

Abstract
The conversion of normal cells to cancer cells involves a shift from catabolic to anabolic metabolism involving increased glucose uptake and the diversion of glycolytic intermediates into nucleotides, amino acids and lipids needed for cell growth. An underappreciated aspect of nutrient uptake is the utilization of serum lipids. We investigated the dependence of human cancer cells on serum lipids and report here that Ras-driven human cancer cells are uniquely dependent on serum lipids for both proliferation and survival. Removal of serum lipids also sensitizes Ras-driven cancer cells to rapamycin – indicating that the enhanced need for serum lipids creates a synthetic lethal phenotype that could be exploited therapeutically. While depriving humans of serum lipids is not practical, suppressing uptake of lipids is possible. Suppressing macropinocytosis in Ras-driven cancer cells also created sensitivity to suppression of the mammalian/mechanistic target of rapamycin complex 1 (mTORC1). It is speculated that this property displayed by Ras-driven cancer cells represents an Achilles' heel for the large number of human cancers that are driven by activating Ras mutations.
Introduction
An emerging hallmark of cancer is the “metabolic transformation” that occurs to accommodate the needs of a proliferating population of cells (1). The conversion of normal cells to cancer cells involves a shift from catabolic to anabolic metabolism involving increased glucose uptake and the diversion of glycolytic intermediates into nucleotides, amino acids and lipids needed for cell growth (1–4). In addition to glucose, cancer cells utilize glutamine as a nitrogen source for nucleotides and as a carbon source (5). Cancer cells also need essential amino acids that mammalian cells cannot synthesize. An underappreciated aspect of nutrient uptake is the utilization of exogenously supplied fatty acids (6). Cells grown in culture are provided with media that is supplemented with glucose, essential amino acids, and glutamine as nutrients for cell growth. However, mammalian cell do not synthesize all of the unsaturated lipids needed for membrane biosynthesis – there are “essential fatty acids” that must also be present in the medium (6). Conventional growth media used for culturing mammalian cells do not contain lipids – they are provided in the serum that typically supplement culture media.
One of the emerging fields of cancer therapeutics is the possibility of targeting the special metabolic needs of cancer cells (7). There has been considerable enthusiasm about the possibility of interfering with both glucose (8) and glutamine (5) utilization as therapeutic options for human cancers. However, while interfering with fatty acid synthesis in cancer cells has received attention (9), there has been very little reported on the utilization of exogenously supplied lipids and the therapeutic options. mTOR – the mammalian/mechanistic target of rapamycin – integrates signals that respond to nutrients and promotes cell cycle progression and cell survival (10). We have previously reported that suppression of mTOR in the absence of serum results in apoptosis in cancer cells harboring mutant Ras genes (11–13). In this report, we identify an enhanced need for exogenously supplied serum lipids in Ras-driven human cancer cell lines that creates a synthetic lethality (14) for suppressing mTOR. This finding suggests that the increased need for serum lipids by Ras-driven cancers may represent an Achilles' heel that could be therapeutically targeted in what may be as many as 30% of all human cancers.
Materials and Methods
Cells, cell culture conditions
The MDA-MB-231, Calu-1, BJ, MCF7, BxPC3, T24, HT29, Panc-1, HCT116 cell lines used in this study were obtained from American Type Culture Collection. No authentication was performed by the authors. Cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% FBS (Sigma). BxPC3 cell line was maintained in Roswell Park Memorial Institute (RPMI) (Sigma) medium supplemented with 10% fetal bovine serum (FBS). Delipidated FBS was obtained from Gemini Bio Products (900–123).
Materials
Reagents were obtained from the following sources. Antibodies against Cleaved PARP, actin, Akt, P-Akt (Ser473), P-Akt (Thr308), S6 kinase, P-S6 kinase (Thr389), 4EBP1, P-4EBP1 (Thr37-46), FASN, SCD1, ACL were obtained from Cell Signaling; antibodies against KRas were obtained from Abcam. MTT reagent was obtained from Sigma. Rapamycin was obtained from LC Labs, and 5-(N-ethyl-N-isopropyl) amiloride (EIPA) was obtained from Sigma.
