

Abstract
Objective:
To investigate association of genetic risk factors for late-onset Alzheimer disease (LOAD) with risk of posterior cortical atrophy (PCA), a syndrome of visual impairment with predominant Alzheimer disease (AD) pathology in posterior cortical regions, and with risk of “posterior AD” neuropathology.
Methods:
We assessed 81 participants with PCA diagnosed clinically and 54 with neuropathologic diagnosis of posterior AD vs 2,523 controls for association with 11 significant single nucleotide polymorphisms (SNPs) from published LOAD risk genome-wide association studies.
Results:
There was highly significant association with APOE ε4 and increased risk of PCA (p = 0.0003, odds ratio [OR] = 3.17) and posterior AD (p = 1.11 × 10−17, OR = 6.43). No other locus was significant after corrections for multiple testing, although rs11136000 near CLU (p = 0.019, OR = 0.60) and rs744373 near BIN1 (p = 0.025, OR = 1. 63) associated nominally significantly with posterior AD, and rs3851179 at the PICALM locus had significant association with PCA (p = 0.0003, OR = 2.84). ABCA7 locus SNP rs3764650, which was also tested under the recessive model because of Hardy-Weinberg disequilibrium, also had nominally significant association with PCA risk. The direction of association at APOE, CLU, and BIN1 loci was the same for participants with PCA and posterior AD. The effects for all SNPs, except rs3851179, were consistent with those for LOAD risk.
Conclusions:
We identified a significant effect for APOE and nominate CLU, BIN1, and ABCA7 as additional risk loci for PCA and posterior AD. Our findings suggest that at least some of the genetic risk factors for LOAD are shared with these atypical conditions and provide effect-size estimates for their future genetic studies.
Posterior cortical atrophy (PCA) is a syndrome characterized by predominant visual deficits in the absence of primary ocular disease1,2 and parieto-occipital pathology. Neuropathologic series of PCA revealed Alzheimer disease (AD) pathology in the majority of cases3–7 (introduction in appendix e-1 on the Neurology® Web site at Neurology.org). CSF8,9 and amyloid neuroimaging8,10,11 biomarkers in subjects with PCA follow patterns similar to those in AD, also suggestive of underlying AD neuropathology (appendix e-1 introduction).
Given the existing neuropathology, biomarker, and imaging data, we predicted that PCA would share some genetic risk factors with AD but may also have different genetic risk factors that drive its distinct clinicopathologic features. Genetic studies on PCA are scarce, with limited sample sizes and main focus of APOE. The 2 largest published studies to evaluate the frequency of genetic factors in PCA were done in fewer than 30 subjects,5,12 with conflicting results, making it impossible to reach reliable conclusions (appendix e-1 introduction).
Understanding the genetic risk factors in atypical AD is of paramount importance in the identification of pathophysiologic mechanisms that are both common with and distinct from typical AD. In this study, we sought to investigate whether any of the well-replicated genetic risk variants in AD13−17 were also common to clinical PCA and/or posterior AD neuropathology. Our results have implications for the genetic components of these atypical focal cortical AD subtypes and also their future studies.
METHODS
Participants.
