

Abstract
Ten protein kinase C (PKC) isozymes play divergent roles in signal transduction. Because of sequence similarities, it is particularly difficult to generate isozyme-selective small molecule inhibitors. In order to identify such a selective binder, we derived a pharmacophore model from the peptide EAVSLKPT, a fragment of PKCε that inhibits the interaction of PKCε and receptor for activated C-kinase 2 (RACK2). A database of 330 000 molecules was screened in silico, leading to the discovery of a series of thienoquinolines that disrupt the interaction of PKCε with RACK2 in vitro. The most active molecule, N-(3-acetylphenyl)-9-amino-2,3-dihydro-1,4-dioxino[2,3-g]thieno[2,3-b]quinoline-8-carboxamide (8), inhibited this interaction with a measured IC50 of 5.9 μM and the phosphorylation of downstream target Elk-1 in HeLa cells with an IC50 of 11.2 μM. Compound 8 interfered with MARCKS phosphorylation and TPA-induced translocation of PKCε (but not that of PKCδ) from the cytosol to the membrane. The compound reduced the migration of HeLa cells into a gap, reduced invasion through a reconstituted basement membrane matrix, and inhibited angiogenesis in a chicken egg assay.
Introduction
Protein kinase C (PKC) is a family of serine/threonine-specific protein kinases. The PKC isozymes can be classified into three groups: (i) the conventional α, βI, βII, γ; (ii) the novel δ, ε, θ, η, and (iii) the atypical λ/ι (mouse/human) and ζ. PKC isozymes seem to play important roles in the activation of signal transduction pathways leading to synaptic transmissions, the activation of ion fluxes, secretion, proliferation, cell cycle control, differentiation, and tumorigenesis. Because of their role in a complex network of signal transduction pathways, different isoforms have divergent, sometimes opposite roles within a given biological process. As a result, no simple, unique function can be assigned to a given PKC isozyme.
The PKCε isozyme has been reported to participate in neoplastic transformation,1 cardiac hypertrophy,2 protection from ischemic insult,2,3 nociceptor function,4 macrophage activation,5 diabetes,6 and alcohol consumption.7 A PKCε isozyme-specific inhibitor would be a valuable tool for analyzing the function of PKCε and is expected to have pharmaceutical potential for cancer, stroke, drug addiction, or pain.8,9 Typically, kinase inhibitors interact with the ATP-binding site, which is well conserved among different kinase families and is even more so within isoforms of a given kinase. This poses a serious hurdle for the development of isozyme-specific inhibitors, as there are about 500 kinases encoded by the human genome.10 Although several selective kinase inhibitors have been reported, it was later found that they also inhibit other targets. For example, the marketed drug imatinib was developed as an inhibitor of the oncoprotein Bcr-Abl. However, it has turned out to inhibit also other tyrosine kinases such as Kit and platelet-derived growth factor receptor, as well as non-kinase targets. Similarly, relatively unspecific inhibitors initially intended as PKCβ-selective, such as ruboxistaurine11,12 and enzastaurine,13 are in clinical trials for diabetic retinopathy and cancer, respectively. Rottlerin was described as specific inhibitor of PKCδ. However, additional modes of action have also been observed more recently for this compound.14
Because of the high degree of conservation among the ATP-binding sites in PKC isozymes, it is obviously very difficult to develop monospecific inhibitors. While monospecificity is not a prerequisite for a successful drug, secondary activities create ambiguity in the interpretation of biochemical, pharmacological, and clinical results.