Lipid mix supplementation
Fatty acid mix was obtained from Invitrogen (11905) and was supplied to cells as 1:200 dilution complexed with 10% bovine serum albumin (BSA) (Sigma) in 2 to 1 ratio for the final concentration of lipids in the media of 0.375 mg/L. The exact composition of the fatty acid mixture is provided in Table S1. Palmitic acid (Sigma) was diluted in Pluronic F-68 (Gibco, 24040) and supplied to the cells in the complex with BSA to the final concentration of lipids of 0.2 mg/L. The reduced level of lipid used was due to cytotoxicity of higher concentrations palmitic acid.
Transient transfections
Plasmids for transient transfections were obtained from the following sources: constitutively active KRas (Missouri S&T cDNA Resource Center, RASK2000C0), pcDNA3.1 verctor (Invitrogen). For transient transfections, cells were plated at 3×103 cells/6-well plate (cell proliferation assay) or at 30% confluence/60-mm plate (flow cytometry assay), overnight, and transfected using Polyfect (Qiagen) according to manufacturer's instructions. 18 hours post-transfection, cells were shifted to conditions as described in the text and figure legends.
Cell proliferation
Cells 6-well plates at 3,000 cells per well in 2 ml of full serum media overnight and shifted to various lipid conditions the next day. Media was not changed throughout the course of the experiment. After 5 days, cell number was determined by counting viable (adherent) and non-viable (floating) cells using hemocytometer. The MTT cell viability/growth assay, which measures NAD(P)H dependent anabolic activity was performed according to the vendors (Sigma) instructions.
PLD activity
PLD was determined by accumulation of the transphosphatidylation product [3H]-phosphatidylbutanol as described previously (15). Lipid membranes were labeled with [3H]-myristic acid (60 Ci/mmol; 1.5 μCi/ml; Perkin-Elmer) for 4 hours. 1-BtOH was added for 20 min before lipids where collected. Lipids were extracted and separated by thin layer chromatography along with phosphatidylbutanol standard (Enzo Life Sciences, BMLST401-0050). The phosphatidylbutanol fraction was identified through co-migration with standards and the levels of the PLD product [3H]-phosphatidylbutanol was determined by scintillation counting.
Western blot analysis
Proteins were extracted from cultured cells using M-PER (Thermo Scientific, 78501) and Western blot analysis of extracted proteins was performed using ECL (Thermo Scientific) as described previously (15).
Flow cytometric analysis
Cells were washed and collected by trypsinazation. Recovered cells were re-suspended in a solution containing 7 ml of 2% BSA in phosphate buffered saline, 5mM EDTA, 0.1% NaN3 and fixed by drop wise addition of 3ml of 100% ethanol. Fixed cells were collected and re-suspended in 500 μl of sorting buffer containing 2% BSA in phosphate buffered saline, 0.1% Triton-X 100, 5mM EDTA, 40μg/ml propidium iodide, 100μg/ml RNAse A, and incubated at 37C for 30 min. The cells were filtered through 70μm mesh to remove cell aggregates. The DNA content was analyzed by flow cytometry (FACSCalibur; Becton Dickinson), and percentages of cells within each phase of the cell cycle were determined using WinCycle software (Phoenix Flow Systems).
Results
Lipid deprivation leads to increased phospholipase D activity in Ras-driven human cancer cell lines
We previously reported that serum withdrawal led to an increase in phospholipase D (PLD) activity that was largely restricted to cancer cells harboring Ras mutations (11). PLD generates phosphatidic acid (PA) from phosphatidylcholine, which is required for the stability and activity of mTOR complexes (15, 16). mTORC1 is a sensor of nutrients that regulates both cell cycle progression and survival (17). It has been suggested that activation of the mTOR signaling node is the most commonly dysregulated signal in human cancers (1, 18). Since PA is at the center of membrane phospholipid biosynthesis, we recently proposed that PA impacts upon mTOR as an indicator of sufficient precursors for membrane synthesis in dividing cells and that cancer cells have co-opted PLD to sustain cell proliferation and survival of cancer cells by providing the PA needed to keep mTOR active (4). Thus, we speculated that the increased PLD activity observed in KRas-driven cancer cells could be a response to insufficient lipids in serum. To test this hypothesis, we examined the impact of serum lipids on the PLD activity stimulated by the withdrawal of lipids from MDA-MB-231 breast, Calu-1 lung, and T24 bladder cancer cells – all of which harbor mutant Ras genes and were previously shown to elevate their PLD activity is response to serum withdrawal (11). Replacing 10% FBS with 10% delipidated FBS resulted in an increase in PLD activity in all three cell lines that was almost as robust as that observed with complete withdrawal of FBS (Fig. 1). If a mixture of fatty acids (FAs) consisting of saturated, mono-unsaturated, and poly-unsaturated FAs (Table S1), was substituted for the FBS, elevated PLD activity was still observed – indicating that the lack of serum growth factors also contributed to the elevated PLD activity. Importantly, if the delipidated FBS was combined with the FA mixture, PLD activity was restored to the basal level observed in the presence of serum. These data demonstrate that the elevated PLD activity observed in response to serum withdrawal in Ras-driven human cancer cells is due in part to the absence of serum lipids.
