

Abstract
Objective:
To investigate the genetic and physiologic basis of the neuromuscular symptoms of hypotonia-cystinuria syndrome (HCS) and isolated PREPL deficiency, and their response to therapy.
Methods:
We performed molecular genetic, histochemical, immunoblot, and ultrastructural studies, investigated neuromuscular transmission in vitro in a patient with isolated PREPL deficiency, and evaluated the effect of pyridostigmine in this patient and in 3 patients with the HCS.
Results:
HCS is caused by recessive deletions involving the SLC3A1 and PREPL genes. The major clinical features of HCS are type A cystinuria, growth hormone deficiency, muscle weakness, ptosis, and feeding problems. The proband with isolated PREPL deficiency had myasthenic symptoms since birth and a positive edrophonium test but no cystinuria. She and 1 of 3 patients with HCS responded transiently to pyridostigmine during infancy. The proband harbors a paternally inherited nonsense mutation in PREPL and a maternally inherited deletion involving both PREPL and SLC3A1; therefore, the PREPL deficiency determines the phenotype. We detected no PREPL expression in the patient's muscle and endplates. Electrophysiology studies revealed decreased quantal content of the endplate potential and reduced amplitude of the miniature endplate potential without endplate acetylcholine receptor deficiency or altered endplate geometry.
Conclusion:
Isolated PREPL deficiency is a novel monogenic disorder that causes a congenital myasthenic syndrome with pre- and postsynaptic features and growth hormone deficiency. The myasthenic symptoms in PREPL deficiency with or without cystinuria may respond to pyridostigmine in early life. We attribute the myasthenia to abrogated interaction of PREPL with adaptor protein 1.
The hypotonia-cystinuria syndrome (HCS) (Online Mendelian Inheritance in Man [OMIM] # 606407) is characterized by cystinuria in combination with severe neonatal hypotonia, fluctuating ptosis, facial paresis, dysarthria and feeding problems suggesting a myasthenic disorder, and growth hormone deficiency. Other features are viscous saliva and hypergonadotrophic hypogonadism, and half of the patients need special education.1,2 The hypotonia and feeding problems improve during the first year of life, but ptosis, nasal voice, dysarthria, facial weakness, and mild axial and proximal muscle weakness persist. Hyperphagia with tendency to obesity develops during childhood. HCS is caused by autosomal recessive deletions involving 2 contiguous genes on chromosome 2p21: SLC3A1, which encodes the heavy subunit of the cystine dibasic and neutral amino acid transporter (OMIM * 104614), and PREPL, the prolyl endopeptidase-like gene (OMIM * 609557) (figure 1G). Seventeen HCS families with 9 different deletions ranging from 23.8 to 127.2 kb have been reported to date.1–4
Figure 1. Clinical and genetic features.

(A, B) Patient at age 6 months. Note marked hypotonia with frog-leg posture and head drop. (C, D) Positive response to pyridostigmine at age 11 weeks. (E) PCR fragments in the patient, the parents, and a control. Upper lane: junction fragment, present only in the patient and her mother; middle lane: PCR with forward primer in the nondeleted and reverse primer in the deleted region of SLC3A1; lower lane: PCR with forward primer in the deleted and reverse primer in the nondeleted region of PREPL. Only the normal allele will produce a band in the 2 lower lanes. (F) Sequence chromatograms show a single base pair (bp) deletion (first bp after the vertical bar) in the patient's genomic DNA (gDNA) and complementary DNA (cDNA) and in the father's gDNA and cDNA, not present in the mother. cDNA primers were designed to amplify only the nondeleted allele so that only the paternal allele is sequenced in the patient's cDNA, clearly demonstrating the single bp deletion. (G) Overview of 2p21 region showing the identified deletions. The patient with isolated PREPL deficiency was compound heterozygous for the nonsense mutation (red arrow) and deletion J. Deletions A, B, and H were homozygous in the patients with hypotonia-cystinuria syndrome.