All participants were recruited at Mayo Clinic Jacksonville in Florida or Mayo Clinic Rochester in Minnesota. Eighty-one participants had clinical diagnosis of PCA, 5 of whom also had pathologic diagnosis; 54 had only neuropathologic diagnosis as “posterior AD,” but no clinical information. Clinical diagnosis of PCA was made according to the core criteria previously suggested5 as follows: presentation of visual complaints in the absence of significant primary ocular disease; relative preservation of anterograde memory and insight early in the disorder; insidious onset and gradual progression; disabling visual impairment throughout the disorder; absence of stroke or tumor; and absence of early parkinsonism and hallucinations. All participants with pathologic diagnoses were evaluated by one neuropathologist (D.W.D.). For the neuropathologic diagnosis, senile plaques (per ×10 field) and neurofibrillary tangles (NFTs) (per ×40 field) are counted in the midfrontal, superior temporal, inferior parietal, motor, visual (Brodmann area [BA] 17) and entorhinal cortices, in addition to 2 sectors of the amygdala. NFTs are also counted in the nucleus basalis of Meynert. Visual association cortex (BA18) is also scanned in all cases and counted in those with severe NFT pathology. All subjects with neuropathologic diagnosis of posterior AD have disproportionate severity of NFT in BA17 and BA18 compared with typical AD cases, but could have additional pathologies including Lewy bodies, vascular disease, hippocampal sclerosis, or progressive supranuclear palsy. The overall pattern of NFT severity in BA17 and BA18 is greater than that in the frontal cortex in posterior AD, such that this diagnosis is not simply a function of the overall disease severity, but reflects disproportionate, focal involvement of the visual cortices. Thus, patients with posterior AD had neuropathology characteristics of PCA.1 The disproportionate involvement of the posterior cortices was not determined by objective criteria, but subjectively. All neuropathologically diagnosed patients had Braak stage of 5.5 or 6, except for one patient with a Braak stage of 4. Our rationale for including posterior AD cases in our cohort stems from prior neuropathologic studies of PCA, which consistently identified higher tangle counts in posterior cortical regions.1
Elderly control participants were recruited either at Mayo Clinic Rochester for the Mayo Clinic Study of Aging or Alzheimer's Disease Research Center, or at Mayo Clinic Jacksonville for the Alzheimer's Disease Research Center or Sib-Pair Study, which is enriched for cognitively normal siblings aged 80 years or older. Only one sibling was included per sibship. All control participants were evaluated by a neurologist and were cognitively normal at last visit with a Clinical Dementia Rating of zero. All participants were Caucasian–North Americans from the United States. Only participants with complete age, sex, ethnicity, and APOE genotype information were included in the study. Participants were not screened for mutations in the early-onset familial AD genes APP, PSEN1, and PSEN2, or the familial frontotemporal dementia genes MAPT, GRN, and C9orf72.
Genotyping.
All participants were genotyped for the most significant single nucleotide polymorphisms (SNPs) identified from late-onset AD (LOAD) risk genome-wide association studies (GWAS).13–17 The genotyped SNPs and their closest genes are as follows: APOE rs429358 (tagging SNP for APOE ε4), APOE rs7412 (tagging SNP for APOE ε2), CLU rs11136000, CR1 rs3818361, PICALM rs3851179, BIN1 rs744373, ABCA7 rs3764650, MS4A6A rs610932, EPHA1 rs11767557, CD33 rs3865444, and CD2AP rs9349407. The genotyping was done using TaqMan SNP Genotyping Assays in an ABI PRISM 7900HT Sequence Detection System with 384-Well Block Module from Applied Biosystems (Foster City, CA). The genotype data were analyzed using the SDS software version 2.2.3 (Applied Biosystems).
Statistical analysis.
Association of genetic variants with risk of clinical PCA or posterior AD only or combined was assessed using multivariable logistic regression analysis applied in PLINK18 (http://pngu.mgh.harvard.edu/purcell/plink/). All analyses included age, sex, and APOE ε4 dosage as covariates, except for the APOE rs429358 association test, in which only age and sex were included as covariates. Age is defined as age at onset of symptoms for participants primarily recruited via their clinical diagnosis (n = 76) or age at death for those with primary neuropathologic recruitment (n = 57). In addition, for one participant recruited from the clinic, who lacked age-at-onset information, age at death was used; for another from the neuropathologic recruitment, who lacked age-at-death data, age at onset was used. An additive model was tested for all genetic variants. A recessive model was also tested for ABCA7 rs3764650, given the significant deviation from Hardy-Weinberg equilibrium for this SNP in the combined cases. Minor allele frequency (MAF) estimates for each SNP were compared between the participants with clinical PCA and those with posterior AD using proportions meta-analysis, with Cochran Q test p value <0.05 indicating significant difference in MAF estimates.
Minimum detectable odds ratios (ORs) were estimated for the combined cohort for an additive effect, using allele frequencies estimated in the controls and effect-size estimates published for LOAD risk,16,19–23 for 80% power and α = 0.05. We also obtained empirical p values using 10,000 random permutations in PLINK to account for multiple testing. Finally, the difference in effect sizes between participants with combined clinical PCA and posterior AD vs participants with LOAD was tested using polytomous logistic regression analyses in SAS (SAS Institute, Cary, NC) for the SNPs with nominally significant associations in PCA. For these analyses, effect-size estimates for LOAD were obtained using a cohort of 696 participants with diagnosis of LOAD who were compared against the same control group used for the PCA risk estimates. These participants with LOAD were from Mayo Clinic Rochester (n = 608) or Jacksonville (n = 88), diagnosed clinically according to National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association24 criteria and followed longitudinally with clinical and cognitive measures.