Figure 1.
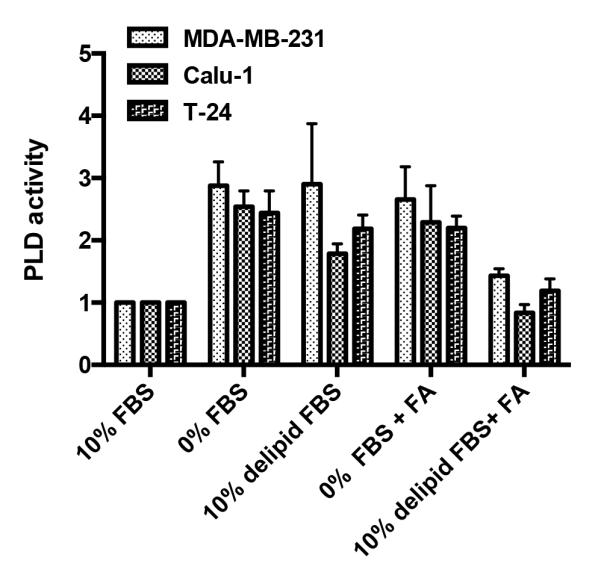
Elevated PLD activity in response to serum withdrawal in Ras-driven Cancer cell lines is dependent on withdrawal of both growth factors and lipids. MDA-MB-231, T24, and Calu-1 cells were plated at 70% confluence overnight, and shifted for 24 h to various media conditions containing 10% FBS, 0% FBS, 10% delipidated FBS, 0% FBS with BSA-FA mixture, 10% delipidated FBS with BSA-FA mixture. After 20 hr, [3H]-myristic acid was added for 4 hr to label lipids. 1-BtOH was added for 20 min, and the amount of the PLD catalyzed transphosphatidylation product, phosphatidyl-butanol, was determined as described in Experimental Procedures. Values were normalized to the levels of PLD activity in full growth serum conditions, which were given a value of 100%. Error bars represent S.D. values for at least two independent experiments.
Ras-driven cancer cells are uniquely dependent on exogenous fatty acids for proliferation and survival
Elevated PLD activity promotes survival in response to stress in Ras-driven cancer cells (11). To test whether the increased PLD activity in Ras-driven cell lines yields a proliferative advantage, we investigated the lipid dependence for cell proliferation and viability for a subset of human cancer cell lines. MDA-MB-231 breast and Calu-1 lung cancer cells both harbor KRas mutations; whereas MCF-7 breast cancer cells and the human diploid fibroblast BJ hTERT cell line does not. Cells were plated and after 24 hr, shifted to the indicated conditions. After 5 days, viable and non-viable cells were counted. For all three cancer cell lines, there were very few viable cells after 5 days when put in medium lacking FBS (Fig. 2A). In contrast, almost all of the BJ cells were still viable. There were fewer cells observed in the absence of serum than in the controls due to G1 cell cycle arrest of these cells in the absence of serum (19). If the cells were put in medium containing 10% delipidated FBS, virtually all of the Ras-driven MDA-MB-231 and Calu-1 cancer cells, were non-viable, whereas the MCF7 and BJ cells were mostly viable – indicating a significant difference in the way that Ras-driven cancer cells respond to the lack of serum lipids. Most strikingly, when the FA mixture was provided to the KRas-driven MDA-MB-231 and Calu-1 cells, there was a dramatic increase in cell viability relative to the viability observed with delipidated FBS – indicating a greater dependence on lipids than growth factors for both proliferation and survival (Fig. 2A). Both the MCF7 and BJ cells had lower cell numbers with the FA mixture than with the delipidated FBS – indicating a greater dependence on growth factors than lipids for proliferation. The combination of delipidated FBS and the FA mixture restored full proliferation and survival to all cell lines. Of interest, a lipid mixture that contained only palmitic acid, the sixteen carbon saturated fatty acid produced by de novo fatty acid synthesis, did not enhance either survival or proliferation – indicating that the longer chain unsaturated fatty acids present in the FA mixture were critical. Similarly, BSA, which was included as a carrier for the FA's, did not improve survival of the MDA-MB-231 or Calu-1 cells in 10% delipidated FBS. The pattern of a greater dependence of lipids than growth factors for Ras-driven cancer cells was also observed with a larger panel of human cancer cell lines (Supplementary Fig. S1).