Recessive deletion syndromes involving more genes at the 2p21 chromosomal region have been described.5–7 These are clinically different and more severe, probably because of mitochondrial involvement. When C2orf34, distal to PREPL, is involved, the syndrome is referred to as atypical HCS, and when PPM1B, proximal to SLC3A1, is also involved, the syndrome is designated as 2p21 deletion syndrome.
PREPL is ubiquitously expressed, with highest levels in brain, kidney, and muscle, in decreasing order.8 Because mutations in SLC3A1 cause isolated cystinuria type A, we hypothesized that the neuromuscular and other noncystinuric manifestations of HCS are caused by PREPL deficiency.1,7 In atypical HCS, the involvement of SLC3A1 is only responsible for cystinuria, because the phenotype associated with a deletion restricted to PREPL and C2orf34 is very similar to that of atypical HCS but without cystinuria.9
Herein, we report a patient with isolated PREPL deficiency, show it causes a novel myasthenic syndrome, elucidate its structural and electrophysiologic features, and assess the effects of pyridostigmine treatment in this patient as well as in 3 patients with the HCS.
METHODS
Standard protocol approvals, registrations, and patient consents.
The human studies reported herein were approved by the Institutional Review Boards of the Hospitals of Leuven University and the Mayo Clinic, and each participant or their parent gave informed consent to participate in the study.
Molecular genetics.
Total blood RNA was obtained with the PAXgene system (Qiagen Benelux, Venlo, the Netherlands). Complementary DNA (cDNA) was produced from RNA templates with the iScript Select cDNA synthesis kit (Bio-Rad Laboratories, Nazareth, Belgium). Genomic DNA was isolated from blood by routine techniques. The combined deletion of PREPL and SLC3A1 was detected by microarray-based comparative genomic hybridization performed by Athena Diagnostics (Worcester, MA). To map the borders of the deletion, we used quantitative PCR with different primer sets around the predicted borders of the deletion. A junction fragment was then amplified by standard PCR techniques. To formally prove heterozygosity, we chose different primer sets to amplify either the junction fragment or the normal allele (primers available on request). Sanger sequencing was used to sequence PREPL cDNA, genomic DNA, and the junction fragment. The reference sequence was GRCh37, NM_006036.4.
PREPL expression monitored by immunoblotting and immunohistochemistry.
Extracts of frozen muscle from patient and control specimens were prepared for immunoblotting as previously described.10 After transfer to nitrocellulose membrane (Life Technologies, Carlsbad, CA), the blots were probed at 1:200 dilution with a mouse monoclonal antibody (Santa Cruz Biotechnology, Dallas, TX) directed against a C-terminal PREPL epitope, and the bands were detected with an appropriate secondary antibody as described.11 We immunolocalized PREPL in frozen sections of muscle harboring endplates (EPs) with the same antibody at 1:20 dilution and with a DyLight 488 (green) anti-mouse second antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:200 dilution. PREPL expression at the EPs was ascertained by colocalization with rhodamine-labeled α-bungarotoxin.12
EP studies.
These were performed on the anconeus muscle. Acetylcholinesterase and the acetylcholine receptor (AChR) were localized in frozen sections as previously described.12 EPs were localized for electron microscopy and quantitatively analyzed by established methods.13 Peroxidase-labeled α-bungarotoxin was used for the ultrastructural localization of AChR.14 The number of AChRs per EP was measured with [125I]α-bungarotoxin.15 The miniature EP potential (MEPP), miniature EP current (MEPC), and EP potential (EPP) amplitudes and the number of quanta released by nerve impulse (m) were measured as previously reported.16–18 The number of quanta available for release (n), and the probability of quantal release (p) were determined as previously described.19
Evaluation of response of patients with HCS to pyridostigmine.
