions in which the density matrix is expanded, such that the projection of the density matrix onto the subspace formed by atom-based and atom pair-based subsets of basis functions is maximally diagonal. In the current calculations the basis functions in question are NGWFs (32), transformed to NBOs using the NBO 5 program (30), recently interfaced to ONETEP, as described in ref. 31. This procedure is carried out in such a manner that the final NBOs are then categorized into largely occupied bonding and lone-pair orbitals, and largely vacant antibonding and Rydberg orbitals. Although normally applied to Kohn–Sham density-functional theory, to date, the NBO generation procedure is independent of the model (and so the Hamiltonian and self-energy) generating the density matrix, and so we may apply it to the density matrix integrated from the DFT + DMFT full Green’s function, to our knowledge, for the first time. The resulting many-body NBOs largely retain the familiar profile of DFT-based NBOs, in this study, but their occupancies may be expected to deviate further from integer values due to quantum-mechanical and finite-temperature multireference effects captured within DFT + DMFT. We computed the energy of MbCO and MbO2 as a function of the Hund’s exchange coupling J. Defining 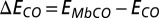 and
and 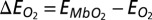 , the binding energy difference
, the binding energy difference 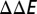 is given by
is given by 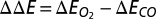 . When
. When 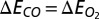 , the MbO2- and MbCO-binding energies are identical. In comparisons with the experimental relative free energy of binding, we have assumed that the relative change in entropy of the two ligands upon binding is zero, which is a reasonable approximation for two sterically similar diatomic ligands.
, the MbO2- and MbCO-binding energies are identical. In comparisons with the experimental relative free energy of binding, we have assumed that the relative change in entropy of the two ligands upon binding is zero, which is a reasonable approximation for two sterically similar diatomic ligands.
Acknowledgments
We thank T. Saha-Dasgupta, N. Hine, G. Kotliar, L. Lee, P. Littlewood, and A. Millis for helpful discussions. We gratefully acknowledge the support of the NVIDIA Corporation with the donation of the Tesla K20 GPU used for this research. D.J.C. is supported by a Marie Curie International Outgoing Fellowship within the Seventh European Community Framework Programme. Calculations were performed on BlueGene/Q at the Science and Technology Facilities Council Hartree Centre under Project HCBG005 and on the Cambridge High Performance Computing Service, funded by Engineering and Physical Sciences Research Council Grants EP/J017639/1 and EP/F032773/1.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Weiss JJ. Nature of the iron–oxygen bond in oxyhaemoglobin. Nature. 1964;203:182–183. [PubMed] [Google Scholar]
- 2.Weiss JJ. Nature of the iron–oxygen bond in oxyhaemoglobin. Nature. 1964;202:83–84. doi: 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- 3.Goddard WA, III, Olafson BD. Ozone model for bonding of an O2 to heme in oxyhemoglobin. Proc Natl Acad Sci USA. 1975;72(6):2335–2339. doi: 10.1073/pnas.72.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Ikeda-Saito M, Shaik S. Nature of the Fe-O2 bonding in oxy-myoglobin: Effect of the protein. J Am Chem Soc. 2008;130(44):14778–14790. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 5.Ribas-Ariño J, Novoa JJ. The mechanism for the reversible oxygen addition to heme. A theoretical CASPT2 study. Chem Commun (Camb) 2007;(30):3160–3162. doi: 10.1039/b704871h. [DOI] [PubMed] [Google Scholar]
- 6.Radoń M, Pierloot K. Binding of CO, NO, and O2 to heme by density functional and multireference ab initio calculations. J Phys Chem A. 2008;112(46):11824–11832. doi: 10.1021/jp806075b. [DOI] [PubMed] [Google Scholar]
- 7.Reed CA, Cheung SK. On the bonding of FeO2 in hemoglobin and related dioxygen complexes. Proc Natl Acad Sci USA. 1977;74(5):1780–1784. doi: 10.1073/pnas.74.5.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Momenteau M, Reed CA. Synthetic heme dioxygen complexes. Chem Rev. 1994;94(3):659–698. [Google Scholar]
