

Abstract
Objective:
To determine the genes underlying Dravet syndrome in patients who do not have an SCN1A mutation on routine testing.
Methods:
We performed whole-exome sequencing in 13 SCN1A-negative patients with Dravet syndrome and targeted resequencing in 67 additional patients to identify new genes for this disorder.
Results:
We detected disease-causing mutations in 2 novel genes for Dravet syndrome, with mutations in GABRA1 in 4 cases and STXBP1 in 3. Furthermore, we identified 3 patients with previously undetected SCN1A mutations, suggesting that SCN1A mutations occur in even more than the currently accepted ∼75% of cases.
Conclusions:
We show that GABRA1 and STXBP1 make a significant contribution to Dravet syndrome after SCN1A abnormalities have been excluded. Our results have important implications for diagnostic testing, clinical management, and genetic counseling of patients with this devastating disorder and their families.
Dravet syndrome (Online Mendelian Inheritance in Man #607208), previously known as severe myoclonic epilepsy of infancy, is an infantile-onset epileptic encephalopathy characterized by a distinctive electroclinical and developmental course culminating in intellectual disability and refractory seizures. The genetic basis of this disorder is attributed to heterozygous disease-causing mutations in the sodium channel α1 subunit gene, SCN1A, in 75% of patients; 90% of mutations arise de novo.1,2 A small proportion of girls and one mosaic male, with a phenotype resembling Dravet syndrome, have mutations of protocadherin 19, PCDH19.3,4 Two patients with heterozygous truncating GABRG2 mutations and 2 case reports with homozygous SCN1B mutations have also been described.5–8 Finally, recently, 3 patients with de novo CHD2 mutations and several overlapping features of Dravet syndrome were reported.9 These mutations, however, are rare, and the genetic etiology of most patients with Dravet syndrome without mutations in SCN1A remains to be solved. Here we employ a whole-exome sequencing (WES) and targeted resequencing approach for gene discovery in SCN1A-negative patients with Dravet syndrome.
METHODS
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from all patients and in the case of minors, their parents or legal guardians. This study was approved by the human research ethics committees at Austin Health, the University of Washington, and the Christian-Albrechts University, as well as the Commission for Medical Ethics at the University of Antwerp.
Patients: WES cohort.
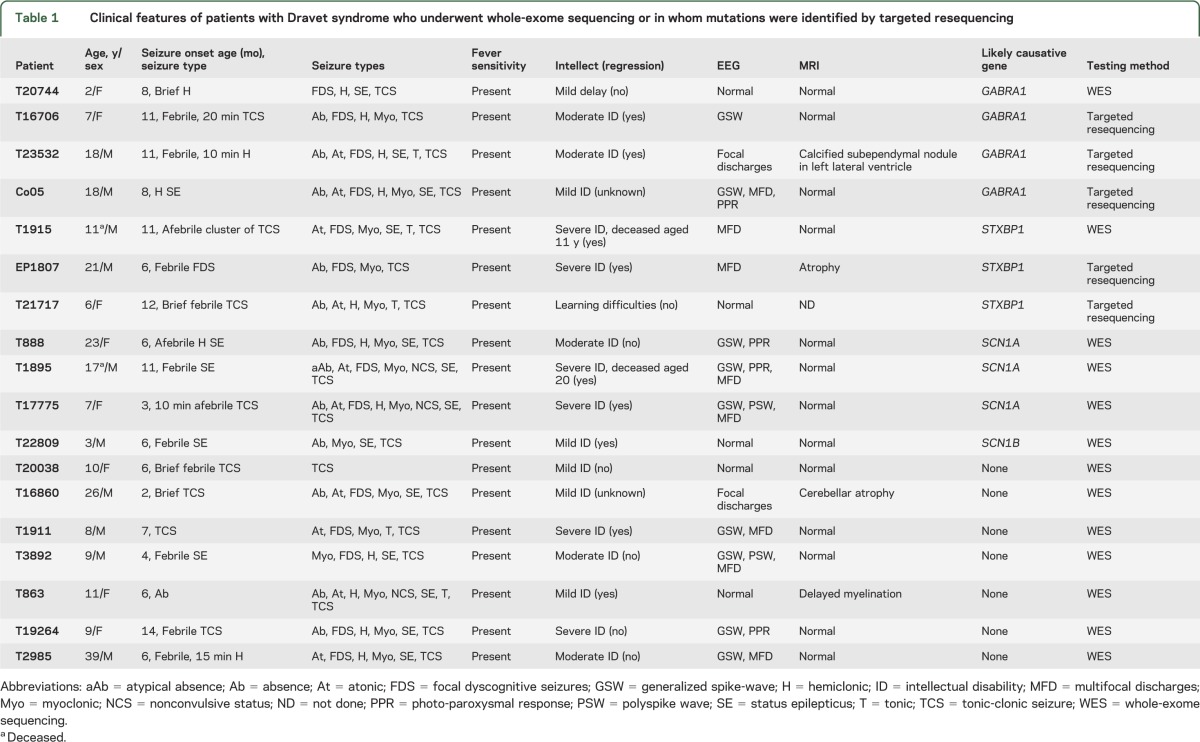
Probands with Dravet syndrome were recruited from the epilepsy clinic at Austin Health, from the practices of the investigators, and by referral for epilepsy genetics research from Australia and New Zealand. A diagnosis of Dravet syndrome was based on the following criteria: onset less than 15 months of age with convulsive seizures (hemiclonic or generalized) that were often prolonged and triggered by fever. Other seizure types emerged over time, including focal, myoclonic, absence seizures, and drop attacks. Development was normal in the first year of life with later slowing and intellectual disability.