RESULTS
Among our cases, mean age was higher in the 54 participants with posterior AD compared with the 81 participants with clinical PCA, as expected, given that age of death vs age at onset were used, respectively (table 1). Although there was a higher proportion of females and APOE ε4 carriers in the posterior AD group compared with clinical PCA group, these differences were not statistically significant. In the combined 135 cases, there was a higher percentage of females, increased proportion of APOE ε4 carriers, and younger mean age compared with the 2,523 elderly controls, all of which were statistically significant.
Table 1.
Participant demographics
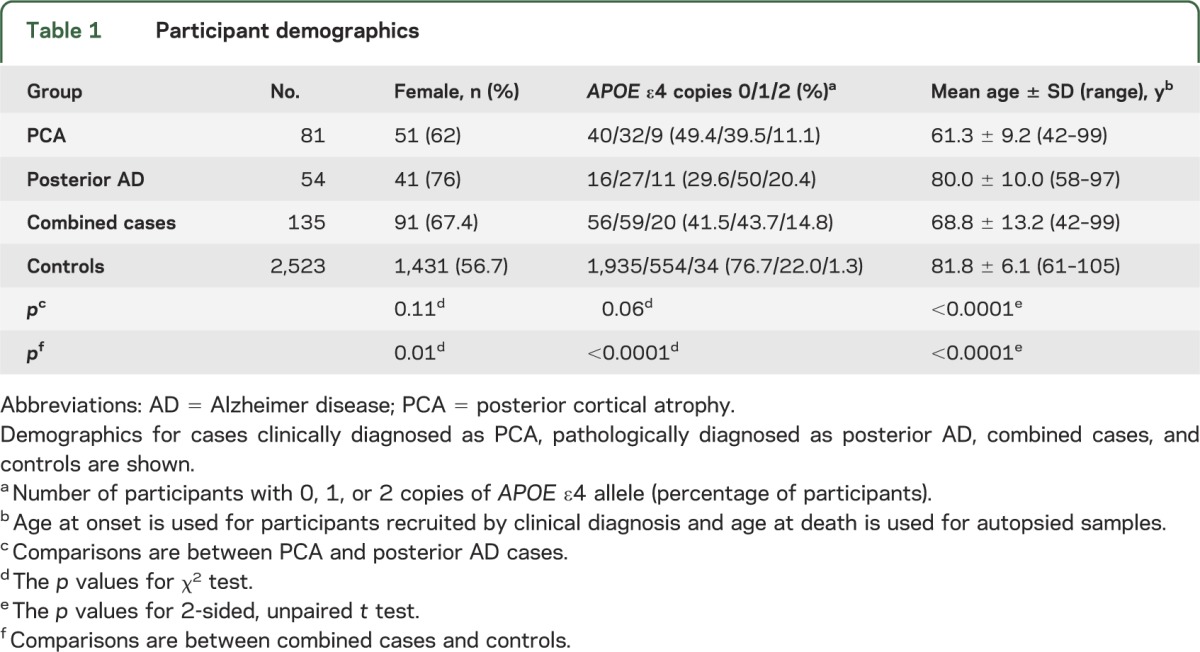
Sex and age at onset or death were included as covariates in all genetic associations. As expected from the demographics, increased age and being male had protective effects in the analysis of the combined case-control cohort (data not shown).
APOE ε4–tagging SNP rs429358 has a highly significant association with risk of clinical PCA (p = 0.0003) (table 2), with OR estimates (3.17, 95% confidence interval [CI] = 1.70–5.90) similar to the estimate published for LOAD20 (OR = 4.36). APOE ε4 association with posterior AD was even stronger (p = 1.11 × 10−17, OR = 6.43, 95% CI = 4.20–9.84). These associations would remain significant after corrections for multiple testing of 11 SNPs, and as expected, show significance in the combined cohort even when a genome-wide correction, evidently too stringent for our 11 tests, is applied (p = 4.90 × 10−19, OR = 4.74).
Table 2.