- 9.Vojtechovský J, Chu K, Berendzen J, Sweet RM, Schlichting I. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys J. 1999;77(4):2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson SA, et al. Iron L-edge X-ray absorption spectroscopy of oxy-picket fence porphyrin: Experimental insight into Fe-O2 bonding. J Am Chem Soc. 2013;135(3):1124–1136. doi: 10.1021/ja3103583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones RD, Summerville DA, Basolo F. Synthetic oxygen carriers related to biological systems. Chem Rev. 1979;79:139–179. [Google Scholar]
- 12.Olson JS, Phillips GN., Jr Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J Biol Inorg Chem. 1997;2(4):544–552. [Google Scholar]
- 13.Cole DJ, O’Regan DD, Payne MC. Ligand discrimination in myoglobin from linear-scaling DFT+U. J. Phys Chem Lett. 2012;3(11):1448–1452. doi: 10.1021/jz3004188. [DOI] [PubMed] [Google Scholar]
- 14.Sigfridsson E, Ryde U. Theoretical study of the discrimination between O2 and CO by myoglobin. J Inorg Biochem. 2002;91(1):101–115. doi: 10.1016/s0162-0134(02)00426-9. [DOI] [PubMed] [Google Scholar]
- 15.De Angelis F, Jarzęcki AA, Car R, Spiro TG. Quantum chemical evaluation of protein control over heme ligation: CO/O2 discrimination in myoglobin. J Phys Chem B. 2005;109(7):3065–3070. doi: 10.1021/jp0451851. [DOI] [PubMed] [Google Scholar]
- 16.Scherlis DA, Cococcioni M, Sit P, Marzari N. Simulation of heme using DFT + U: A step toward accurate spin-state energetics. J Phys Chem B. 2007;111(25):7384–7391. doi: 10.1021/jp070549l. [DOI] [PubMed] [Google Scholar]
- 17.Kulik HJ, Cococcioni M, Scherlis DA, Marzari N. Density functional theory in transition-metal chemistry: A self-consistent Hubbard U approach. Phys Rev Lett. 2006;97(10):103001. doi: 10.1103/PhysRevLett.97.103001. [DOI] [PubMed] [Google Scholar]
- 18.O’Regan DD, Payne MC, Mostofi AA. Subspace representations in ab initio methods for strongly correlated systems. Phys Rev B. 2011;83(24):245124. [Google Scholar]
- 19.O’Regan DD, Hine NDM, Payne MC, Mostofi AA. Linear-scaling DFT+U with full local orbital optimization. Phys Rev B. 2012;85(8):085107. [Google Scholar]
- 20.Georges A, Kotliar G, Krauth W, Rozenberg MJ. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev Mod Phys. 1996;68(1):13–125. [Google Scholar]
- 21.Lin N, Marianetti CA, Millis AJ, Reichman DR. Dynamical mean-field theory for quantum chemistry. Phys Rev Lett. 2011;106(9):096402. doi: 10.1103/PhysRevLett.106.096402. [DOI] [PubMed] [Google Scholar]
- 22.Jacob D, Haule K, Kotliar G. Dynamical mean-field theory for molecular electronics: Electronic structure and transport properties. Phys Rev B. 2010;82(19):195115. [Google Scholar]
- 23.Hine NDM, Haynes PD, Mostofi AA, Skylaris C-K, Payne MC. Linear-scaling density-functional theory with tens of thousands of atoms: Expanding the scope and scale of calculations with ONETEP. Comput Phys Commun. 2009;180(7):1041–1053. [Google Scholar]
- 24.Weber C, et al. Vanadium dioxide: A Peierls-Mott insulator stable against disorder. Phys Rev Lett. 2012;108(25):256402. doi: 10.1103/PhysRevLett.108.256402. [DOI] [PubMed] [Google Scholar]
- 25.Weber C, et al. Importance of many-body effects in the kernel of hemoglobin for ligand binding. Phys Rev Lett. 2013;110(10):106402. doi: 10.1103/PhysRevLett.110.106402. [DOI] [PubMed] [Google Scholar]
- 26.Nozawa T, Yamamoto T, Hatano M. Infrared magnetic circular dichroism of myoglobin derivatives. Biochim Biophys Acta. 1976;427(1):28–37. doi: 10.1016/0005-2795(76)90282-8. [DOI] [PubMed] [Google Scholar]
- 27.Churg AK, Makinen MW. The electronic structure and coordination geometry of the oxyheme complex in myoglobin. J Chem Phys. 1978;68(4):1913–1925. [Google Scholar]
- 28.Makinen MW, Churg AK, Glick HA. Fe-O2 bonding and oxyheme structure in myoglobin. Proc Natl Acad Sci USA. 1978;75(5):2291–2295. doi: 10.1073/pnas.75.5.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev. 1988;88(6):899–926. [Google Scholar]
- 30.Glendening ED, et al. 2009. The NBO 5.9 manual. Available at www.chem.wisc.edu/∼nbo5/. Accessed November 1, 2013.