The 13 patients subject to WES had been previously screened for SCN1A point mutations using denaturing high-performance liquid chromatography (dHPLC) (n = 4) or bidirectional sequencing (n = 9). Small exonic deletion/duplications had also been excluded using SCN1A multiplex ligation-dependent probe amplification and all patients were negative for large copy number variants (reference 10 and unpublished data).
WES and analysis.
The exome sequencing libraries of 34 individuals, including 10 parent–proband trios, 1 mother–proband pair, and 2 unrelated probands were prepared using the SeqCap EZ Human Exome Library v2.0 (Roche, Nimblegen). Libraries were sequenced on an Illumina HiSeq, using a 50 bp paired-end read protocol as per the manufacturer's recommendations. Reads were aligned to the human genome (hg19) using the Burrows-Wheeler Aligner,11 removing all potential PCR duplicates. The Genome Analysis Toolkit12 was used for base quality recalibrations, realignment around known indels, variant calling, and filtering to retrieve only high-quality variants. We considered only rare, disruptive (missense, nonsense, splice, frameshift) variants that were not present in the ESP6500 control dataset (see URLs in the appendix) for further analysis.
Patients: Targeted resequencing (WES) cohort.
We performed targeted resequencing of candidate genes in a cohort of 67 Dravet and Dravet-like patients. All 67 of these patients had been screened for SCN1A mutations previously by the various collaborating institutions. In addition, we performed SCN1A mutation screening using molecular inversion probes and high-throughput sequencing. Only SCN1A-negative patients were included in the validation cohort (n = 67).
Targeted resequencing of candidate genes.
We selected 15 candidate genes (STXBP1, GABRA1, SCN1B, ATP6VOC, SLC8A1, CLSNT1, NKAIN3, NOL11, RIMS2, KIF1B, CDK5RAP3, ABTB2, STK31, KDM2B, SPATA13) from the WES analysis for mutation screening in a validation cohort of 67 SCN1A-negative Dravet probands. From the 13 cases in whom WES was performed, we identified candidate genes belonging to one of 3 categories, based on the presence of a rare variant in that gene. Three candidate genes (STXBP1, GABRA1, SCN1B) were previously implicated in epileptic encephalopathies or other epilepsies. An additional 5 genes (ATP6VOC, SLC8A1, CLSNT1, NKAIN3, NOL11) were selected as candidates as we identified a rare, de novo variant in a single proband. Finally, we selected 7 genes with variants that segregated in a recessive manner in a single proband (RIMS2, KIF1B, CDK5RAP3, ABTB2, STK31, KDM2B, SPATA13). We used molecular inversion probes to “capture” exonic regions and 5 flanking intronic base pairs of target genes, and performed massively parallel sequencing and variant detection as described previously.13,14
GABAA mutagenesis and in vitro transcription.
Human GABAA complementary DNA (cDNA) was cloned into the pGEMHE vector containing a T7 promoter for in vitro transcription. The Gly251Ser mutation was generated using QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) with primers forward 5′-GAAGAGAAAGATTAGCTACTTTGTTATTCAAACATACCTGCC and reverse 5′-GGCAGGTATGTTTGAATAACAAAGTAGCTAATCTTTCTCTTC. Gly251Ser mutation is underlined. The GABRA1 (Gly251Ser) pGEMHE plasmid was verified by DNA sequencing. cRNA was made using linearized cDNA template and in vitro transcription performed using the mMessage mMachine kit (Applied Biosystems/Ambion, Austin, TX).
GABA modulation of wild-type and mutant receptors.