Figure 2.

Ras-driven cancer cells are uniquely dependent on exogenous fatty acids. A, MDA-MB-231, Calu-1, MCF7 and BJ cells were plated and shifted to media conditions as indicated and cell number was determined 5 days later. BSA, the carrier protein for the FA mix and palmitic acid (PA) was included as a control. Each measurement was normalized against the cell number for cells grown in 10% FBS, which was given a value of 100%. Attached viable cells are in blue, detached non-viable cells are in red. Error bars represent S.D. values for at least two independent experiments. B, Changes in G1 and S phase cell population were evaluated by flow cytometry. MDA-MB-231, Calu-1, MCF7 and BJ cells were plated at 30% confluence in 10% FBS and shifted to 0% FBS with or without BSA-FA mix. DNA content/cell was evaluated 48 hours later using flow cytometric analysis as described in Experimental Procedures. Values were normalized to cell cycle profile in full serum condition and relative difference is plotted as indicated. C, BJ cells were transfected with constitutively active KRas (V12G) or empty vector as described in Experimental Procedures, and shifted to full growth serum media or to 0% serum media with or without BSA-FA mix at 18 hours post-transfection. 72 hours later, cell number was determined. Each measurement was normalized against relative cell number in 10% FBS, which was given a value of 100%. Error bars represent S.D. values for at least two independent experiments.
We next compared the effect of serum deprivation in the absence and presence of FAs on cell cycle progression using flow cytometry. All four cell lines (BJ, MCF7, MDAMB-231, and Calu-1) had increased G1 DNA content when cells were shifted from 10% FBS to 0% FBS (Fig. 2B, left panel). However, if the cells were shifted to 0% FBS in the presence of the FA mix, the BJ and MCF7 cells still arrested in G1, whereas the KRas-driven MDA-MB-231 and Calu-1 cells did not significantly accumulate in G1. Similarly, there was a reduction of cells in S-phase for all cell lines when placed in 0% FBS (Fig. 2B, right panel). However if the FA mix was provided, there was still a reduction in S-phase cells in the BJ and MCF7 cells, but not in the MDA-MB-231 and Calu-1 cells. These data demonstrate that the MDA-MB-231 and Calu-1 cells harboring KRas mutations continue to cycle in absence of serum growth factors but need exogenously supplied lipids. In contrast, the MCF7 and BJ cells accumulated in G1 in the absence of growth factors and the addition of lipids did not make a difference. Thus, KRas-driven cancer cells have a greater dependence on exogenously supplied lipids and a lesser dependence on serum growth factors than the BJ and MCF7 cells.
To further demonstrate that mutant Ras promotes a dependence on lipids, we transfected the BJ cells with a plasmid expressing oncogenic KRas (KRasV12G). Parental BJ cells transiently transfected with KRas-expressing and empty vector plasmids were shifted to medium containing 0% FBS and 0% FBS containing the FA mixture and the relative cell numbers were determined three days later. The cells transfected with the KRasV12G plasmid had significantly fewer cells in the absence of FBS than the control BJ cells transfected with vector alone (Fig. 2C). However, if FAs were included with the medium lacking FBS, then the oncogenic Ras-transfected BJ cells grew as well as the parental BJ cells – indicating that expression of KRasV12G sensitized the BJ cells to the lack of lipids in the media.