- 31.Lee LP, Cole DJ, Payne MC, Skylaris C-K. Natural bond orbital analysis in the ONETEP code: Applications to large protein systems. J Comput Chem. 2013;34(6):429–444. doi: 10.1002/jcc.23150. [DOI] [PubMed] [Google Scholar]
- 32.Skylaris C-K, Mostofi AA, Haynes PD, Diéguez O, Payne MC. Nonorthogonal generalized wannier function pseudopotential plane-wave method. Phys Rev B. 2002;66(3):035119. [Google Scholar]
- 33.Tomczak JM, Haule K, Kotliar G. Signatures of electronic correlations in iron silicide. Proc Natl Acad Sci USA. 2012;109(9):3243–3246. doi: 10.1073/pnas.1118371109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veiseh O, Gunn JW, Zhang M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv Drug Deliv Rev. 2010;62(3):284–304. doi: 10.1016/j.addr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Active sites of transition-metal enzymes with a focus on nickel. Curr Opin Struct Biol. 1998;8(6):749–758. doi: 10.1016/s0959-440x(98)80095-x. [DOI] [PubMed] [Google Scholar]
- 36.Maier TA, Pruschke T, Jarrell M. Angle-resolved photoemission spectra of the Hubbard model. Phys Rev B. 2002;66(7):075102. [Google Scholar]
- 37.Hine NDM, Haynes PD, Mostofi AA, Payne MC. Linear-scaling density-functional simulations of charged point defects in Al2O3 using hierarchical sparse matrix algebra. J Chem Phys. 2010;133(11):114111. doi: 10.1063/1.3492379. [DOI] [PubMed] [Google Scholar]
- 38.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 39.Hill Q, Skylaris CK. Including dispersion interactions in the ONETEP program for linear-scaling density functional theory calculations. Proc R Soc A. 2009;465(465):669–683. [Google Scholar]
- 40.Dias da Silva LGGV, Tiago ML, Ulloa SE, Reboredo FA, Dagotto E. Many-body electronic structure and Kondo properties of cobalt-porphyrin molecules. Phys Rev B. 2009;80(15):155443. [Google Scholar]
- 41.Aichhorn M, Daghofer M, Evertz HG, von der Linden W. Low-temperature Lanczos method for strongly correlated systems. Phys Rev B. 2003;67(16):161103. [Google Scholar]
- 42.Slater JC. The ferromagnetism of nickel. Phys Rev. 1936;49(7):537–545. [Google Scholar]
- 43.Kanamori J. Superexchange interaction and symmetry properties of electron orbitals. J Phys Chem Solids. 1959;10(2-3):87–98. [Google Scholar]
- 44.Imada M, Fujimori A, Tokura Y. Metal-insulator transitions. Rev Mod Phys. 1998;70(4):1039. [Google Scholar]
- 45.Pruschke T, Zölfl M. Electronic structure and ordered phases in transition metal oxides: Application of the dynamical mean-field theory. In: Kramer B, editor. Advances in Solid State Physics. Vol 40. Berlin: Springer; 2000. pp. 251–265. [Google Scholar]
- 46.Millis AJ. Optical conductivity and correlated electron physics. Strong interactions in low dimensions. PCMLD. 2004;25:195. [Google Scholar]
- 47.Halpern V, Bergmann A. Calculation of electronic Green's functions using nonorthogonal basis functions: Application to crystals. J Phys C Solid State Phys. 1972;5(15):1953. [Google Scholar]
- 48.Ratcliff LE, Hine NDM, Haynes PD. Calculating optical absorption spectra for large systems using linear-scaling density functional theory. Phys Rev B. 2011;84(16):165131. [Google Scholar]
- 49.Read AJ, Needs RJ. Calculation of optical matrix elements with nonlocal pseudopotentials. Phys Rev B Condens Matter. 1991;44(23):13071–13073. doi: 10.1103/physrevb.44.13071. [DOI] [PubMed] [Google Scholar]
- 50.Di Marco I, et al. Correlation effects in the total energy, the bulk modulus, and the lattice constant of a transition metal: Combined local-density approximation and dynamical mean-field theory applied to Ni and Mn. Phys Rev B. 2009;79(11):115111. [Google Scholar]
- 51.Pourovskii LV, Amadon B, Biermann S, Georges A. Self-consistency over the charge density in dynamical mean-field theory: A linear muffin-tin implementation and some physical implications. Phys Rev B. 2007;76(23):235101. [Google Scholar]
- 52.Galitskii VM, Migdal AB. Application of quantum field theory methods to the many body problem. Sov Phys JETP. 1958;7(1):96–104. [Google Scholar]