We employed a different approach to specifically target PKCε signaling by preventing the binding of PKCε to its adaptor protein, the PKCε-specific receptor of activated C-kinase 2 (RACK2, β′COP).15,16 It has been shown that adaptor proteins bind and translocate activated PKC isozymes to subcellular sites in proximity to their substrates.17 RACK2 binds to activated PKCε. The peptide EAVSLKPT, corresponding to amino acids 14–21 of PKCε, selectively inhibits the translocation of PKCε but not that of other PKC isozymes by binding to RACK2.3,18 It has also been shown that the PKCε antagonist EAVSLKPT inhibits protection from hypoxia-induced cell death of cardiac myocytes.18 One example of an inhibitor of such protein–protein interactions is aurothiomalate, which interferes with the interaction between PKCι and its adaptor molecule Par6. The compound blocks oncogenic PKCι signaling and growth of human lung cancer cells.19
The objective of the current work was to develop a small molecule peptidomimetic of EAVSLKPT that prevents the PKCε/RACK2 interaction. It has been shown that small molecule inhibitors of protein–protein interactions are useful for pharmacological purposes.20,21 A cell-permeable inhibitor of PKCε signal transduction could help to explain the exact function of PKCε and could probably also be developed further for pharmaceutical purposes.
Results
Molecular Modeling
The lack of any known nonpeptidic inhibitor of the PKCε/RACK2 interaction and the very limited structural data available render rational drug discovery of an inhibitor of this protein–protein interaction a difficult task. A crystal structure is only available for the N-terminal-C2-like domain of PKCε (PDB entry 1GMI), but no structural data are available on RACK2. The PKCε protein segment EAVSLKPT, which as a peptide is known to disrupt PKCε/RACK2 interaction, is highly solvent-exposed and expected to undergo conformational changes during the interaction with RACK2. The interface is large (with respect to the size of small molecule inhibitors), only vaguely characterized, and interaction hot spots remain speculative. Conformations observed for EAVSLKPT (in particular Glu14 and Lys19; numbering according to the crystal structure) in the crystal structure are likely to be influenced by crystal packing.
Lacking known ligands and structural data for modeling, several pharmacophore models were derived directly from EAVSLKPT in its crystal structure conformation (Figure 1A). One pharmacophore model obtaining comparably high specificity is reported in Figure 1B. It consists of three hydrogen bonding features and one hydrophobic feature. The latter attempts to resemble the hydrophobic area presented to RACK2 by Val16, while the hydrogen bonding features characterize the side chain properties of Glu14, Ser17, and Lys19. The rationale for selecting these features was their surface exposure and, consequently, an increased likelihood of being involved in the interaction with RACK2. In order to account for protein flexibility (as far as possible and without becoming overly unspecific), the tolerance spheres of the pharmacophore feature and projection points were set to 1.5 and 2.0 Å, respectively (the latter are not indicated in Figures 1B and 1D). This pharmacophore model was used for virtual screening of the Asinex Gold and Platinum collections (Asinex Ltd., Moscow, Russia), which contain approximately 330 000 compounds in total.
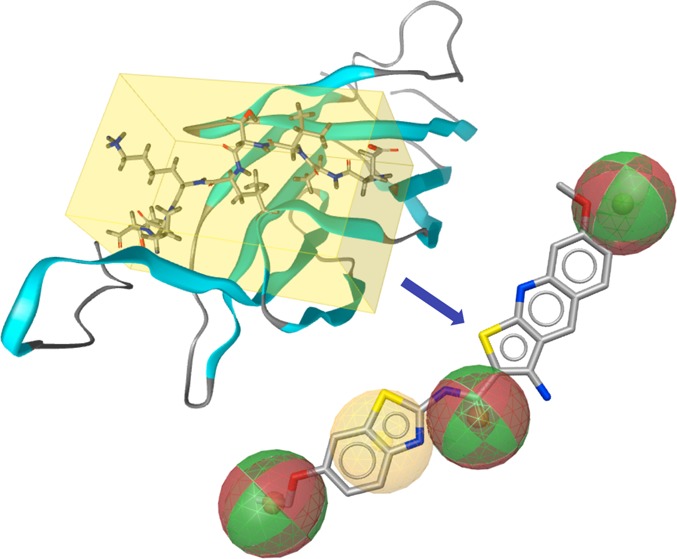
Figure 1.

