

Significance
Atmospheric secondary organic aerosol (SOA) has important impacts on climate and air quality, yet models continue to have difficulty in accurately simulating SOA concentrations. Nearly all SOA models are tied to observations of SOA formation in laboratory chamber experiments. Here, a comprehensive analysis of new experimental results demonstrates that the formation of SOA in laboratory chambers may be substantially suppressed due to losses of SOA-forming vapors to chamber walls, which leads to underestimates of SOA in air-quality and climate models, especially in urban areas where anthropogenic SOA precursors dominate. This analysis provides a time-dependent framework for the interpretation of laboratory chamber experiments that will allow for development of parameterized models of SOA formation that are appropriate for use in atmospheric models.
Abstract
Secondary organic aerosol (SOA) constitutes a major fraction of submicrometer atmospheric particulate matter. Quantitative simulation of SOA within air-quality and climate models—and its resulting impacts—depends on the translation of SOA formation observed in laboratory chambers into robust parameterizations. Worldwide data have been accumulating indicating that model predictions of SOA are substantially lower than ambient observations. Although possible explanations for this mismatch have been advanced, none has addressed the laboratory chamber data themselves. Losses of particles to the walls of chambers are routinely accounted for, but there has been little evaluation of the effects on SOA formation of losses of semivolatile vapors to chamber walls. Here, we experimentally demonstrate that such vapor losses can lead to substantially underestimated SOA formation, by factors as much as 4. Accounting for such losses has the clear potential to bring model predictions and observations of organic aerosol levels into much closer agreement.
Most of the understanding concerning the formation of secondary organic aerosol (SOA) from atmospheric oxidation of volatile organic compounds (VOCs) over the past 30 y has been developed from data obtained in laboratory chambers (1). SOA is a major component of particulate matter smaller than 1 μm (2) and consequently has important impacts on regional and global climate and human health and welfare. Accurate simulation of SOA formation and abundance within 3D models is critical to quantifying its atmospheric impacts. Measurements of SOA formation in laboratory chambers provide the basis for the parameterizations of SOA formation (3) in regional air-quality models and global climate models (4). A number of studies indicate that ambient SOA concentrations are underpredicted within models, often substantially so, when these traditional parameterizations are used (e.g., 5, 6). Some of this bias has been attributed to missing SOA precursors in emissions inventories, such as so-called intermediate volatility organic compounds, to ambient photochemical aging of semivolatile compounds occurring beyond that in chamber experiments (7) or to aerosol water/cloud processing (8). The addition of a more complete spectrum of SOA precursors into models has not, however, closed the measurement/prediction gap robustly. For example, recent analysis of organic aerosol (OA) concentrations in Los Angeles revealed that observed OA levels, which are dominated by SOA, exceed substantially those predicted by current atmospheric models (9), in accord with earlier findings in Mexico City (10).
Here, we demonstrate that losses of SOA-forming vapors to chamber walls during photooxidation experiments can lead to substantial and systematic underestimation of SOA. Recent experiments have demonstrated that losses of organic vapors to the typically Teflon walls of a laboratory chamber can be substantial (11), but the effects on SOA formation have not yet been quantitatively established. In essence, the walls serve as a large reservoir of equivalent OA mass that compete with the particulate SOA for SOA-forming compounds.
Toluene Photooxidation Experiments
The effect of vapor wall loss on SOA formation has been comprehensively assessed based on results from a series of 18-h toluene photooxidation experiments conducted in the California Institute of Technology (Caltech) environmental chamber under both high- and low-NOx conditions (SI Appendix, Toluene Photooxidation Experiments and Table S1). Toluene is a component of motor vehicle emissions and an important SOA precursor (1). Initial [VOC]/[NOx] ratios were 5.4 ± 0.3 ppbC/ppb, similar to current conditions in Los Angeles. In these experiments, the ratio of initial seed particle surface area (SA) to chamber wall SA was systematically varied by changing the concentration and diameter of (NH4)2SO4 seed aerosol and keeping all other conditions the same [i.e., hydroxyl radical (OH) and toluene concentration]. In this manner, the influence of seed SA on SOA formation can be isolated.