Association of LOAD risk SNPs with PCA, posterior AD, and combined cases
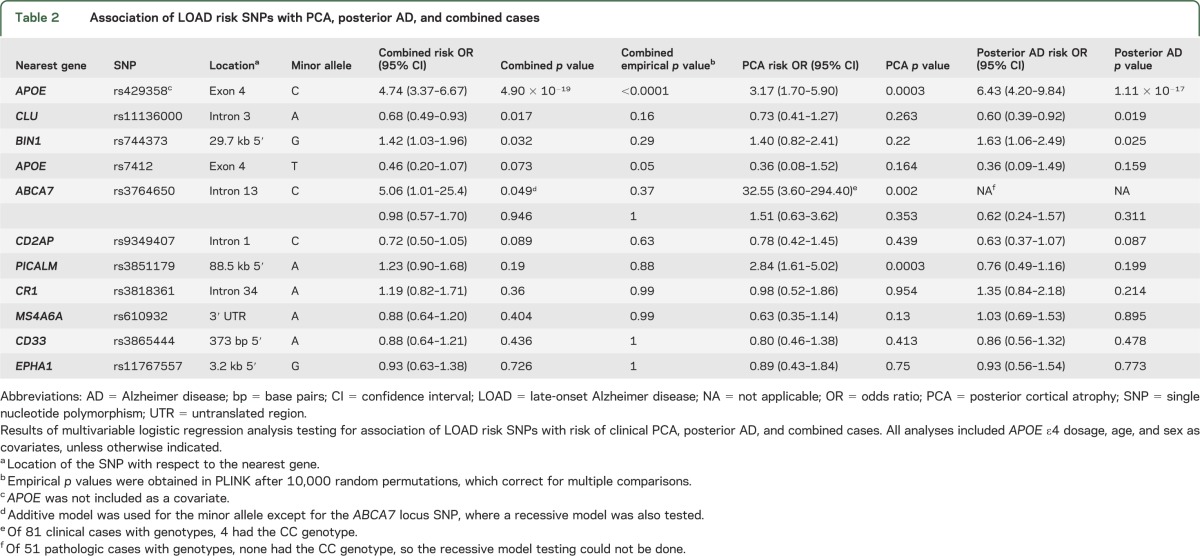
No other SNPs achieved the same level of significance, although rs11136000 at CLU (p = 0.019) and rs744373 at BIN1 (p = 0.025) were nominally significant in posterior AD. It is important that the minor alleles of these variants had effects that were in the same direction as that for LOAD risk, with a protective OR estimate for CLU (OR = 0.60, 95% CI = 0.39–0.92) and a risk effect for BIN1 locus SNPs (OR = 1.63, 95% CI = 1.06–2.49). Both SNPs had the same direction of effect for clinical PCA association (rs11136000 OR = 0.73, rs744373 OR = 1.40), but neither reached nominal significance for this group. As expected, both SNPs were nominally significant in the combined case-control analysis. PICALM rs3851179 variant was significantly associated with clinical PCA (p = 0.0003), but with a risky OR estimate (OR = 2.84), which is not consistent with the published protective effect in LOAD and also inconsistent with the estimate obtained in posterior AD (OR = 0.76). No other SNPs had nominal significance with the additive model, although the LOAD protective APOE ε2–tagging SNP rs7412 had suggestive protective effects in clinical PCA, posterior AD, and in the combined cohort (p = 0.073, OR = 0.46, 95% CI = 0.20–1.07). Given the multiple testing problem, we obtained empirical p values for the combined case-control cohort correcting for multiple comparisons. Only APOE ε4 rs429358 achieved significance with corrected empirical p value <0.0001, with marginal significance for APOE ε2 rs7412 (p = 0.05).