Oocytes from adult female Xenopus laevis were prepared as previously described.6 Fifty nanoliters of cRNA encoding the wild-type (WT) human A1, B2, and G2L and mutant A1 (Gly251Ser) GABA receptor subunits (12 ng/μL; stocks confirmed spectrophotometrically and by gel analysis) were injected into the cytoplasm of stage 5 or 6 oocytes using the Roboocyte Robot (Multi Channel Systems, Reutlingen, Germany) and stored for 2 days prior to experimentation. Two-electrode voltage clamp recordings were made in 96-well plates using the Roboocyte automated platform. Oocytes were impaled using recording heads with 2 glass electrodes containing 1.5 M potassium acetate and 0.5 M KCl and held at a membrane potential of −80 mV. Oocytes were continually perfused with a ND96 solution (96 mM NaCl, 2 mM KCl, 0.1 mM CaCl2, and 5 mM HEPES, pH 7.5) using a Gilson 222 XL Liquid Handler and Gilson Minipuls 3 Peristaltic Pump (Gilson Medical Electronics, Middleton, WI). To construct a dose-response curve, oocytes were exposed to a 30-second application of test γ-aminobutyric acid (GABA) (Sigma Aldrich, Sydney, Australia) (range 1 μM–1 mM) followed by a 60-second wash in ND96 and then a 15-second application of a maximum dose of GABA (1 mM). Only 1 test concentration and 1 maximum concentration of GABA was applied per oocyte. The effect of the test GABA concentration on an individual oocyte was expressed as a percentage of the maximal GABA response in the same oocyte. These percentages were then averaged from many oocytes (range 8–20 oocyes per test dose). Maximum current at 1 mM GABA was also averaged over many oocytes (100 for WT, 97 for Gly251Ser mutation).
RESULTS
We performed WES in 13 SCN1A-negative Dravet syndrome probands (clinical features in table 1), including 10 parent–proband trios, 1 mother–proband pair, and 2 unrelated probands, to identify novel genetic causes for this devastating disorder. On average, we generated 3.8 Gb of mapped sequence data per individual and 92% of bases had >8× coverage across all samples. On average, ∼27,000 raw variants were identified in each individual. We prioritized only disruptive (nonsynonymous, splice, frameshift) variants that were not present in the ESP6500 control dataset (see URLs in the appendix) for further analysis and initially applied a de novo model for gene discovery in these patients.
Table 1.
Clinical features of patients with Dravet syndrome who underwent whole-exome sequencing or in whom mutations were identified by targeted resequencing
De novo variants.