Fig. 1 shows the observed time-dependent SOA yields, defined as the mass of SOA formed per mass of VOC reacted, determined from the evolution of the particle size distributions (SI Appendix, Fig. S1) as a function of initial seed SA for the high- and low-NOx experiments. The SOA concentrations have been corrected for physical deposition of particles to the walls (SI Appendix, Particle Wall Loss Correction), which is the appropriate correction to use here as our analysis explicitly accounts for loss of vapors to the walls, discussed further below. Except for the unseeded experiment, the SOA yield for each low-NOx experiment is reasonably constant with time (from 4 to 18 h) and COA at any given SA. For each high-NOx experiment there is a clear increase in the yield with COA and time. The toluene SOA yield at a given COA is generally lower for high-NOx than for low-NOx conditions at a comparable initial seed SA. These differences in the time-dependent yields indicate differences in the chemical pathways leading to SOA formation between low- and high-NOx conditions. Most importantly, the absolute amount of SOA formed for the same initial conditions increases with seed SA, consistent with refs. 12 and 13, with an indication of a plateau being reached at the highest seed SA. The SOA formed in the unseeded experiments is particularly small. Typical laboratory chamber experiments use initial seed SA < 103 μm2 cm−3, which corresponds to seed-to-wall ratios of <1 × 10−3 in most chambers. These experiments definitively demonstrate that SOA yields vary with seed SA. This variability may partially explain some of the differences in SOA yields reported in the literature.
Fig. 1.

Hourly averaged lower bound SOA yields over the course of a toluene photooxidation experiment as a function of initial ammonium sulfate seed surface area for (A) high-NOx and (B) low-NOx conditions. Symbol color indicates the SOA mass concentration and symbol size the time after lights were turned on. The filled circles are from the current experiments and the open diamonds from ref. 21. The dashed gray line and × are the end-of-experiment yields from the optimized best-fit SOM simulations.
SOA Modeling and the Influence of Vapor Wall Loss
The increase in yield with seed SA is consistent with loss of vapors to the chamber walls, and likely results from an increase in the rate of mass transfer of vapor species to the particles relative to the walls. Quantitative understanding of the role of vapor wall loss is necessary to characterize the extent to which SOA yields in chamber experiments are underestimated relative to the atmosphere. The loss of “extremely low volatility” organic compounds to chamber walls has recently been implicated as important for understanding SOA formation for a different chemical system, α-pinene + O3 (13, 14). The competition between vapor condensation to particles versus to chamber walls is quantitatively examined here using a time-dependent, parameterizable model of SOA formation, the statistical oxidation model (SOM; refs. 15, 16; SI Appendix, The Statistical Oxidation Model). The SOM accounts for vapor wall losses based on observations showing that wall losses of semivolatile species in Teflon chambers are reversible (11). Such wall loss is modeled dynamically and depends on the equivalent OA mass of the chamber walls (Cw, mg m−3), the first-order vapor wall loss rate coefficient (kw, s−1), and the vapor saturation concentration of compounds i (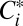 , μg m−3). kw reflects the combined effects of turbulent mixing in the chamber, molecular diffusion of vapor molecules through the near-wall boundary layer, and any penetration into chamber walls. kw is likely to be chamber specific, as the extent of turbulent mixing depends on specific chamber operating conditions, but is reasonably independent of compound identity (11). Values of kw for a range of gases were estimated in one study (17) to range from ∼2 × 10−5 to 10−3 s−1, corresponding to timescales of many hours to 10 min. The largest kw values are appropriate only for chambers with active mixing, which the Caltech chamber does not have (SI Appendix, Vapor Wall Loss). Observations (11) suggest that Cw varies somewhat with compound identity, ranging from ∼2 to 24 mg m−3, compared with chamber OA concentrations that are usually 1–3 orders of magnitude smaller; a value of 10 mg m−3 is used here as the base case. Gas-particle partitioning is modeled dynamically assuming absorptive partitioning, including correction for noncontinuum effects and imperfect accommodation, the latter of which is characterized by the mass-accommodation coefficient, α. The accommodation coefficient reflects the overall transfer of vapor molecules into the particle bulk, and is likely dependent on the chemical makeup of both the vapor and particle phases and processes that occur at the particle surface. This formulation is more general than most previous analyses of SOA formation in environmental chambers, which typically assume instantaneous gas-particle equilibrium.