An infant (HCS1) was evaluated using the Alberta Infant Motor Scale and 2 older patients (HCS2 and HCS3) with the 6-Minute Walk Test, handheld myometry of selected muscle groups, maximal inspiratory and expiratory pressures, and video-recordings of ptosis and speech. To exclude spontaneous improvement or training effects, we stopped the pyridostigmine for at least 24 hours before reevaluating the patients.
RESULTS
Clinical data.
The patient with isolated PREPL deficiency was born after 39 weeks of gestation. Her birth weight was normal, but she had marked hypotonia and feeding difficulties. At age 11 weeks, she had eyelid ptosis, a tented upper lip, proximal-predominant muscle weakness that varied during the day, normal tendon reflexes, and a strongly positive response to edrophonium and pyridostigmine (figure 1, C and D). She had a normal metabolic workup, no cystinuria, and a normal brain MRI. Tests for anti-AChR and anti-MuSK antibodies were negative. She was treated with pyridostigmine and discharged with a nasogastric tube that was later changed to a J-tube. At age 6 months, she was further investigated at the Mayo Clinic. She was still markedly hypotonic (figure 1, A and B). EMG studies when not taking pyridostigmine showed no decrement at 2 Hz, a finding also noted in myasthenic infants whose small muscle fibers have an increased input resistance.17 She continued to improve and was weaned off pyridostigmine at age 12 months without deterioration. She walked at 17 months, but her gait was waddling and she preferred using a walker. Language development was normal. The serum insulin-like growth factor (IGF)-1 level was repeatedly low (<25 ng/mL), but the level of its binding protein IGFBP-3 was normal (1.5 μg/mL), and a growth hormone stimulation test showed growth hormone deficiency, indicating that these features of HCS are caused by the PREPL deficiency.
We also treated 3 previously identified patients with HCS (for clinical and genetic features, see table e-1 on the Neurology® Web site at Neurology.org), at 1,2 11,1 and 121 years of age with pyridostigmine. Only the infant improved, with an impressive change in the Alberta Infant Motor Scale score within a day, but, similar to the infant with isolated PREPL deficiency, there was no deterioration when pyridostigmine was stopped 2 months later (see figure e-1). The 2 older children did not respond to pyridostigmine.
Molecular genetics.
Microarray-based comparative genomic hybridization detected a heterozygous deletion in the patient at 2p21 estimated to encompass base pairs (bp) 44524771–44557935. Quantitative PCR confirmed the deletion and showed the same deletion in the mother. To fine-map the deletion, we generated and sequenced a junction fragment (figure 1E) and found a novel deletion, CAGGATCTCCTTCTGCCA (44525557) … (44559126) CCCAGGCTGGAGTGCAGTGGCACGATCTAGGCTCACTGCATGCTC. The deletion encompasses 33,568 bp and includes exons 5 to 10 of SLC3A1 and exons 9 to 14 of PREPL (figure 1G, deletion J). A 278-bp fragment at the border of the deletion was 82% complementary in SLC3A1 and PREPL. Within this fragment, a 22-bp fragment, beginning exactly at the breakpoint, was 100% complementary to SLC3A1 and PREPL. In addition, the patient harbors a paternally inherited c.807delT (p.Met270X) mutation in PREPL exon 6 (see figure 1F).
PREPL expression in muscle and at EPs.
To ascertain that PREPL expression was absent from the patient's muscle fibers and EPs, we immunostained frozen sections of patient and control muscles harboring EPs with an antibody against the C-terminal region of PREPL (green signal) and demonstrated AChR at the EPs with rhodamine-labeled α-bungarotoxin (red signal). This revealed PREPL in control muscle fibers and EPs (figure 2, A–C) and its absence in the patient's muscle fibers and EPs (figure 2, D and E). Immunoblots of extracts of patient and control muscles confirmed absence of PREPL in the patient's muscle (see figure e-2).
Figure 2. PREPL expression (A–E) and EP ultrastructure (F–I).