Under a de novo disease model, we identified 15 rare, disruptive variants in 9 individuals, including 2 individuals (T1895, T1911) who were originally sequenced as singletons and whose mutations were confirmed as occurring de novo using Sanger sequencing in the parents (table 2). Five of these de novo variants occurred in known epilepsy genes. Unexpectedly, 3 variants were detected in SCN1A that were not previously identified by Sanger DNA sequencing in 2 and dHPLC in the third. Furthermore, we detected a single mutation in 2 genes previously implicated in other epilepsy syndromes, GABRA1 and STXBP1 (table 2). We identified 3 additional probands with GABRA1 mutations (figure 1A) and 2 patients with de novo STXBP1 mutations by targeted resequencing in 67 patients with a clinical diagnosis of Dravet syndrome (table 2).
Table 2.
Rare, disruptive variants of interest in 13 patients with Dravet syndrome detected by WES and 67 patients by candidate gene targeted resequencing
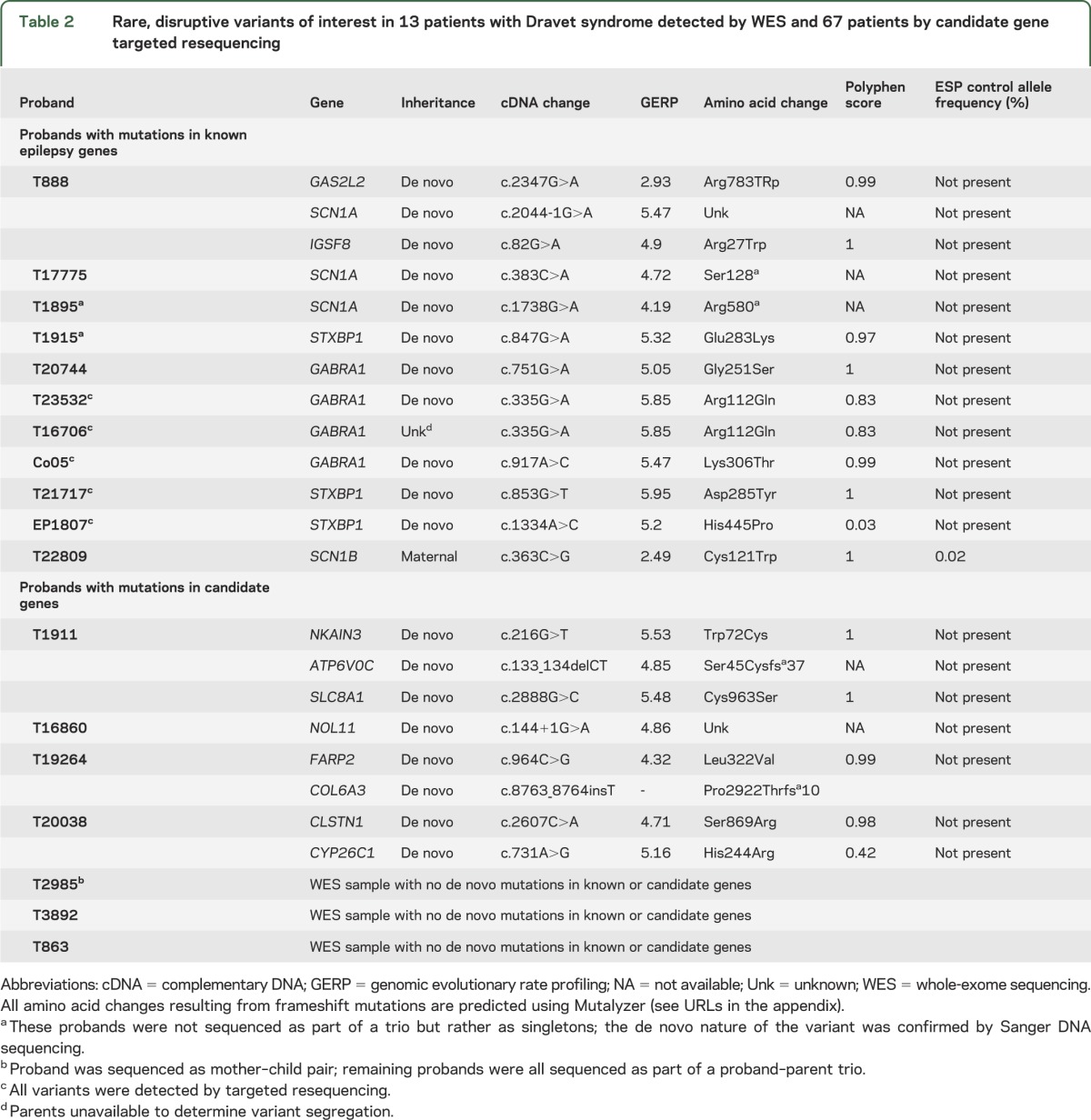
Figure 1. GABRA1 mutations in epilepsy and effects on γ-aminobutyric acid (GABA) response.

(A) The amino acid locations of the 8 mutations identified in patients with Dravet syndrome (purple) and other epilepsy syndromes (green). There is no clear genotype–phenotype correlation with respect to either nature or localization of the mutation and severity of phenotype. (B) Maximal current response (1 mM GABA) of the wild-type (WT) and p.Gly251Ser mutant. (C) GABA dose-response curves of the WT and p.Gly251Ser mutant in Xenopus laevis oocytes. CAE = childhood absence epilepsy; EE = epileptic encephalopathy; GGE/FS = genetic generalized epilepsies/febrile seizures; JME = juvenile myoclonic epilepsy.
Of the 10 trios who underwent WES, 3 probands had no candidate de novo mutations that passed our filtering criteria, whereas 4 subjects had de novo mutations in one or more genes that are not known epilepsy genes (table 2). Each gene was only implicated in 1 patient, with unique de novo events in 8 genes. In order to validate these genes in Dravet syndrome, we prioritized 5 candidate genes (ATP6VOC, SLC8A1, CLSTN1, NKAIN3, NOL11) for further study. We excluded FARP2, COL6A3, and CYP26C1 as candidate genes given that they encode proteins with no obvious neuronal function or had multiple putatively truncating mutations in the ESP control dataset. Targeted resequencing in these 5 candidate genes in the validation cohort (n = 67) revealed no additional rare, de novo pathogenic variants.
Inherited mutations in known epilepsy genes.
We identified all disruptive variants in known epilepsy genes with an allele frequency in the ESP dataset of <1% in the 8 patients with Dravet syndrome who underwent WES, but did not carry de novo mutations in known epilepsy genes (table e-1 on the Neurology® Web site at Neurology.org). Of interest, we detected a maternally inherited c.363C>T (p.Cys121Trp) in SCN1B in T22809; this individual had no candidate de novo mutations (figure e-1). This mutation has been described in families with other types of epilepsy.15,16
Autosomal recessive model for Dravet syndrome.