, μg m−3). kw reflects the combined effects of turbulent mixing in the chamber, molecular diffusion of vapor molecules through the near-wall boundary layer, and any penetration into chamber walls. kw is likely to be chamber specific, as the extent of turbulent mixing depends on specific chamber operating conditions, but is reasonably independent of compound identity (11). Values of kw for a range of gases were estimated in one study (17) to range from ∼2 × 10−5 to 10−3 s−1, corresponding to timescales of many hours to 10 min. The largest kw values are appropriate only for chambers with active mixing, which the Caltech chamber does not have (SI Appendix, Vapor Wall Loss). Observations (11) suggest that Cw varies somewhat with compound identity, ranging from ∼2 to 24 mg m−3, compared with chamber OA concentrations that are usually 1–3 orders of magnitude smaller; a value of 10 mg m−3 is used here as the base case. Gas-particle partitioning is modeled dynamically assuming absorptive partitioning, including correction for noncontinuum effects and imperfect accommodation, the latter of which is characterized by the mass-accommodation coefficient, α. The accommodation coefficient reflects the overall transfer of vapor molecules into the particle bulk, and is likely dependent on the chemical makeup of both the vapor and particle phases and processes that occur at the particle surface. This formulation is more general than most previous analyses of SOA formation in environmental chambers, which typically assume instantaneous gas-particle equilibrium.
Optimal values for kw and α have been determined for the sets of low- and high-NOx experiments by comparing observed and simulated best-fit time-dependent COA profiles for a wide range of kw and α. The kw/α pair that provides for the best overall agreement with the time-dependent SOA formation observed across all experiments at a given NOx condition, excluding the nucleation experiments, is considered the optimal solution (Fig. 2 A and B) (SI Appendix, Optimizing kwand α and Fig. S3). For low-NOx experiments, the optimal kw = 2.5 × 10−4 s−1 and α = 2 × 10−3. The independently determined values for high-NOx experiments are similar: kw = 2.5 × 10−4 s−1 and α = 1 × 10−3. The simulations provide good overall agreement with the observed seed SA dependence only for a combination of a small α and a kw on the order of 10−4 s−1, indicating that both parameters are important to describing chamber SOA formation. (It should be noted that a reasonable fit for an individual experiment can be obtained for many kw/α pairs, including when instantaneous gas-particle equilibrium is assumed, i.e., when α = 1. This is not meant to imply that the absolute values of these parameters are not important, but that only in analyzing the combined datasets at multiple seed SA can optimal kw and α values be uniquely established.) The determined optimal kw values are consistent with theoretical estimates (SI Appendix, Vapor Wall Loss and Fig. S4) and some observations (11), but larger than some previous observations in the Caltech chamber (18), most likely reflecting the limited time resolution of those observations, which might not have allowed for separation of filling and mixing of the chamber from wall loss, but potentially also reflecting differences between chemical systems.
Fig. 2.

Observed (points) and simulated (lines) SOA concentrations for each photochemical oxidation experiment performed for different initial inorganic seed surface area for (A) low-NOx and (B) high-NOx conditions. The dashed lines indicate the experiment to which the SOM was explicitly fit, and the solid lines indicate simulation results based on those fits. (C) The wall loss bias factor, Rwall, as a function of seed surface area. Filled symbols use the optimal kw/α pair and the corresponding best-fit SOM parameters determined from A and B. Open symbols assume α = 1 and each experiment was individually fit using the optimal kw. The error bars indicate the 1σ SD in Rwall for each experiment over the period when COA > 0.5 μg m−3.