Oncogenic Ras prevents induction of stearoyl-CoA desaturase-1 levels upon serum and lipid withdrawal
The above results clearly indicate a differential response to lipid deprivation in cells expressing mutant Ras. Key enzymes in the generation of fatty acids needed for membrane biosynthesis include fatty acid synthase (FASN), ATP citrate lyase (ACL), and stearoyl-CoA desaturase-1 (SCD1) (9). We examined the effect of serum and lipid withdrawal on the levels of these three enzymes in the BJ, MCF7, MDA-MB-231, and Calu-1 cells (Fig. 3A). While no significant changes in expression levels of FASN and ACL were detected in response to serum or lipid withdrawal, there was a dramatic increase in the level of SCD1 in the BJ and MCF7 cells. In contrast, the MDA-MB-231 and Calu-1 cells did not elevate SCD1 expression in response to serum and lipid deprivation (Fig. 3A). Similarly, if the KRasV12G expressing plasmid was transfected into BJ cells, it suppressed the elevated expression of SCD1 observed in response to serum withdrawal (Fig. 3B). Although the effect is more pronounced for serum withdrawal than lipid withdrawal, the lack of response in cells with oncogenic KRas indicates that these cells have a disabled response to serum and lipid deprivation.
Figure 3.

Oncogenic Ras leads to ablation of stearoyl-CoA desaturase-1 levels. A, MDA-MB-231, Calu-1, MCF7 and BJ cells were plated and shifted to the indicated media conditions as indicated for 18 hours at which time lysates were collected and immunoblotted with the indicated antibodies. Two exposure times are shown for SCD1 in order to reveal the weak response by the MDA-MB-231 cells. B, BJ cells were transfected as in 2C, and shifted to serum free conditions for 18 hours at which time the levels of FASN, ACC, and SCD1 were evaluated by Western blot analysis. Data are representative of two independent experiments.
Withdrawal of serum lipids creates synthetic lethality for rapamycin in cancer cells harboring mutant KRas
We previously reported that in the absence of serum, MDA-MB-231, as well as most other cancer cells, are killed by rapamycin doses (20 μM) capable of suppressing phosphorylation of the mTORC1 substrate eukaryotic initiation factor 4E-binding protein-1 (4E-BP1) (12, 13). The high doses of rapamycin are required for complete dissociation of mTOR from its companion protein Raptor. At conventional nano-molar doses, mTOR is only partially dissociated from Raptor such that mTORC1 can still phosphorylate 4E-BP1, but cannot recognize and phosphorylate S6 kinase (20). The effect of high dose rapamycin treatment was shown not to be due to off-target effects (12, 15, 21). In the presence of serum, MDA-MB-231 cells were protected by TGF-β present in serum, which prevented apoptosis by inducing G1 cell cycle arrest (13, 20). However, while the MDA-MB-231 cells could be partially protected from the apoptotic effect rapamycin by TGF-β alone, these cells still displayed significant sub-genomic DNA indicating some level of apoptosis (13). Thus, there was apparently something else in the serum that along with TGF-β contributed to survival. We therefore examined the effect of serum lipids on the effect of rapamycin on cell viability of MDA-MB-231, Calu-1, BJ, and MCF7 cells. As reported previously, in the absence of serum, rapamycin (20 μM) induced cleavage of the caspase 3 substrate poly-ADP-ribose polymerase (PARP) in the MDA-MB-231 and Calu-1 cells – indicating apoptotic cell death (Fig. 4A). PARP cleavage was not observed in the presence of 10% FBS. PARP cleavage was not observed with rapamycin treatment in the BJ or MCF7 cells in either the presence or absence of serum. Most significantly, if the MDA-MB-231 or Calu-1 cells were deprived of lipids, by incubating in 10% delipidated serum, rapamycin still induced PARP cleavage – indicating that the lack of lipids created a synthetic lethal phenotype for rapamycin treatment. If the FA mix was provided in the absence of FBS, rapamycin still induced apoptosis – consistent with our previous observation that TGF-β was required for suppressing rapamycin-induced apoptosis in the presence of serum. If the FA mixture was combined with the delipidated serum, PARP cleavage was suppressed. Stimulation of apoptosis, indicated by induction of PARP cleavage in the delipidated serum, required the high dose of rapamycin (Fig. 4B) that causes a complete dissociation of mTOR from the mTORC1 companion protein Raptor and suppresses 4E-BP1 phosphorylation (12). We also examined the effect of rapamycin on MDA-MB-231 cells deprived of lipids on cell viability/growth using the MTT assay. The high dose rapamycin reduced cell viability in the absence of FBS (Fig. 4C), in delipidated FBS, and in the presence of the FA mixture – but not in the presence of delipidated FBS + FA mixture. The effect of rapamycin on cell viability on MCF7 and BJ cells was substantially less than that observed for the MDA-MB-231 cells (Fig. 4C). The loss of cell viability, like PARP cleavage, required the high dose rapamycin treatment (Fig. 4D). Thus, the KRas-driven cancer cells are sensitized to rapamycin by depriving cells of either lipids or growth factors.