Given that 2 recessive cases of Dravet syndrome have been reported,7,8 we applied this disease inheritance model in the 7 probands without mutations in known epilepsy genes and identified 15 genes with variants that followed an autosomal recessive pattern (variant allele frequency <1%) (table e-2). Targeted resequencing was performed in 7 candidate genes (RIMS2, KIF1B, CDK5RAP3, ABTB2, STK31, KDM2B, and SPATA13). We excluded the remaining genes as they have been implicated in unrelated disorders (MLL2, PDE6B, PCNT) or have no known or obvious neuronal function (VWA5B2, OAS3, DCHS2, DNAH3, DNAH11). We found no instances of autosomal recessive inheritance in our validation cohort (n = 67).7,8
Dose response of the GABRA1 mutant p.Gly251Ser to GABA.
To assess the effect of the p.Gly251Ser GABAA mutation on neuronal function, we measured GABA-mediated currents in X laevis oocytes expressing mutant (p.Gly251Ser) GABAA (e-Methods). Maximum current values recorded at −1 mM GABA dosage showed a 2.6-fold reduction in the amplitude of GABA-induced currents in vitro for the p.Gly251Ser mutant (max I ± SEM: 2,621 ± 142, n = 97) compared to WT (max I ± SEM: 7,010 ± 325, n = 100) (figure 1B). Furthermore, the GABA dose-response curves showed a 5-fold decrease in sensitivity to GABA of the p.Gly251Ser mutant compared to WT (figure 1C).
DISCUSSION
We applied massively parallel sequencing approaches in 67 SCN1A-negative patients with Dravet syndrome to identify novel de novo genetic causes for this devastating disorder. Overall, we show that GABRA1 and STXBP1 are new causes for Dravet syndrome. Furthermore, we identified 3 patients with undetected SCN1A mutations, despite previous mutation screening. This finding verifies the efficacy of our WES approach in gene discovery and the accuracy of clinical diagnosis. Furthermore, we propose that Dravet syndrome is due to mutations in SCN1A more often than the generally reported estimate of 75%,1 as some mutations pass undetected using conventional mutation detection techniques.
Overall, we identified 4 novel GABRA1 mutations in patients with Dravet syndrome. The clinical presentation was typical for Dravet syndrome, with the only uncommon feature being atonic drop attacks in 2 of the 4 cases. Three of the 4 variants arose de novo, while the inheritance of the fourth variant, a c.335 G>A, p.Arg112Gln in proband T16706, could not be determined as parents were unavailable. However, this same mutation (p.Arg112Gln) arose de novo in another proband (T23532), suggesting this is a recurrent pathogenic mutation resulting in Dravet syndrome. This finding redefines GABRA1 from a gene associated with mild genetic generalized epilepsies and febrile seizures17–19 to a gene also implicated in severe epilepsies such as Dravet syndrome (figure 1A). Our results are supported by the recent identification of a de novo mutation in GABRA1 in a patient with epileptic encephalopathy.20
We propose that the p.Gly251Ser GABRA1 mutation reduces the ability of mutant receptors to contribute to phasic inhibition as demonstrated by the decreased sensitivity to GABA and a significantly reduced amplitude of GABA-induced currents. It is likely that the additional 2 missense GABRA1 mutations (p.Arg112Gln, Lys306Thr) act in a similar fashion. Overall, these studies suggest that seizures in these patients are the result of impaired functioning of GABA inhibition in the brain.
We also describe 3 Dravet syndrome patients with de novo missense mutations in STXBP1. Our 3 patients had a Dravet phenotype with onset in the first year of life; however, 2 had both tonic and atonic seizures. Both seizure types are rare early in Dravet syndrome, although tonic seizures are reported in older patients.21 Status epilepticus was only seen in 1 case. Heterozygous de novo STXBP1 mutations cause early-onset epileptic encephalopathies and neurodevelopmental disorders. Of the >50 patients with STXBP1 encephalopathy described, the majority present by 3 months of age, with Ohtahara syndrome or other early-onset epileptic encephalopathies.22–32 Our patients had onset from 6 to 12 months, which is later than usually seen in STXBP1 encephalopathy. It is typically associated with epileptic spasms, and notably these were not observed in our patients with Dravet syndrome. The wide range of STXBP1 mutations show no genotype–phenotype correlation with respect to mutations and clinical presentation (figure e-2).
STXBP1 plays a role in the release of neurotransmitters into the synapse, via regulation of syntaxin. The 3 de novo missense mutations described here all lead to the replacement of a charged residue with a neutral amino acid. These alterations are likely to destabilize the STXBP1 protein or affect binding to syntaxin, as has been shown previously for the Cys180Tyr missense mutation,22 though experimental validation needs to be performed.