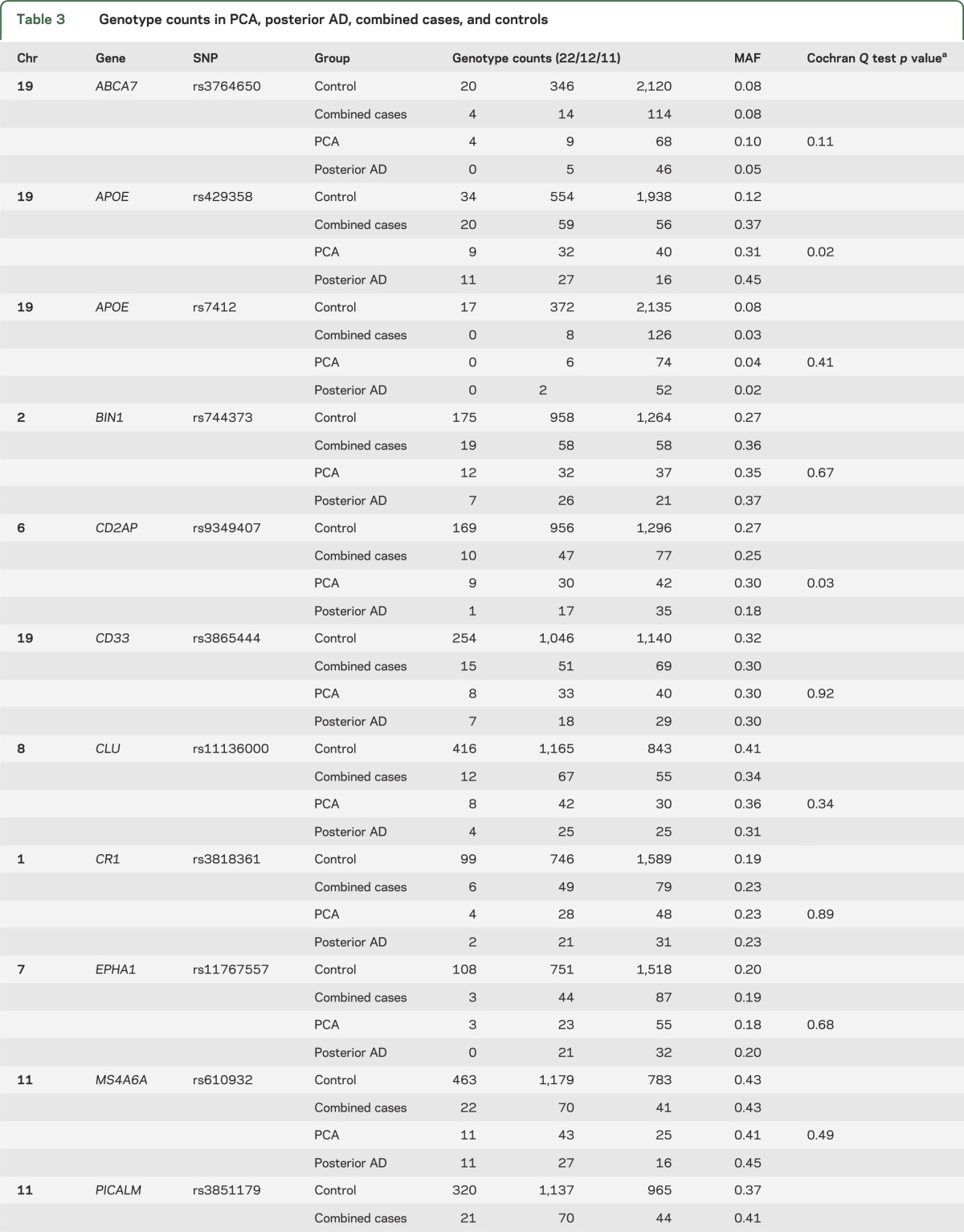
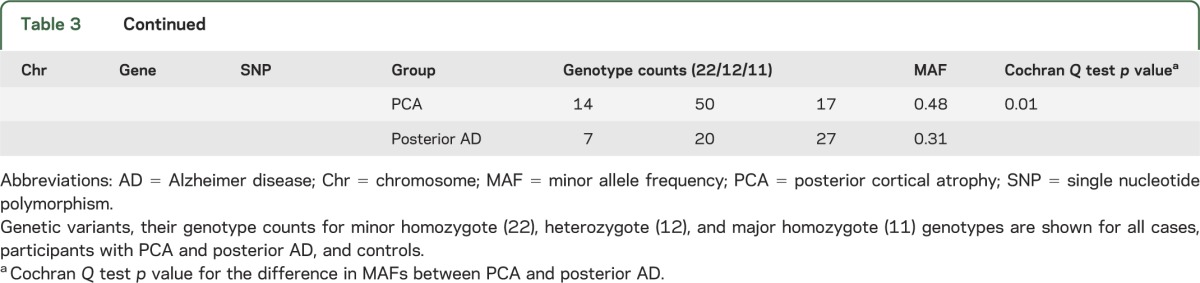
Upon review of the genotype frequencies for the tested SNPs in combined cases vs control participants (table e-1), we determined that there were significantly fewer heterozygote participants for the ABCA7 locus SNP rs3764650 than expected in the cases, leading to deviation from Hardy-Weinberg equilibrium (p = 0.005). There were no other SNPs with significant Hardy-Weinberg disequilibrium in either cases or controls. Given that a recessive mode of inheritance can result in Hardy-Weinberg disequilibrium in subjects with disease,25 we repeated the ABCA7 locus SNP association test under a recessive model. This test could only be done in the clinical PCA cohort, because the smaller posterior AD cohort did not have any rs764650 minor homozygotes. Although significant with a risky OR estimate (p = 0.002, OR = 32.55) and a direction of effect consistent in LOAD, there were only 4 minor homozygotes in the clinical PCA cohort (table e-1).