Figure 4.

Protection of rapamycin-induced apoptosis by serum requires both lipids and growth factors. A, MDA-MB-231, MCF7 and BJ cells were plated at 70% confluence overnight and shifted to media conditions as indicated containing 20uM rapamycin. Cell lysates were collected 4 hours post-treatment and the level of cleaved PARP was determined by Western blot analysis. The data are representative of at least two independent experiments. B, MDA-MB-231 cells were plated at 70% confluence overnight and shifted to 10% delipidated FBS media conditions containing increasing doses of rapamycin. Cell lysates were collected at 4 and 24 hours post-treatment the levels of cleaved PARP were determined as in (A). C, MDA-MB-231, MCF7 and BJ cells were plated in duplicate at approximately 1000 cells per well in 96-well plate overnight, and shifted to various media conditions as indicated containing 20uM rapamycin. MTT reagent was added according manufacture's instructions, and absorbance values were determined at the indicated times. Values were normalized to absorbance value of 10% FBS 20uM rapamycin treated set, which was given a total value of 100%. Error bars, S.D. values for at least two independent experiments. D, MDA-MB-231 cell were plated as in (B) and shifted to 10% delipidated FBS conditions containing increasing dose of rapamycin as indicated. MTT reagent was added and absorbance levels were determined at the indicated times as in C. Values were normalized to 10% delipidated FBS condition set, which was given a total value of 100%. Error bars, S.D. values for at least two independent experiments.
Suppression of macropinocytosis sensitizes Ras-driven cancer cells to rapamycin
The sensitivity of Ras-driven cancer cells deprived of serum lipids to rapamycin suggests a means to target the many cancers that harbor Ras mutations. While ridding the serum of lipids in a human is problematic, it could be possible to block the uptake of serum lipids. Dafna Bar-Sagi and colleagues reported previously that mutant Ras stimulates macropinocytosis (22). We therefore wanted to examine whether blocking macropinocytosis would also sensitize the KRas-driven MDA-MB-231 and Calu-1 cells to rapamycin. Macropinocytosis can be suppressed by 5-(N-ethyl-N-isopropyl) amiloride (EIPA) (23, 24). We therefore examined the effect of EIPA on the rapamycin sensitivity of MDA-MB-231, Calu-1, MCF7, and BJ cells in the presence of 10% FBS. EIPA treatment made the MDA-MB-231 and Calu-1 cells sensitive to the apoptotic effect of rapamycin (Fig. 5A), while having little or no effect on the MCF7 or BJ cells. We also examined whether EIPA could sensitize the BJ cells expressing activated KRas cells to rapamycin treatment. The BJ-KRas cells displayed PARP cleavage when treated with the combination of EIPA and 20 μM rapamycin (Fig. 5B). Another indicator of apoptotic cell death is the appearance of sub-genomic DNA. We compared the levels of sub-genomic DNA in the BJ and MDA-MB-231 cells treated with EIPA and rapamycin. The combination of EIPA and rapamycin induced a substantial increase in sub-genomic DNA in the MDAMB-231, but not the BJ cells (Fig. 5C). The increase in sub-genomic DNA observed in Fig. 5C corresponded with large drops in the percentage of cells in S-phase and G2/M cells relative to the drop in G1 cells (Table S2) – which is consistent with our previous work where we demonstrated that the cells killed by rapamycin were in S-phase (13). The data provided in Fig. 5 demonstrate that EIPA, like lipid deprivation, creates a synthetic lethal phenotype for rapamycin in KRas-driven cancer cells – demonstrating that it could be possible to exploit the apparent acute need of lipids of KRas-driven cancer cells.
Figure 5.