(A, B) PREPL (green signal) and α-bungarotoxin (red signal) localization in control muscle fibers and endplates (EPs). (C) Merged image of A and B. (D) PREPL localization in patient muscle fibers and EPs. Note absence of PREPL expression. (F) Ultrastructure of well-developed EP harboring abundant synaptic vesicles. Asterisks indicate vesicles focused on active zones. (G) Normal density and distribution of the acetylcholine receptor on junctional folds revealed by peroxidase-labeled α-bungarotoxin. (H) Abnormal nerve terminal filled with neurofilaments surrounded by degenerate organelles. (I) An EP harbors autophagic vacuoles in the junctional sarcoplasm (X). A postsynaptic region is denuded of its nerve terminal (x). The asterisk indicates a nearby nerve sprout. Bar in D = 25 μm for panels A–E. Bars in F and G = 0.5 μm; bar in H = 1.3 μm; bar in I = 2 μm.
Structural studies.
In frozen sections, the patient EPs showed robust expression of AChR (figure 2E) and acetylcholinesterase. It is interesting that in transverse sections, the patient's EPs appeared large relative to the size of the muscle fibers (figure 2E), and in longitudinal sections, the patient's EPs were 20 to 22 μm long (figure e-3), which was similar to the length of EPs in adult control muscles (20.7 ± 1.2 μm, mean ± standard error; n = 17).
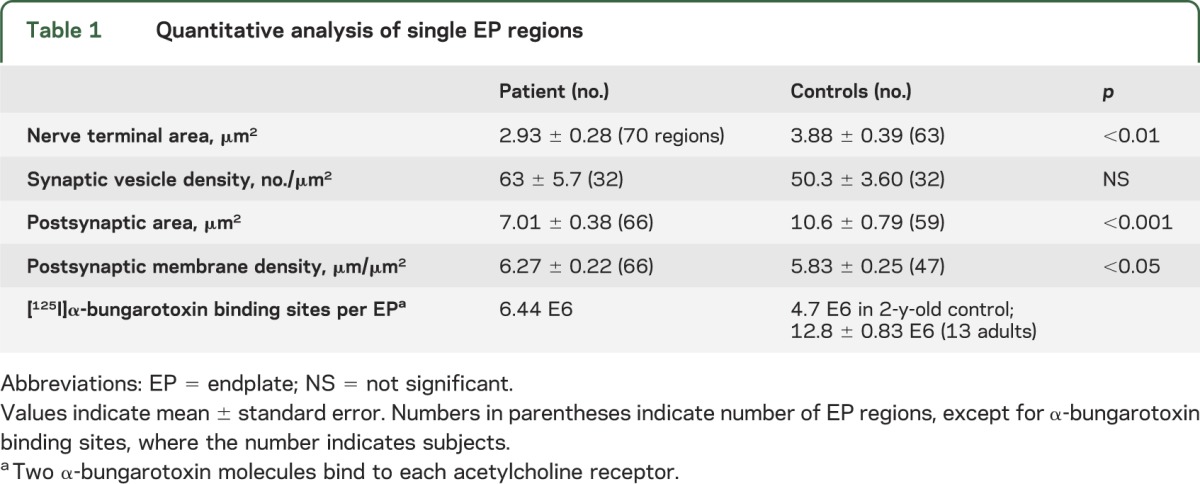
Qualitative inspection of electron microscopy images revealed well-developed EPs with nerve terminals harboring abundant synaptic vesicles (figure 2F) reacting strongly for AChR (figure 2G). Quantitative analysis of 66 regions of 35 EPs showed that the mean nerve-terminal and postsynaptic areas were, respectively, 75% and 66% of the corresponding adult control values. These findings are likely related to the smaller size of the patient muscle fibers. The postsynaptic membrane density was higher than normal. The synaptic vesicle density in the nerve terminals was normal (table 1). One EP displayed a degenerating nerve terminal (figure 2H); another EP harbored autophagic vacuoles in the junctional sarcoplasm and an abandoned postsynaptic region (figure 2I).