The direction of association was consistent between clinical PCA and posterior AD for APOE ε4, APOE ε2, CLU, and BIN1 loci, although the effect-size estimates were larger for the APOE ε4, CLU, and BIN1 associations in the pathologic posterior AD group (table 2). Of the 11 tested variants, the MAF in clinical PCA vs posterior AD was significantly different for only APOE ε4, PICALM, and CD2AP (table 3). APOE ε4 MAF for the clinical PCA group (MAF = 0.31) is similar to the estimates for typical LOAD (MAF in Caucasians from AlzGene26 = 0.38), and both are smaller than the MAF estimates for posterior AD (MAF = 0.45).
Table 3.
Genotype counts in PCA, posterior AD, combined cases, and controls
DISCUSSION
In this study, we assessed the association of 11 well-replicated genetic risk variants for LOAD with risk of clinical PCA and posterior AD neuropathology. All of our participants with clinical PCA fit the previously suggested clinical criteria,5 but only 5 had a pathologic evaluation. The participants with posterior AD in our study had disproportionate tangle neuropathology in the visual cortices in comparison to frontal regions, as is characteristic of PCA,1 but lacked clinical information. Despite strong clinicopathologic correlations between PCA and AD pathology3–7 in the literature, with consistent identification of higher tangle counts in posterior cortical regions,1 because we did not have both clinical and neuropathologic diagnoses for most patients within our series, we primarily assessed the clinically and pathologically diagnosed cases separately, but also performed formal comparisons of MAFs for all tested variants and exploratory combined case-control analyses.
Several findings emerge from this study: first, the risk of APOE ε4 is demonstrated unequivocally in this cohort of 81 clinical PCA, 54 posterior AD, and combined 135 cases, where the strength of association exceeds genome-wide significance in the pathologic cohort and even stronger in the combined analysis. Although earlier studies had conflicting results regarding APOE frequency in PCA, they had small sample sizes ranging from 8 to 27 participants.1,5,12 Despite being the largest published cohort in PCA and posterior AD to date, our study is modest in size for a genetic association study. Nevertheless, given the estimated effect size for APOE in LOAD, our estimated power to detect this gene in PCA is sufficient. We also identified a marginally significant protective OR for APOE ε2 in the combined cohort with equivalent effect sizes in the clinical and pathologic participants, which is consistent with its direction of effect in LOAD, further strengthening a role for the APOE locus in PCA and posterior AD that is similar to LOAD.
The association for APOE ε4 was stronger in posterior AD, with OR estimates that are greater than both PCA and LOAD. There may be several explanations for this, including the presence of participants without AD pathology in the clinical PCA cohort, which might decrease the OR estimates. Lack of neuropathology on all participants is a weakness in this study that is common to almost all case-control studies of complex diseases, because restricting cohorts to those with neuropathology, while ideal, would in reality impose limitations on sample size. Another possibility is that APOE ε4 may be preferentially driving the posterior AD pathology, thus resulting in stronger effect sizes for this pathologic cohort.
Second, we identified nominally significant associations for CLU and BIN1 loci in posterior AD, with protective and risk effects, respectively, for the minor alleles of the tested variants, which is consistent with their direction of OR in LOAD. The direction of these effects was also similar in clinical PCA and, although the OR estimates were smaller compared with posterior AD, there were no statistically significant differences in the MAFs of these variants between these clinical and pathologic cohorts. The slightly stronger effects in posterior AD could be attributable to a “cleaner” diagnosis afforded by pathologic cohorts in general, rather than a posterior AD pathology–specific effect, as seems possible for APOE ε4.
We also detected increased risk of PCA with the minor allele of the ABCA7 rs3764650 SNP using a recessive model, although this association is based on 4 minor homozygotes observed in clinical PCA vs 20 in the controls and therefore requires replication. PICALM rs3851179 was the only other SNP with nominally significant association in the clinical PCA group, although the direction of effect is opposite to that seen in typical LOAD and posterior AD. Given these, the biological significance of this finding, if any, remains to be established. Using permutations, we determined the genetic associations for APOE ε4 and APOE ε2 to be significant, although none of the other variants achieved significance with empirical p values or after multiple testing corrections, thus requiring replication in larger series.