Blockage of macropinocytosis mimics lipid deprivation and sensitizes Ras-driven tumors to rapamycin. A, MDA-MB-231, Calu-1, MCF7 and BJ cells were plated overnight and shifted to 10% FBS conditions containing EIPA (10 μM) or rapamycin (20 μM) as indicated. Cell lysates were collected 18 hours post-treatment and levels of cleaved PARP as in 4A. B, BJ cells were transfected with constitutively active KRas (V12G) or empty vector and treated with EIPA or rapamycin in 10% FBS condition as described in A. Cell lysates were collected 18 hours after treatment at which time the levels of cleaved PARP were determined as in A. Data in (A) and (B) are representative of least two independent experiments. C, MDA-MB-231 and BJ cells were plated at 40% confluence and treated as in B for 48 hours, after which collected and subjected to flow cytometric analysis. Total sub-genomic DNA is plotted as indicated. Error bars represent S.D. values for at least two independent experiments.
Discussion
The data presented here reveal an enhanced requirement for lipids in human cancer cells harboring activating Ras mutations. Depriving these cells of lipids leads to what we would call a “replicative cell death” – continued attempts to proliferate in the absence of sufficient nutrients. Whereas most cells are capable of synthesizing fatty acids for membrane phospholipids from glucose (9), the Ras-driven cancer cells apparently have a greater need for exogenously supplied lipids. This unique property of Ras-driven cancer cells is apparently an Achilles' heel for Ras-driven cancer cells in that suppressing mTOR in these cells induces apoptosis if the uptake of lipids is suppressed.
Of significance, two very recent reports have also identified the need for exogenously supplied protein as an amino acid supply (24) and for lipids (25) in Ras-transformed cells. The uptake of albumin served as a source of glutamine for needed for cell growth (24). We used albumin as a carrier for exogenously supplied lipids to promote cell survival and rapamycin resistance. The use of albumin alone did not promote survival by itself (Fig. 2) – indicating that while Ras-transformed cells depend on scavenged proteins as an amino acid source for cell growth, it was the lipids that were critical for survival and resistance to rapamycin. The lipid requirement for exogenous lipids by Ras-transformed cells was dependent on an unsaturated fatty acid (25). Consistent with this, the saturated fatty acid palmitic acid was not able to substitute for the mixture of fatty acids used in this study, which consisted with several unsaturated fatty acids.
The dependence of Ras-transformed cells on exogenously supplied nutrients over standard de novo synthetic pathways appears to be a Ras-driven program shift. In response to the lack of serum lipids, cells lacking mutant Ras showed a dramatic increase in the level of SCD1 (Fig. 3). This is consistent with the recent report from the Rabinowitz and Thompson labs where they showed that Ras-transformed cells were resistant to inhibition of SCD1 (25). Although they did not look at SCD1 protein levels, their study also demonstrated that SCD1 was not important for the growth of Ras-transformed cells. It is likely that the lack of increased SCD1 expression in the Ras-transformed cells is a reflection of the apparent scavenger program stimulated by Ras that involves macropinocytosis (22). In this regard, the elevated PLD activity observed in KRas-driven cancer cell in response to lipid deprivation may be of significance. We have proposed that the PA requirement for mTOR complex stability and activity represents a means for sensing the presence of sufficient lipids for cell growth (4) – consistent with mTORC1's role in sensing other nutrients such as essential amino acids and glucose (10). PA is at the center of membrane phospholipid biosynthesis and is an ideal indicator of lipid sufficiency. The major pathway for PA synthesis from fatty acids is through the acylation of glucose-derived glycerol-3-phosphate (4). In the absence of de novo lipid biosynthesis and exogenously supplied lipids, you create a need for PA from a different source. We have proposed previously that the elevated PLD activity in cancer cells deprived of serum is an attempt to promote survival by keeping mTOR complexes intact and active (4, 11, 15). The KRas-driven cancer cells may be especially dependent on elevated PLD activity because of the deactivated de novo FA synthesis machinery.