Table 1.
Quantitative analysis of single EP regions
Intracellular microelectrode studies and EP AChR content.
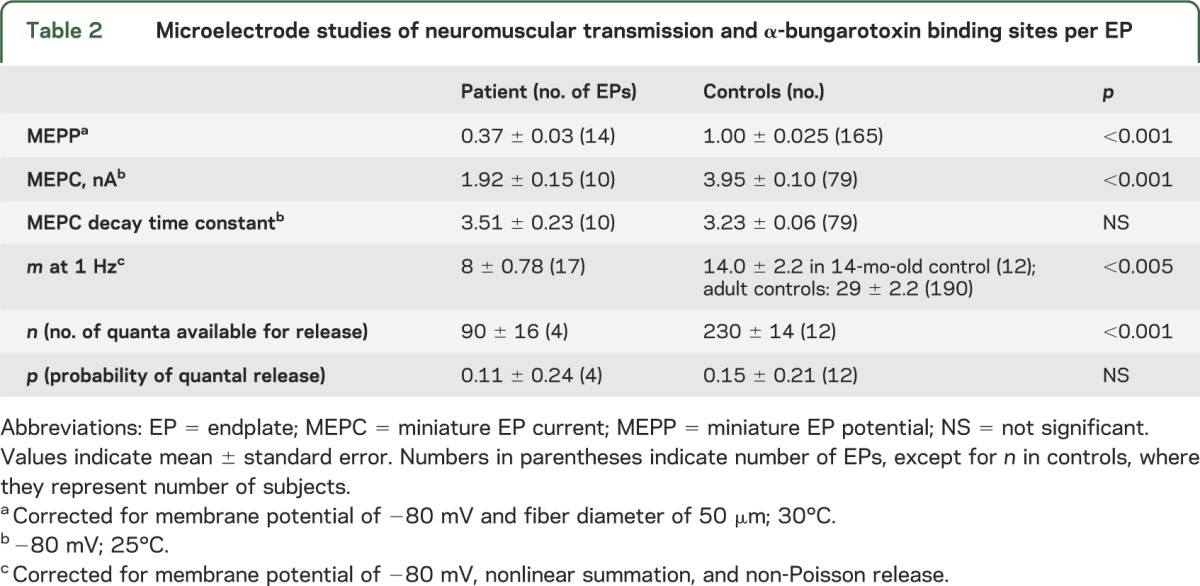
At age 6 months, the MEPP and MEPC amplitudes were reduced to approximately 40% of normal. The MEPC decay time constant was normal, indicating normal AChR channel kinetics. The quantal content of the EPP (m) was approximately half of that of a 16-month-old control. On increasing the Ca2+ concentration in the perfusing fluid from 2 to 5 mM, m increased by an amount comparable to that observed in normal controls. The reduced m was due to reduction of the number of quanta available for release (n), whereas the probability of quantal release (p) was normal. The number of [125I]α-bungarotoxin binding sites per EP was higher than that of a 2-year-old normal control (table 2).
Table 2.
Microelectrode studies of neuromuscular transmission and α-bungarotoxin binding sites per EP
DISCUSSION
We here show that the myasthenic symptoms in HCS are due to PREPL deficiency and that isolated PREPL deficiency is a monogenic cause of a novel myasthenic syndrome. The proband is compound heterozygous for a paternal nonsense mutation in PREPL (p.Met270X) and a maternal deletion mutation involving PREPL and SLC3A1. Because this patient retains a functional SLC3A1 allele, her phenotype is determined by loss of both PREPL alleles. Interestingly, the borders of the SLC3A1-PREPL deletion harbor a 278-bp region with high homology between SLC3A1 and PREPL that would facilitate nonallelic homologous recombination and provide a mechanism for the novel deletion. That the patient also has growth hormone deficiency indicates that PREPL is essential for normal growth hormone secretion.