The small optimal α values required to reproduce the observed seed SA dependence likely reflect mass-transfer limitations within the particle phase, which can occur for highly viscous SOA particles (19). When α ∼10−3, mass accommodation is relatively slow and the vapors and particles cannot be assumed to be in instantaneous equilibrium. The timescale associated with reaching gas-particle equilibrium (τg-p,eqm) varies with seed SA and size (SI Appendix, Gas-Particle Partitioning Timescales and Fig. S5). Given α = 10−3, τg-p,eqm ranges from ∼230 min for the smallest seed SA to only ∼30 min for the largest seed SA. Thus, at the smallest seed SA τg-p,eqm is similar to the timescale determined for gas wall loss (1/kw = 67 min) and the influence of gas wall loss is more pronounced.
Importantly, loss of condensable vapors to the chamber walls leads to a low bias in the observed SOA formed even for the experiments with the highest seed SA. To quantitatively assess this bias, simulations have been performed using the best-fit SOM parameters determined for the optimal kw/α pair above, but with kw = 0 (no wall loss). The ratio Rwall = CSOA(kw = 0)/CSOA(kw,optimal) has been calculated for each experiment and quantifies the bias in the SOA yield due to traditionally unaccounted for vapor wall losses. The magnitude of Rwall depends on the experiment considered, decreasing with increasing seed SA and reaching a plateau at large seed SA (Fig. 2C). For the seeded experiments, Rwall averaged over the period when COA > 0.5 μg m−3 varied from 3.6 (±0.6) to 2.1 (±0.2) for low NOx and from 4.2 (±0.9) to 2.1 (±0.2) for high NOx in going from lowest to highest seed SA, and where the uncertainties are 1 SD over the averaging period. These ratios correspond to end-of-experiment corrected mass yields of 1.6 (low NOx) and 0.93 (high NOx), substantially exceeding the values currently used in chemical transport models for toluene (∼0.47 for low NOx and ∼0.12 for high NOx; ref. 4). Given an O:C ratio for toluene + OH SOA of ∼0.7 (20), the carbon yields would be 0.94 (low NOx) and 0.55 (high NOx). [The calculated end of experiment O:C are 0.67 (low NOx) and 0.91 (high NOx).] This implies that ∼6% and ∼55% of the product carbon mass remains in the gas phase for low- and high-NOx conditions, respectively. The oxidation process is a balance between functionalization and fragmentation. In the absence of fragmentation, the carbon yield would asymptote to 100%. The low-NOx carbon yield in the absence of vapor wall loss is close to 100% and the SOA yield is approximately independent of the total OA mass, which together indicate that fragmentation plays only a minor role. Fragmentation is comparably more important under high-NOx conditions.
Interestingly, Rwall values similar to those determined for the optimal kwall/α pair are obtained when vapor wall loss is accounted for (i.e., kw ≠ 0) but when it is assumed that α = 1 (i.e., that gas-particle equilibration is effectively instantaneous) during fitting of each individual experiment (Fig. 2C). This indicates that the magnitude of α is not key to fitting of an individual experiment, but when the experiments are taken together as a combined dataset α is indeed key to matching the observed dependence on seed surface area. Related, α is not key to there being an influence of vapor wall loss on the overall SOA yield, which is controlled more so by the magnitude of kw, although at lower seed SA for a given kw, the derived Rwall increases as α decreases. The finding that large Rwall values are obtained even when α = 1 indicates that the general conclusions here regarding vapor wall loss are robust with respect to knowledge of α. This is important because other chemical systems might not exhibit as slow mass accommodation but could still be affected by vapor wall loss. Clearly, loss of condensable vapors to the chamber walls can suppress SOA yields relative to those that are relevant for the atmosphere.