We identified no de novo mutations in proband T22809; however, this individual was shown to carry a maternally inherited c.363C>T (p.Cys121Trp) in SCN1B. This mutation has been described in genetic epilepsy with febrile seizures plus (GEFS+) in 41 affected individuals from 4 families. The affected individuals displayed heterogeneous epilepsy phenotypes ranging from mild (febrile seizures [FS]) to moderate (temporal lobe epilepsy) and severe (epilepsy with myoclonic-atonic seizures).15,16 GEFS+ families may include individuals with Dravet syndrome who also inherit a dominant familial mutation.33 The p.Cys121Trp mutation affects a highly conserved residue and putatively disrupts a disulphide bridge in the extracellular domain of the protein. p.Cys121Trp mutants induce a hyperexcitable state in vitro.34 The p.Cys121Trp SCN1B mutation potentially contributes to the presentation of Dravet syndrome in proband T22809 and causes the FS in his mother. Interestingly, it was nonpenetrant in his maternal grandmother, which is in keeping with the low penetrance observed in GEFS+ families.35 Other unaffected SCN1B c.363C>T (p.C121W) carriers (n = 6) have been reported,16 and the variant is present in controls, suggesting other genetic or nongenetic factors modify the epilepsy phenotype. These observations recapitulate those seen in other patients with Dravet syndrome, where ∼3–5% of cases have inherited a pathogenic SCN1A variant, typically from a more mildly affected parent with GEFS+.36 Recently, 2 reports of recessive SCN1B mutations causing Dravet syndrome have been published, although one had an atypical phenotype,37 but no heterozygous dominant mutations have been reported.7,8 Our patient did not carry additional mutations in SCN1B, nor did we identify additional SCN1B mutations in our validation cohort (n = 67) by targeted resequencing of the gene. Collectively, these results suggest that SCN1B may play a susceptibility role in the pathogenesis of Dravet syndrome, though further investigations are required.
We show that the genetic etiology of SCN1A-negative Dravet syndrome can, in part, be attributed to de novo mutations in GABRA1 and STXBP1. Of note, mutation screening of GABRA1 in cohorts of patients with genetic generalized epilepsy and epileptic encephalopathies have rarely identified pathogenic mutations (data not shown and references 14, 17, 18, and 20). Our finding of 4 GABRA1 mutations in 77 SCN1A-negative patients with Dravet syndrome suggests that GABRA1 mutations may be largely limited, at least in terms of epileptic encephalopathies, to Dravet syndrome, though further studies are needed. Conversely, STXBP1 mutations are seen in other epileptic encephalopathy phenotypes, suggesting considerable phenotypic heterogeneity compared to GABRA1.
GABRA1 and STXBP1 are significant contributors to SCN1A-negative Dravet syndrome that should be tested in patients with Dravet syndrome negative for SCN1A mutations. With identification of further cases with Dravet syndrome due to these genes, specific phenotypic patterns may emerge that distinguish these rarer causes of Dravet syndrome from those due to SCN1A mutations. We would argue that, in SCN1A-negative individuals, targeted resequencing of known epilepsy genes is a more cost-effective and high-throughput approach to diagnostic testing.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for participating in the research study, Dr. Simon Harvey for referral of patients, and the Northwest Genomics Center for WES and analysis (http://nwgc.gs.washington.edu/) and the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be) for genetic analyses.
GLOSSARY
- cDNA
complementary DNA
- dHPLC
denaturing high-performance liquid chromatography
- FS
febrile seizures
- GABA
γ-aminobutyric acid
- GEFS+
genetic epilepsy with febrile seizures plus
- WES
whole-exome sequencing
- WT
wild-type
APPENDIX.
Accession numbers
SCN1A (NM_001165963.1), STXBP1 (NM_003165.3), GABRA1 (NM_000806.5), SCN1B (NM_199037.3), ATP6VOC (NM_001198569.1), SLC8A1 (NM_021097.2), CLSTN1 (NM_001009566.1), NKAIN3 (NM_173688.2), NOL11 (NM_015462), RIMS2 (NM_001100117.2), KIF1B (NM_015074.3), CDK5RAP3 (NM_001278197.1), ABTB2 (NM_145804.2), STK31 (NM_031414.4), KDM2B (NM_032590.4), SPATA13 (NM_001166271.1).