The structural and electrophysiologic features of our patient differentiate the congenital myasthenic syndrome caused by PREPL deficiency from all heretofore identified congenital myasthenic syndromes. For example, the combined occurrence of low MEPP amplitude at rest in the face of robust AChR expression, no kinetic abnormality in AChR, reduced quantal release, and enlarged EP size excludes defects in AChR subunit genes, RAPSN, MUSK, AGRN, ColQ, ChAT, SCN4A, DOK7, GFPT1, DAPGT1, and in other genes subserving protein glycosylation.20,21
The myasthenic features of the patient with isolated PREPL deficiency and of our 3 patients with HCS prompted us to treat them with pyridostigmine. That only the 2 infants—the one with isolated PREPL deficiency and patient HCS1—responded favorably, and that they did not worsen when therapy was stopped after 9 and 2 months, respectively, suggest spontaneous improvement of the myasthenia with age. The reason for this and why the older patients are refractory to pyridostigmine await explanation.
A reduced postsynaptic response to ACh indicated by the reduced amplitudes of the MEPP and MEPC in the face of robust AChR expression and no kinetic abnormality of AChR point to a reduced ACh content of the synaptic vesicles. This could stem from a defect in presynaptic choline transport,22 ACh resynthesis, as in EP choline acetyltransferase deficiency,11 the vesicular ACh transporter,23 or the vesicular proton pump.24 Because the phenotype with reduced presynaptic choline transport is different,22 another mechanism is more likely to be pathogenic. The known function of PREPL is to act as an effector of the clathrin-associated adaptor protein 1 (AP-1) by binding to its μ1A subunit to release AP-1 from target membranes.25 Because the trafficking of the vesicular ACh transporter between the synaptic vesicle membrane and the cytosol depends on AP-1,26 absence of PREPL provides a plausible explanation for reduced filling of the synaptic vesicles with ACh.
A reduced number of quanta (n) available for release with a normal probability (p) of release can result from small synaptic contacts,27 paucity of synaptic vesicles,28 impaired transport of the synaptic vesicles to the active zones, or a defect in the molecular machinery governing exocytosis.29 Nerve terminal size at single EP regions in our patient was only mildly reduced, but the individual EPs were enlarged, the synaptic vesicle density was in the high-normal range (table 1), and abundant synaptic vesicles abutted on the presynaptic active zones (figure 2A), pointing to a defect in the late stages of exocytosis, for instance soluble N-ethylmaleimide-sensitive factor atachment protein receptor (SNARE)-dependent vesicle fusion. Enlargement of the EPs might serve to compensate for reduced quantal release for it also occurs in Lambert-Eaton syndrome in which quantal release is markedly decreased (unpublished observations by Andrew Engel).
Recently, the SNARE content of membranous vesicles delivering cargo to and from the trans-Golgi network was shown to depend on AP-1.30 Because PREPL enhances the membrane dissociation of AP-1,25 absence of PREPL may affect the SNARE content of vesicles emerging from the trans-Golgi network. However, it remains unclear how this would reduce delivery of SNAREs to the primitive synaptic vesicles assembled in the anterior horn cells for delivery to the nerve terminals by axonal transport.31,32
The degenerative changes observed at 2 EP regions were of special interest because they suggest that PREPL may also be required for maintaining normal EP architecture. Because these changes were infrequent, they are unlikely to be clinically significant at age 6 months, but the possibility exists that they were more abundant in the neonatal period when the patient's weakness was most severe.