The rapamycin sensitivity of Ras-transformed cells deprived of lipids or with suppressed macropinocytosis may be a reflection of the impact of lipid deprivation on cell cycle progression. We reported previously that in the absence of serum, rapamycin induced apoptosis in the MDA-MB-231 cells, but in the presence of serum, rapamycin induced a TGF-β-dependent G1 cell cycle arrest that protected the cells from apoptosis (13, 26). However, if cells were synchronized in early S-phase, then the cells were killed by rapamycin – even in the presence of serum/TGF-β. These studies indicated that if cells get past the TGF-β-dependent G1 cell cycle checkpoint and enter S-phase, then suppression of mTORC1 activates an apoptotic program. Consistent with our previous studies (13), the increase in sub-genomic DNA observed in Fig. 5C corresponded with large drops in the percentage of cells in S-phase and G2/M cells relative to the drop in G1 cells (Table S2). While these data are too preliminary to draw any firm conclusions about why rapamycin kills KRas-driven cancer cells when deprived of lipids, the data are consistent with an apoptotic effect on cells that have progressed into S-phase. It is possible that depriving KRas-driven cancer cells, where de novo FA synthesis is suppressed (25), of serum lipids leads to the arrest or slowed progression through S-phase. Once a cell has entered S-phase, the cell has committed to replicating its genome and doubling its mass. If mTORC1 is suppressed in S-phase cells – telling the cell that there are not sufficient raw materials to finish the job – then a default apoptotic program is activated rather than try to remedy the situation at this phase of the cell cycle. While there is still much to be learned about the impact of mTORC1 suppression on cells in S-phase, it is clear that in KRas-driven cancer cells, depriving cells of lipids, creates a synthetic lethal phenotype for rapamycin treatment that could create therapeutic strategies for targeting the large number of human cancers that harbor Ras mutations.
Supplementary Material
Acknowledgements
The authors thank Laurel Eckhardt, Mitchel Goldfarb, Deepak Menon, and Mahesh Saqcena for the helpful discussions.
Grant Support This work was supported by a grant from the National Cancer Institute (R01-CA046677) to D.A. Foster; and a pilot award to D.A. Foster from Research Centers in Minority Institutions (RCMI) RR-03037 from the National Center for Research Resources of the National Institutes of Health, which supports infrastructure and instrumentation at Hunter College.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
REFERENCES
- 1.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster DA. Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol Metab. 2013 doi: 10.1016/j.tem.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–24. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das UN. Essential fatty acids and their metabolites as modulators of stem cell biology with reference to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2011;30:311–24. doi: 10.1007/s10555-011-9316-x. [DOI] [PubMed] [Google Scholar]
- 7.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–8. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamanaka RB, Chandel NS. Targeting glucose metabolism for cancer therapy. J Exp Med. 2012;209:211–5. doi: 10.1084/jem.20120162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Rev Cancer. 2007;7:763–77. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 10.Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, et al. Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol Chem. 2006;281:15862–8. doi: 10.1074/jbc.M600660200. [DOI] [PubMed] [Google Scholar]
- 12.Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, et al. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex1 and suppressing phosphorylation of 4E-BP1. Cell Cycle. 2011;10:3948–56. doi: 10.4161/cc.10.22.18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gadir N, Jackson DN, Lee E, Foster DA. Defective TGF-β signaling sensitizes human cancer cells to rapamycin. Oncogene. 2008;27:1055–62. doi: 10.1038/sj.onc.1210721. [DOI] [PubMed] [Google Scholar]
- 14.Reinhardt HC, Jiang H, Hemann MT, Yaffe MB. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112–9. doi: 10.4161/cc.8.19.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–20. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 17.Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr. Opin Cell Biol. 2009;21:209–18. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 18.Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging. 2011;3:1130–41. doi: 10.18632/aging.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One. 2013;8:e74157. doi: 10.1371/journal.pone.0074157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yellen P, Chatterjee A, Preda A, Foster DA. Inhibition of S6 kinase suppresses the apoptotic effect of eIF4E ablation by inducing TGF-β-dependent G1 cell cycle arrest. Cancer Lett. 2013;333:239–43. doi: 10.1016/j.canlet.2013.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YJ, Zheng YF, Wang NF, Lu S, Pang T, Yang Q, et al. Significance of urinary nucleosides in diagnosis of gastric carcinoma. Ai Zheng. 2003;22:537–40. [PubMed] [Google Scholar]
- 22.Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–8. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- 23.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, et al. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–22. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad. Sci USA. 2013;110:8882–7. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Gendre O, Sookdeo A, Duliepre SA, Utter M, Frias M, Foster DA. Suppression of AKT phosphorylation restores rapamycin-based synthetic lethality in SMAD4-defective pancreatic cancer cells. Mol Cancer Res. 2013;11:474–81. doi: 10.1158/1541-7786.MCR-12-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.