Web resources
BWA-v0.5.6 (http://bio-bwa.sourceforge.net/)
GATK-v2.2-9 (http://www.broadinstitute.org/gatk/)
Seattle seq-v134 (http://snp.gs.washington.edu/SeattleSeqAnnotation/)
National Heart Lung and Blood Institute (NHLBI) Exome sequencing project (http://evs.gs.washington.edu/EVS/)
Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/)
Allen brain atlas (http://www.brain-map.org/)
Mutalyzer (https://mutalyzer.nl/index)
Online Mendelian Inheritance in Man (http://www.omim.org)
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Gemma L. Carvill designed the study, performed experiments and data analysis, and wrote the manuscript. Sarah Weckhuysen performed experiments and data analysis and performed phenotypic analysis. Jacinta M. McMahon performed phenotypic analysis. Corinna Hartmann performed experiments and data analysis. Rikke S. Møller performed phenotypic analysis. Helle Hjalgrim performed phenotypic analysis. Joseph Cook assisted with data analysis method development. Eileen Geraghty performed experiments. Brian J. O’Roak developed the MIPs methodology and analysis. Steve Petrou performed experiments and data analysis. Alison Clarke performed experiments and data analysis. Deepak Gill performed phenotypic analysis. Lynette G. Sadleir performed phenotypic analysis. Hiltrud Muhle performed phenotypic analysis. Sarah von Spiczak performed phenotypic analysis. Marina Nikanorova performed phenotypic analysis. Bree L. Hodgson performed experiments and data analysis. Elena V. Gazina performed experiments and data analysis. Arvid Suls performed experiments and data analysis. Jay Shendure developed the MIPs methodology and analysis. Leanne M. Dibbens performed experiments and data analysis. Peter De Jonghe performed phenotypic analysis. Ingo Helbig performed phenotypic analysis. Samuel F. Berkovic performed phenotypic analysis and critically reviewed the manuscript. Ingrid E. Scheffer supervised and designed the study, performed phenotypic analysis, and wrote the manuscript. Heather C. Mefford supervised and designed the study and wrote the manuscript.
STUDY FUNDING
Supported by funding from the NIH (NINDS 5R01NS069605). H.C.M. is a recipient of the Burroughs Wellcome Fund Career Award for Medical Scientists. I.E.S. and S.F.B. are supported by a National Health and Medical Research Council of Australia (NHMRC) Program Grant and Practitioner Fellowship (I.E.S.). L.G.S. is supported by a Health and Research Council of New Zealand project grant. The research is supported by the Fund for Scientific Research Flanders (FWO), Methusalem excellence grant of the Flemish Government, University of Antwerp, the Interuniversity Attraction Poles (IAP) program P6/43 of the Belgian Science Policy Office (BELSPO) and the Eurocores program EuroEPINOMICS of the European Science Foundation. The VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be) performed genetic analyses. A.S. is a postdoctoral fellow of the Fund for Scientific Research Flanders (FWO).
DISCLOSURE
G. Carvill, S. Weckhuysen, J. McMahon, C. Hartmann, R. Møller, H. Hjalgrim, J. Cook, E. Geraghty, Bree L. Hodgson, and Leanne M. Dibbens report no disclosures relevant to the manuscript. B. O’Roak is an inventor on patent PCT/US2009/30620: Mutations in contactin associated protein 2 are associated with increased risk for idiopathic autism. Steve Petrou, A. Clarke, D. Gill, L. Sadleir, H. Muhle, S. von Spiczak, M. Nikanorova, E. Gazina, and A. Suls report no disclosures relevant to the manuscript. J. Shendure is a member of the scientific advisory board or serves as a scientific consultant for Adaptive Biotechnologies, Ariosa Diagnostics, Stratos Genomics, GenePeeks, Gen9, Good Start Genetics, and Rubicon Genomics, gave expert testimony in Life Technologies v. Illumina and Johns Hopkins University v. 454 Life Sciences, and has received patent royalties from Life Technologies, Illumina, and Gen9. P. De Jonghe has research funded by the Fund for Scientific Research Flanders (FWO) and received speaker's fee from Biocodex. I. Helbig reports no disclosures relevant to the manuscript. S. Berkovic has received grant(s) from the National Health and Medical Research Council; has received honoraria from UCB; has a patent for PCDH19 testing planned; has received payment for development of educational presentations from UCB Pharma, Novartis Pharmaceuticals, Sanofi-Aventis, and Jansen Cilag; has a patent for SCN1A testing held by Bionomics and licensed to various diagnostic companies, with no financial return; and was a consultant to Bionomics and Athena diagnostics over 3 years ago. I. Scheffer has served on scientific advisory boards for UCB and Janssen-Cilag EMEA; serves on the editorial boards of the Annals of Neurology and Epileptic Disorders; may accrue future revenue on pending patent WO61/010176 (filed: 2008): Therapeutic Compound; has received speaker honoraria from GlaxoSmithKline, Athena Diagnostics, UCB, Biocodex, and Janssen-Cilag EMEA; has received funding for travel from Athena Diagnostics, UCB, Biocodex, GlaxoSmithKline, and Janssen-Cilag EMEA; and receives/has received research support from the National Health and Medical Research Council of Australia, NIH, Australian Research Council, Health Research Council of New Zealand, CURE, American Epilepsy Society, US Department of Defense Autism Spectrum Disorder Research Program, the Jack Brockhoff Foundation, the Shepherd Foundation, Perpetual Charitable Trustees, and The University of Melbourne. H. Mefford has received a grant from the NIH/NINDS, Burroughs Wellcome Fund, and was a consultant for the Simons Foundation (SFARI Gene Advisory Board). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet syndrome. Epilepsia 2011;52(suppl 2):24–29 [DOI] [PubMed] [Google Scholar]