In our study, we also provide formal comparisons of MAFs for all tested variants in the participants with pathologic posterior AD vs those with PCA, which identified significant differences for APOE ε4, as well as PICALM and CD2AP loci variants. We also depict estimates of minimal detectable ORs and a comparison with LOAD risk OR estimates for all variants (table e-2). These results should be of utility in guiding future genetic studies of PCA and posterior AD.
Given that the majority of subjects with PCA have AD pathophysiology in clinicopathologic series3–7 and that most subjects with PCA in antemortem studies display CSF biomarker profiles8,9 and amyloid brain imaging8,10,11 consistent with those in AD, it is perhaps not surprising that there are genetic risk factors that are shared between LOAD vs PCA and posterior AD. It is entirely possible that LOAD GWAS include subjects that may qualify for early clinical or pathologic diagnosis of PCA or posterior AD, respectively. That said, given the relative infrequency of PCA and clinical inclusion criteria in LOAD GWAS, which are likely to exclude at least early PCA cases, it is highly unlikely that subjects with PCA have substantial contribution to the current GWAS. We have retrospectively determined that only 14 of approximately 2,000 subjects in the published Mayo Clinic LOAD GWAS27 and 12 of approximately 5,000 subjects in the LOAD GWAS replication studies from Mayo Clinic21–23 would qualify as having PCA. This underscores the importance of pursuing independent studies of this condition, such as this study.
Our study has several strengths, including a cohort with relatively large size and power estimations, assessment of all well-replicated LOAD SNPs, formal comparison of effect-size estimates, with LOAD and neuropathologic diagnosis for 40% of the cases. The weaknesses include lack of clinicopathologic correlations for most cases, determination of disproportionate involvement of posterior cortices in posterior AD subjectively, candidate gene rather than hypothesis-generation study, and limited power. Other limitations include retrospective study design and age difference between the cases and controls. The difference in age is a consequence of the previously established fact that subjects with PCA have younger ages of onset compared with typical LOAD,1 and therefore also compared with the age at last assessment for the elderly control participants in our study. Our cases were not systematically screened for known mutations in early-onset familial AD or frontotemporal dementia, which is another limitation.
Our case-control association study of 81 participants with PCA, 54 with posterior AD, and 2,523 cognitively normal participants identifies a significant effect for APOE and nominates CLU, BIN1, and ABCA7 as potential risk loci for PCA. Additional studies, ideally with prospective study design and of much larger size, are needed to replicate our findings and also to identify other genetic risk factors for these intriguing conditions.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for their participation, without whom these studies would not have been possible.
GLOSSARY
- AD
Alzheimer disease
- BA
Brodmann area
- CI
confidence interval
- GWAS
genome-wide association study
- LOAD
late-onset Alzheimer disease
- MAF
minor allele frequency
- NFT
neurofibrillary tangle
- OR
odds ratio
- PCA
posterior cortical atrophy
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept and design: Carrasquillo, Ertekin-Taner. Acquisition of data: Carrasquillo, Khan, Murray, Krishnan, Nguyen, Ma, Bisceglio, Younkin, Petersen, Dickson, Boeve, Graff-Radford, Ertekin-Taner. Analysis and interpretation of data: Carrasquillo, Aakre, Pankratz, Ertekin-Taner. Drafting of the manuscript: Carrasquillo, Ertekin-Taner. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: Carrasquillo, Aakre, Pankratz, Ertekin-Taner. Obtained funding: Younkin, Petersen, Dickson, Graff-Radford, Ertekin-Taner. Administrative, technical, and material support: Younkin, Petersen, Dickson, Boeve, Graff-Radford, Ertekin-Taner. Study supervision: Dickson, Boeve, Graff-Radford, Ertekin-Taner.
STUDY FUNDING
Support for this research was provided by NIH grants: National Institute on Aging (R01 032990 to N.E.-T. and R01 AG018023 to N.R.G.-R. and S.G.Y.); Mayo Alzheimer's Disease Research Center (P50 AG016574 to R.C.P., D.W.D., N.R.G.-R., S.G.Y., and N.E.-T.); Mayo Alzheimer's Disease Patient Registry (U01 AG006576 to R.C.P.); and National Institute on Aging (AG025711, AG017216, AG003949 to D.W.D.). This project was also generously supported by the Robert and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program (to R.C.P., D.W.D., N.R.G.-R., and S.G.Y.), and by the Palumbo Professorship in Alzheimer's Disease Research (to S.G.Y.). M.M.C. is supported partly by GHR Foundation grants.