Dependence on Experimental Conditions
For a given chamber, the extent to which vapor wall loss affects SOA yield will depend on the combination of (i) the rate of oxidation and duration of a given experiment, (ii) the precursor VOC concentration, (iii) the particular chemical pathways associated with oxidation of a given species, i.e., the precursor identity, and (iv) the seed SA. This is illustrated for the toluene low-NOx system by carrying out a series of 18-h simulations where [OH] is varied from 1 to 100 × 106 molecules cm−3 and [toluene]initial from 1 to 900 ppb for a seed SA = 1,000 μm2 cm−3 using the SOM parameters and optimal kw/α determined above. The calculated Rwall varies with oxidant and precursor concentration (actually, VOC loss rate), with smaller Rwall when oxidation is faster and at larger precursor VOC concentration (Fig. 3 and SI Appendix, Fig. S6). (Note that these calculations do not account for differences that might result from changes in the relative importance of RO2 + HO2 versus RO2 + RO2 reactions as [VOC]initial and [OH] are varied.) Such “rate effects” have been observed for SOA produced in aromatic systems (21). The seed SA dependence is substantially reduced when the [VOC]initial is large, especially at high [OH]. Overall, these dependencies, along with differences in the initial seed SA, may help explain some of the differences (and similarities) in historical aerosol yields measured in different chambers.
Fig. 3.

Calculated SOA yield bias as a function of initial toluene concentration and OH concentration when kw = 2.5 × 10−4 s−1 and Cw = 10 mg m−3. The Rwall values for a given [toluene] and [OH] are indicated by colors and contours, and are averaged over the period when CSOA > 0.5 μg m−3 to the end of an experiment at 18 h. Results are based on the optimal fit of the SOM to the low-NOx experiments.
Vapor Losses and Sensitivity to Cw
The observed time-dependent COA to which the SOM was fit were corrected to account for physical deposition of the particles to the walls. Loss of vapors, excluding growth of suspended particles, was separately accounted for assuming that the vapors continuously interact with the Teflon chamber walls, with Cw = 10 mg m−3 (11). Some previous studies (22, 23) have alternatively assumed that vapors interact only with particles that have deposited to the walls during that experiment, as opposed to with the walls directly, and further that the timescales associated with partitioning between vapors and suspended or wall-deposited particles are the same (SI Appendix, Particle Wall-Loss Correction). In this alternative scenario, the effective Cw is time dependent (and zero at the start of an experiment) and related to the suspended particle concentration and the particle wall loss rate. Most chamber experiments aim to limit the extent of particle deposition, and thus it is reasonable to assume that, in general, the concentration of wall-deposited particles is less than the suspended particle concentration. Further, most modern experiments limit the observed COA to < 0.1 mg m−3, and thus the effective Cw in this alternative case will be substantially smaller than when vapors are assumed to partition into the chamber walls.
It is therefore useful to examine the dependence of the calculated Rwall on the assumed Cw, where an assumed Cw < ∼0.1 mg m−3 corresponds approximately to the wall-deposited particle alternative scenario (22, 23). This has been done for the low-NOx experiments where the SOM was fit to the observations for different assumed Cw, with good fits obtained for all Cw. Above Cw = 0.2 mg m−3 (= 200 μg m−3) the calculated Rwall is constant. Below 0.2 mg m−3 the calculated Rwall falls off, reaching a second plateau at small Cw that is still above unity (Fig. 4). The plateaus at high and low Cw result from the best-fit SOM parameters varying with Cw to compensate for the differing amounts of loss of vapors to the walls and still maintaining the same suspended COA time profile (SI Appendix, Fig. S7). Because measurements demonstrate that vapors are lost directly to Teflon walls (11), this indicates that when vapor wall loss is accounted for assuming that the vapors interact only with wall-deposited particles, the extent of the vapor loss is underestimated. That Rwall is constant above Cw = 0.2 mg m−3 demonstrates that our conclusions are robust with respect to the assumption regarding the exact value of Cw.