Supplementary Material
Glossary
- ACh
acetylcholine
- AChR
acetylcholine receptor
- AP-1
adaptor protein 1
- bp
base pair
- cDNA
complementary DNA
- EP
endplate
- EPP
endplate potential
- HCS
hypotonia-cystinuria syndrome
- IGF
insulin-like growth factor
- MEPC
miniature endplate current
- MEPP
miniature endplate potential
- OMIM
Online Mendelian Inheritance in Man
- SNARE
soluble N-ethylmaleimide-sensitive factor atachment protein receptor
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Luc Régal contributed to patient evaluation, study concept and design, and data acquisition, interpretation, and analysis. X.-M. Shen contributed to study concept and data acquisition and interpretation. Duygu Selcen contributed to patient evaluation, and data acquisition and interpretation. Chantal Vertille contributed to study design, and data acquisition and interpretation. Sandra Meulemans contributed to data acquisition. John W.M. Creemers contributed to study concept and design, and data analysis and interpretation. Andrew G. Engel contributed to patient evaluation, study concept, and data acquisition, analysis, and interpretation.
STUDY FUNDING
Supported by NIH grant NS6277 to A.G.E., an FWO Vlaanderen and European Community Seventh Framework Programme grant 223077 to J.C., and Clinical Doctoral Grant from FWO Vlaanderen to L.R.
DISCLOSURE
L. Régal is supported by Clinical Doctoral Grant by FWO Vlaanderen. X.-M. Shen, D. Selcen, C. Verhille, and S. Meulemans report no disclosures relevant to the manuscript. J. Creemers has been supported by the FWO Vlaanderen and by the FP7 program of the European Community. A. Engel has served as an associate editor of Neurology® during the past year and is supported by a research grant from the NIH. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jaeken J, Martens K, Francois I, et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am J Hum Genet 2006;78:38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Regal L, Aydin HI, Dieltjens AM, Van Esch H, et al. Two novel deletions in hypotonia-cystinuria syndrome. Mol Genet Metab 2012;107:614–616 [DOI] [PubMed] [Google Scholar]
- 3.Martens K, Heulens I, Melumans S, et al. Global distribution of the most prevalent deletions causing hypotonia-cystinuria syndrome. Eur J Hum Genet 2007;15:1029–1033 [DOI] [PubMed] [Google Scholar]
- 4.Eggermann T, Spengler S, Venghaus A, et al. 2p21 deletions in hypotonia-cystinuria syndrome. Eur J Med Gen 2012;55:561–563 [DOI] [PubMed] [Google Scholar]
- 5.Parvari R, Gonen Y, Alshafee I, Buriakovsky S, Regev K, Hershkovitz E. The 2p21 deletion syndrome: characterization of the transcription content. Genomics 2005;86:195–211 [DOI] [PubMed] [Google Scholar]
- 6.Chabrol B, Martens K, Meulemans S, et al. Deletion of C2orf34, PREPL and SLC3A1 causes atypical hypotonia-cystinuria syndrome. J Med Genet 2008;45:314–318 [DOI] [PubMed] [Google Scholar]
- 7.Boonen K, Regal L, Jaeken J, Creemers J. PREPL: a putative novel oligopeptidase-like enzyme by name only? Lessons from patients. CNS Neurol Disord Drug Targets 2011;10:355–360 [DOI] [PubMed] [Google Scholar]
- 8.Martens K, Derua R, Meulmans S, Waelkens E, Jaeken J, Creemers JW. PREPL: a putative novel oligopeptidase propelled into the limelight. Biol Chem 2006;387:879–883 [DOI] [PubMed] [Google Scholar]
- 9.Bartholdi D, Asadollahi R, Oneda B, et al. Further delineation of genotype-phenotype correlation in homozygous 2p21 deletion syndromes: first description of patients without cystinuria. Am J Med Genet A 2013;161:1853–1859 [DOI] [PubMed] [Google Scholar]
- 10.Selcen D, Stilling G, Engel AG. The earliest pathologic alterations in dysferlinopathy. Neurology 2001;56:1472–1481 [DOI] [PubMed] [Google Scholar]