DISCLOSURE
M. Carrasquillo, Q. Khan, M. Murray, S. Krishnan, J. Aakre, V. Pankratz, T. Nguyen, L. Ma, and G. Bisceglio report no disclosures relevant to the manuscript. R. Petersen has been a consultant to GE Healthcare and Elan Pharmaceuticals, has served on a data safety monitoring board in clinical trials sponsored by Pfizer Incorporated and Janssen Alzheimer Immunotherapy and gave a CME lecture at Novartis Incorporated. S. Younkin and D. Dickson report no disclosures relevant to the manuscript. B. Boeve has served as an investigator for clinical trials sponsored by Cephalon, Inc., Allon Pharmaceuticals, and GE Healthcare, and serves on the scientific advisory board of the Tau Consortium. N. Graff-Radford has served as a consultant to Codman and received grant support from Elan Pharmaceutical Research, Pfizer Pharmaceuticals, Medivation, and Forest. N. Ertekin-Taner reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Crutch SJ, Lehmann M, Schott JM, Rabinovici GD, Rossor MN, Fox NC. Posterior cortical atrophy. Lancet Neurol 2012;11:170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol 1988;45:789–793 [DOI] [PubMed] [Google Scholar]
- 3.Victoroff J, Ross GW, Benson DF, Verity MA, Vinters HV. Posterior cortical atrophy: neuropathologic correlations. Arch Neurol 1994;51:269–274 [DOI] [PubMed] [Google Scholar]
- 4.Ala TA, Frey WH, II, Clark HB. Posterior cortical atrophy: neuropathological correlations. Arch Neurol 1996;53:958. [DOI] [PubMed] [Google Scholar]
- 5.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology 2004;63:1168–1174 [DOI] [PubMed] [Google Scholar]
- 6.Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer's disease. Brain 2007;130:2636–2645 [DOI] [PubMed] [Google Scholar]
- 7.Renner JA, Burns JM, Hou CE, McKeel DW, Jr, Storandt M, Morris JC. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology 2004;63:1175–1180 [DOI] [PubMed] [Google Scholar]
- 8.de Souza LC, Corlier F, Habert MO, et al. Similar amyloid-beta burden in posterior cortical atrophy and Alzheimer's disease. Brain 2011;134:2036–2043 [DOI] [PubMed] [Google Scholar]
- 9.Seguin J, Formaglio M, Perret-Liaudet A, et al. CSF biomarkers in posterior cortical atrophy. Neurology 2011;76:1782–1788 [DOI] [PubMed] [Google Scholar]
- 10.Rosenbloom MH, Alkalay A, Agarwal N, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology 2011;76:1789–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer's disease. Brain 2013;136:844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snowden JS, Stopford CL, Julien CL, et al. Cognitive phenotypes in Alzheimer's disease and genetic risk. Cortex 2007;43:835–845 [DOI] [PubMed] [Google Scholar]
- 13.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099 [DOI] [PubMed] [Google Scholar]
- 15.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33, and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33, and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–184 [DOI] [PubMed] [Google Scholar]
- 20.Petersen RC, Waring SC, Smith GE, Tangalos EG, Thibodeau SN. Predictive value of APOE genotyping in incipient Alzheimer's disease. Ann NY Acad Sci 1996;802:58–69 [DOI] [PubMed] [Google Scholar]
- 21.Carrasquillo MM, Belbin O, Hunter TA, et al. Replication of EPHA1 and CD33 associations with late-onset Alzheimer's disease: a multi-centre case-control study. Mol Neurodegener 2011;6:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrasquillo MM, Belbin O, Hunter TA, et al. Replication of BIN1 association with Alzheimer's disease and evaluation of genetic interactions. J Alzheimers Dis 2011;24:751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrasquillo MM, Belbin O, Hunter TA, et al. Replication of CLU, CR1, and PICALM associations with Alzheimer disease. Arch Neurol 2010;67:961–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 25.Nielsen DM, Ehm MG, Weir BS. Detecting marker-disease association by testing for Hardy-Weinberg disequilibrium at a marker locus. Am J Hum Genet 1998;63:1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007;39:17–23 [DOI] [PubMed] [Google Scholar]
- 27.Carrasquillo MM, Zou F, Pankratz VS, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet 2009;41:192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.