Fig. 4.

Calculated vapor wall loss bias, Rwall, as a function of the effective wall mass concentration, Cw, for the low-NOx toluene photooxidation experiments at varying initial seed surface area. For each Cw, the SOM was fit to the experiment with seed surface area = 5.5 × 103 μm2 cm−3 using kw = 2.5 × 10−4 s−1 and α = 2 × 10−3. The determined best-fit SOM parameters were then used to simulate SOA formation for the experiments performed at other seed concentrations but assuming the same Cw.
Consideration of Historical Experiments
To estimate the potential influence of vapor wall losses for systems other than toluene, we calculated SOA yield biases for a variety of other VOCs (16, 20, 21, 23, 24) (SI Appendix, Fitting of Historical Chamber Data). It should be noted that the experimental conditions in the historical experiments are not identical to those for the toluene experiments, especially for high-NOx conditions (SI Appendix, Historical Experiments). Although kw for a given chamber is reasonably independent of the precursor compound, α may depend on the precursor identity. The results for the toluene experiments indicate that smaller α values generally correspond to larger Rwall. Therefore, a conservative, likely lower-bound estimate of Rwall has been obtained for each precursor assuming that α = 1 during fitting and using kw = 1 × 10−4 s−1 (instead of 2.5 × 10−4) to account for potential differences in the chamber used for these historical experiments (SI Appendix, Figs. S8–S10). The use of a smaller kw will decrease Rwall, all other factors being equal. Calculated Rwall values range from as small as 1.1 to as large as 4.1 (Table 1 and SI Appendix, Fig. S11). The typically smaller values for the high-NOx vs. low-NOx experiments reflect the much shorter reaction timescales and higher oxidant and NOx concentrations in the historical high-NOx experiments, compared with the current experiments with toluene. Evidently, the extent to which vapor wall loss will have influenced historical experiments is variable, yet potentially substantial and deserving of further investigation through new experiments and consideration of other datasets. Importantly, the results here indicate that quantitative analysis of SOA formation in chambers requires use of an explicitly time-dependent model that accounts for the simultaneous and competing condensation of vapors onto particles versus onto the chamber walls, representing a major shift from most historical analyses, which did not take time explicitly into account.
Table 1.
Average biases in SOA yields due to vapor wall losses for various VOCs under low- and high-NOx conditions
| VOC* | Low-NOx | High-NOx |
 † †
|
 † †
|
|
| Toluene‡ (this study) | 2.1–3.6 | 2.1–4.2 |
| n-dodecane (16) | 4.1 ± 0.8¶ | 1.16 ± 0.08 |
| 2-methylundecane (16) | 3.7 ± 0.5 | 1.4 ± 0.2 |
| Cyclododecane (16) | 3.0 ± 0.3 | 1.10 ± 0.05 |
| Hexylcyclohexane (16) | 2.4 ± 0.3 | 1.16 ± 0.07 |
| Toluene (21) | 1.9 ± 0.4 | 1.13 ± 0.06 |
| Benzene (21) | 1.8 ± 0.4 | 1.25 ± 0.1 |
| M-xylene (21) | 1.8 ± 0.4 | 1.2 ± 0.1 |
| M-xylene (23) | 1.6 ± 0.3 | |
| Naphthalene (24) | 1.2 ± 0.1 | 1.2 ± 0.1 |
| α-pinene + OH (20) | 1.6 ± 0.3 | 1.3 ± 0.1 |
| Isoprene + OH (20) | 3.1 ± 1.3 | 2.2 ± 0.5 |
For all VOC’s except toluene (this study), kw = 1 × 10−4 s−1 and α = 1. For toluene (this study), kw = 2.5 × 10−4 s−1 and α = 0.002 (low NOx) or 0.001 (high NOx).