- 11.Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA 2001;98:2017–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fambrough DM, Engel AG, Rosenberry TL. Acetylcholinesterase of human erythrocytes and neuromuscular junctions: homologies revealed by monoclonal antibodies. Proc Natl Acad Sci USA 1982;79:1078–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel AG. Quantitative morphological studies of muscle. In: Engel AG, Franzini-Armstrong C, editors. Myology, 2nd ed New York: McGraw-Hill; 1994:1018–1045 [Google Scholar]
- 14.Engel AG, Lindstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 1977;27:307–315 [DOI] [PubMed] [Google Scholar]
- 15.Engel AG. The investigation of congenital myasthenic syndromes. Ann NY Acad Sci 1993;681:425–434 [DOI] [PubMed] [Google Scholar]
- 16.Elmqvist D, Quastel DMJ. A quantitative study of end-plate potentials in isolated human muscle. J Physiol 1965;178:505–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engel AG, Nagel A, Walls TJ, Harper CM, Waisburg HA. Congenital myasthenic syndromes: I: deficiency and short open-time of the acetylcholine receptor. Muscle Nerve 1993;16:1284–1292 [DOI] [PubMed] [Google Scholar]
- 18.Uchitel O, Engel AG, Walls TJ, Nagel A, Bril V, Trastek VF. Congenital myasthenic syndrome attributed to an abnormal interaction of acetylcholine with its receptor. Ann NY Acad Sci 1993;681:487–495 [DOI] [PubMed] [Google Scholar]
- 19.Kamenskaya MA, Elmqvist D, Thesleff S. Guanidine and neuromuscular transmission: II: effect on transmitter release in response to repetitive nerve stimulation. Arch Neurol 1975;32:510–518 [DOI] [PubMed] [Google Scholar]
- 20.Engel AG. Congenital myasthenic syndromes. In: Engel AG, editor. Myasthenia Gravis and Myasthenic Syndromes, 2nd ed New York: Oxford University Press; 2012:173–230 [Google Scholar]
- 21.Engel AG, Shen XM, Selcen D, Sine SM. New horizons for congenital myasthenic syndromes. Ann NY Acad Sci 2012;1275:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barwick KES, Wright J, Al-Turki S, et al. Defective presynaptic choline transport underlies hereditary presynaptic motor neuropathy. Am J Hum Genet 2012;91:1103–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prado VF, Martins-Silva C, de Castro BM, et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 2006;51:601–612 [DOI] [PubMed] [Google Scholar]
- 24.Usdin TB, Eiden LE, Bonner TI, Erickson JD. Molecular biology of the vesicular ACh transporter. Trends Neurosci 1995;18:218–224 [DOI] [PubMed] [Google Scholar]
- 25.Radhakrishnan K, Baltes J, Creemers JWM, Schu P. Trans-Golgi network morphology and sorting is regulated by prolyl-oligopeptidase-like protein PREPL and AP-1 complex subunit μ1A. J Cell Sci 2013;126:1155–1163 [DOI] [PubMed] [Google Scholar]
- 26.Kim MH, Hersh LB. The vesicular acetylcholine transporter interacts with clathrin-associated adaptor complexes AP-1 and AP-2. J Biol Chem 2004;279:12580–12587 [DOI] [PubMed] [Google Scholar]
- 27.Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol 1977;1:315–330 [DOI] [PubMed] [Google Scholar]
- 28.Walls TJ, Engel AG, Nagel AS, Harper CM, Trastek VF. Congenital myasthenic syndrome associated with paucity of synaptic vesicles and reduced quantal release. Ann NY Acad Sci 1993;681:461–468 [DOI] [PubMed] [Google Scholar]
- 29.Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature 2012;490:201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirst J, Borner GHH, Antrobus R, et al. Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr Biol 2012;22:1711–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiene ML, Stadler H. Synaptic vesicles in electromotoneurones: I: axonal transport, site of transmitter uptake and processing of a core proteoglycan during maturation. EMBO J 1987;6:2209–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sgro AE, Bajjalieh SM, Chiu DT. Single-axonal organelle analysis method reveals new protein-motor associations. ACS Chem Neurosci 2013;4:277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.