- 2.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 2001;68:1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Depienne C, Bouteiller D, Keren B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009;5:e1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Depienne C, Gourfinkel-An I, Baulac S, LeGuern E. Genes in infantile epileptic encephalopathies. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, eds. Jasper's Basic Mechanisms of the Epilepsies. Bethesda: National Center for Biotechnology Information; 2012 [PubMed] [Google Scholar]
- 5.Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res 2006;70(suppl 1):S206–S217 [DOI] [PubMed] [Google Scholar]
- 6.Harkin LA, Bowser DN, Dibbens LM, et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 2002;70:530–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patino GA, Claes LR, Lopez-Santiago LF, et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 2009;29:10764–10778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogiwara I, Nakayama T, Yamagata T, et al. A homozygous mutation of voltage-gated sodium channel beta(I) gene SCN1B in a patient with Dravet syndrome. Epilepsia 2012;53:e200–e203 [DOI] [PubMed] [Google Scholar]
- 9.Suls A, Kecskés A, Weber Y, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013;93:967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol 2011;70:974–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Roak BJ, Vives L, Fu W, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. 2012;338:1619–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 1998;19:366–370 [DOI] [PubMed] [Google Scholar]
- 16.Scheffer IE, Harkin LA, Grinton BE, et al. Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 2007;130:100–109 [DOI] [PubMed] [Google Scholar]
- 17.Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 2002;31:184–189 [DOI] [PubMed] [Google Scholar]
- 18.Lachance-Touchette P, Brown P, Meloche C, et al. Novel alpha1 and gamma2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. The Eur J Neurosci 2011;34:237–249 [DOI] [PubMed] [Google Scholar]
- 19.Maljevic S, Krampfl K, Cobilanschi J, et al. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol 2006;59:983–987 [DOI] [PubMed] [Google Scholar]
- 20.Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 2010;51:1043–1052 [DOI] [PubMed] [Google Scholar]
- 22.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008;40:782–788 [DOI] [PubMed] [Google Scholar]
- 23.Hamdan FF, Piton A, Gauthier J, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009;65:748–753 [DOI] [PubMed] [Google Scholar]
- 24.Mignot C, Moutard ML, Trouillard O, et al. STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011;52:1820–1827 [DOI] [PubMed] [Google Scholar]
- 25.Saitsu H, Kato M, Okada I, et al. STXBP1 mutations in early infantile epileptic encephalopathy with suppression-burst pattern. Epilepsia 2010;51:2397–2405 [DOI] [PubMed] [Google Scholar]
- 26.Saitsu H, Kato M, Shimono M, et al. Association of genomic deletions in the STXBP1 gene with Ohtahara syndrome. Clin Genet 2012;81:399–402 [DOI] [PubMed] [Google Scholar]
- 27.Deprez L, Weckhuysen S, Holmgren P, et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology 2010;75:1159–1165 [DOI] [PubMed] [Google Scholar]
- 28.Vatta M, Tennison MB, Aylsworth AS, et al. A novel STXBP1 mutation causes focal seizures with neonatal onset. J Child Neurol 2012;27:811–814 [DOI] [PubMed] [Google Scholar]
- 29.Milh M, Villeneuve N, Chouchane M, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011;52:1828–1834 [DOI] [PubMed] [Google Scholar]
- 30.Sampaio M, Rocha R, Biskup S, Leao M. Novel STXBP1 mutations in 2 patients with early infantile epileptic encephalopathy. J Child Neurol Epub 2013 Mar 26 [DOI] [PubMed]
- 31.Weckhuysen S, Holmgren P, Hendrickx R, et al. Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia 2013;54:e74–e80 [DOI] [PubMed] [Google Scholar]
- 32.Lemke JR, Riesch E, Scheurenbrand T, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012;53:1387–1398 [DOI] [PubMed] [Google Scholar]
- 33.Singh R, Andermann E, Whitehouse WP, et al. Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia 2001;42:837–844 [DOI] [PubMed] [Google Scholar]
- 34.Meadows LS, Malhotra J, Loukas A, et al. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci 2002;22:10699–10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol 1999;45:75–81 [DOI] [PubMed] [Google Scholar]
- 36.Depienne C, Trouillard O, Gourfinkel-An I, et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet 2010;47:404–410 [DOI] [PubMed] [Google Scholar]
- 37.Kim YO, Dibbens L, Marini C, et al. Do mutations in SCN1B cause Dravet syndrome? Epilepsy Res 2013;103:97–100 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.