Calculated for the period when CSOA > 0.5 μg m−3 through the end of a given experiment.
For toluene, the reported Rwall indicate the range of values determined at different initial seed SA.
Uncertainties are 1σ over the averaging period.
Implications
Our results show that the effect of vapor wall loss on SOA yields can be substantial. If reported SOA yields are low by factors of ∼1.1–4.2, as our results suggest they may be, then SOA concentrations simulated in 3D models will be correspondingly low. Although the analysis presented here for the toluene SOA system needs to be comprehensively expanded to other main classes of SOA precursors, beyond the assessment above, it is likely that a lack of proper accounting for vapor wall losses that suppress chamber-derived SOA yields contribute substantially to the underprediction of ambient SOA concentrations in atmospheric models.
Materials and Methods
Chamber Experiments.
Toluene photooxidation experiments were carried out in the new 24-m3 Teflon environmental reaction chambers at Caltech. Liquid toluene was evaporated into the chamber to achieve a concentration of ∼38 ppb gas-phase mixing ratio (= 143 μg m−3). Hydroxyl radicals were generated from photolysis of H2O2 either with (high NOx) or without (low NOx) addition of NO to the chamber. Dried ammonium sulfate seed particles were added via atomization of an aqueous solution of ammonium sulfate solution until the desired seed concentration was obtained. The toluene, seed particles, H2O2 and NOx were allowed to mix in the chamber for 1 h, after which time the black lights were turned on to initiate H2O2 photolysis. Particle number size distributions were measured using a cylindrical differential mobility analyzer coupled to a condensation particle counter. More details are available in the SI Appendix.
SOA Modeling.
The statistical oxidation model (15) was used to analyze the experimental observations. The SOM simulates the multigenerational gas-phase oxidation of a precursor VOC that has NC carbon atoms and NO oxygen atoms as reactions cause the precursor and product species to functionalize, increasing NO, and/or fragment, decreasing NC. Addition of oxygen atoms leads to a decrease in vapor pressure, which drives condensation of the gas-phase species. Mass transfer between the gas and particle phases is treated dynamically. The parameters that describe functionalization, fragmentation, and the decrease in vapor pressure upon oxygen addition are adjusted by fitting to the experimental observations of time-dependent SOA formation. More details are provided in the SI Appendix.
Supplementary Material
Acknowledgments
We thank Alma Hodzic, Julia Lee-Taylor, Sasha Madronich (National Center for Atmospheric Research), Paul Ziemann (University of Colorado Boulder), Paul Wennberg (Caltech), Manabu Shiraiwa (Max Planck Institut für Chemie), and Anthony Wexler (University of California, Davis) for useful discussions. This work was supported by the National Science Foundation Grants ATM-1151062 and AGS-1057183, Department of Energy Grant DE-SC 0006626, and the California Air Resources Board Contract 12-312.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404727111/-/DCSupplemental.
References
- 1.Odum JR, Jungkamp TPW, Griffin RJ, Flagan RC, Seinfeld JH. The atmospheric aerosol-forming potential of whole gasoline vapor. Science. 1997;276(5309):96–99. doi: 10.1126/science.276.5309.96. [DOI] [PubMed] [Google Scholar]
- 2.Jimenez JL, et al. Evolution of organic aerosols in the atmosphere. Science. 2009;326(5959):1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- 3.Odum JR, et al. Gas/particle partitioning and secondary organic aerosol yields. Environ Sci Technol. 1996;30(8):2580–2585. [Google Scholar]
- 4.Carlton AG, et al. Model representation of secondary organic aerosol in CMAQv4.7. Environ Sci Technol. 2010;44(22):8553–8560. doi: 10.1021/es100636q. [DOI] [PubMed] [Google Scholar]
- 5.Heald CL, et al. A large organic aerosol source in the free troposphere missing from current models. Geophys Res Lett. 2005;32:L18809. [Google Scholar]
- 6.Volkamer R, et al. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected. Geophys Res Lett. 2006;33:L17811. doi: 10.1029/2006GL026899. [DOI] [Google Scholar]
- 7.Robinson AL, et al. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science. 2007;315(5816):1259–1262. doi: 10.1126/science.1133061. [DOI] [PubMed] [Google Scholar]
- 8.Ervens B, Turpin BJ, Weber RJ. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmos Chem Phys. 2011;11(21):11069–11102. [Google Scholar]
- 9.Ensberg JJ, et al. Emission factor ratios, SOA mass yields, and the impact of vehicular emissions on SOA formation. Atmos Chem Phys. 2013;14:2383–2397. [Google Scholar]
- 10.Dzepina K, et al. Modeling the multiday evolution and aging of secondary organic aerosol during MILAGRO 2006. Environ Sci Technol. 2011;45(8):3496–3503. doi: 10.1021/es103186f. [DOI] [PubMed] [Google Scholar]
- 11.Matsunaga A, Ziemann PJ. Gas-wall partitioning of organic compounds in a Teflon film chamber and potential effects on reaction product and aerosol yield measurements. Aerosol Sci Technol. 2010;44(10):881–892. [Google Scholar]
- 12.Kroll JH, Chan AWH, Ng NL, Flagan RC, Seinfeld JH. Reactions of semivolatile organics and their effects on secondary organic aerosol formation. Environ Sci Technol. 2007;41(10):3545–3550. doi: 10.1021/es062059x. [DOI] [PubMed] [Google Scholar]
- 13.Ehn M, et al. A large source of low-volatility secondary organic aerosol. Nature. 2014;506(7489):476–479. doi: 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- 14.Kokkola H, et al. The role of low volatile organics on secondary organic aerosol formation. Atmos Chem Phys. 2014;14(3):1689–1700. [Google Scholar]
- 15.Cappa CD, Wilson KR. Multi-generation gas-phase oxidation, equilibrium partitioning, and the formation and evolution of secondary organic aerosol. Atmos Chem Phys. 2012;12:9505–9528. [Google Scholar]
- 16.Cappa CD, et al. Application of the Statistical Oxidation Model (SOM) to secondary organic aerosol formation from photooxidation of C12 alkanes. Atmos Chem Phys. 2013;13:1591–1606. [Google Scholar]
- 17.McMurry PH, Grosjean D. Gas and aerosol wall losses in Teflon film smog chambers. Environ Sci Technol. 1985;19(12):1176–1182. doi: 10.1021/es00142a006. [DOI] [PubMed] [Google Scholar]
- 18.Loza CL, et al. Secondary organic aerosol yields of 12-carbon alkanes. Atmos Chem Phys Discuss. 2013;13(8):20677–20727. [Google Scholar]
- 19.Shiraiwa M, et al. Size distribution dynamics reveal particle-phase chemistry in organic aerosol formation. Proc Natl Acad Sci USA. 2013;110(29):11746–11750. doi: 10.1073/pnas.1307501110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chhabra PS, et al. Elemental composition and oxidation of chamber organic aerosol. Atmos Chem Phys. 2011;11(17):8827–8845. [Google Scholar]
- 21.Ng NL, et al. Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos Chem Phys. 2007;7(14):3909–3922. [Google Scholar]
- 22.Hildebrandt L, Donahue NM, Pandis SN. High formation of secondary organic aerosol from the photo-oxidation of toluene. Atmos Chem Phys. 2009;9(9):2973–2986. [Google Scholar]
- 23.Loza CL, et al. Chemical aging of m-xylene secondary organic aerosol: Laboratory chamber study. Atmos Chem Phys. 2012;12(1):151–167. [Google Scholar]
- 24.Chan AWH, et al. Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: Implications for oxidation of intermediate volatility organic compounds (IVOCs) Atmos Chem Phys. 2009;9(9):3049–3060. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.