Structure-based modeling of the mimic of the PKCε protein fragment EAVSLKPT. (A) X-ray structure of the C2 domain of PKCε (PDB code 1GMI). The protein backbone is illustrated as cartoon, while the protein segment EAVSLKPT is highlighted as sticks in the yellow box. (B) Pharmacophore model derived from EAVSLKPT (extracted from the crystal structure). Hydrogen bonding and hydrophobic features are indicated with red/green and yellow spheres, respectively. (C) Structures of 1 and 8. (D) Alignment of 1 with the pharmacophore model. (A), (B), and (D) were generated using LigandScout 3.0.42
Virtual screening with this pharmacophore model resulted in a list of 468 molecules. The hit list was refined by ranking candidate molecules according to their geometric fit to the pharmacophore model and by visual inspection. Nineteen compounds (Supporting Information Table 1) were selected and purchased for in vitro testing. One of them, 3-amino-N-(6-ethoxy-1,3-benzothiazol-2-yl)-7-methoxythieno[2,3-b]quinoline-2-carboxamide (1), showed significant activity. It disrupted the interaction of PKCε and RACK2 with an IC50 of 25.5 μM (Figure 1C, Figure 1D). Nineteen further thienoquinolines, structurally related to 1, were purchased or synthesized for in vitro testing.
Structure–Activity Relationships Observed for Thienoquinoline-Based Disruptors of the PKCε/RACK2 Interaction
Significant disruption of the PKCε/RACK2 interaction was detected for several tested thienoquinolines (Table 1), with the most active compound being N-(3-acetylphenyl)-9-amino-2,3-dihydro-1,4-dioxino[2,3-g]thieno[2,3-b]quinoline-8-carboxamide (8) (Figure 1C). Acetylphenyl substituents at R1 appear favorable for activity, with a preference for meta- over para-substitution (as exemplified by 8 and 13). Also the amine group at R4 increases activity (see 8 and 11). Compounds with a hydrogen bond acceptor at R3 tend to exhibit significant activity (such as 3, 6, 8, 13).
Table 1. Structure and Activity of Tested Thienoquinolines.
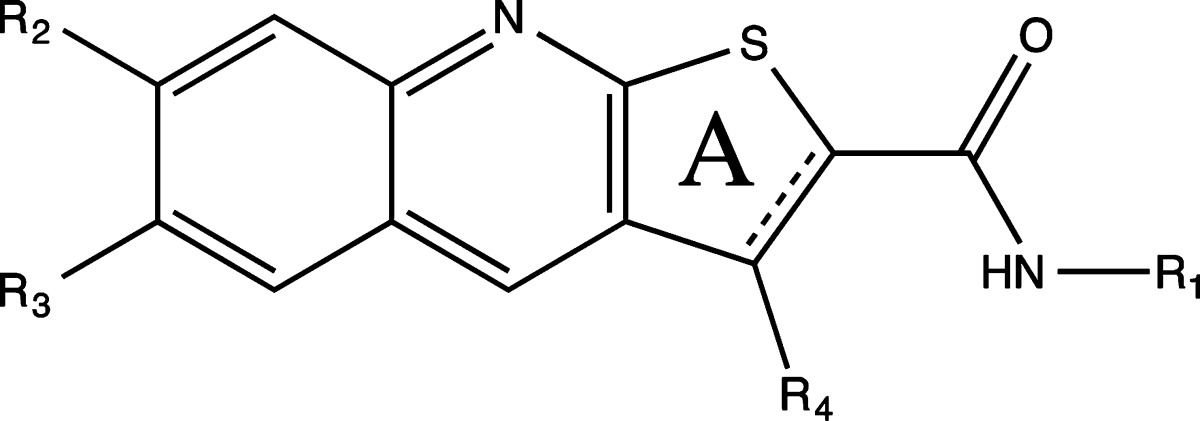
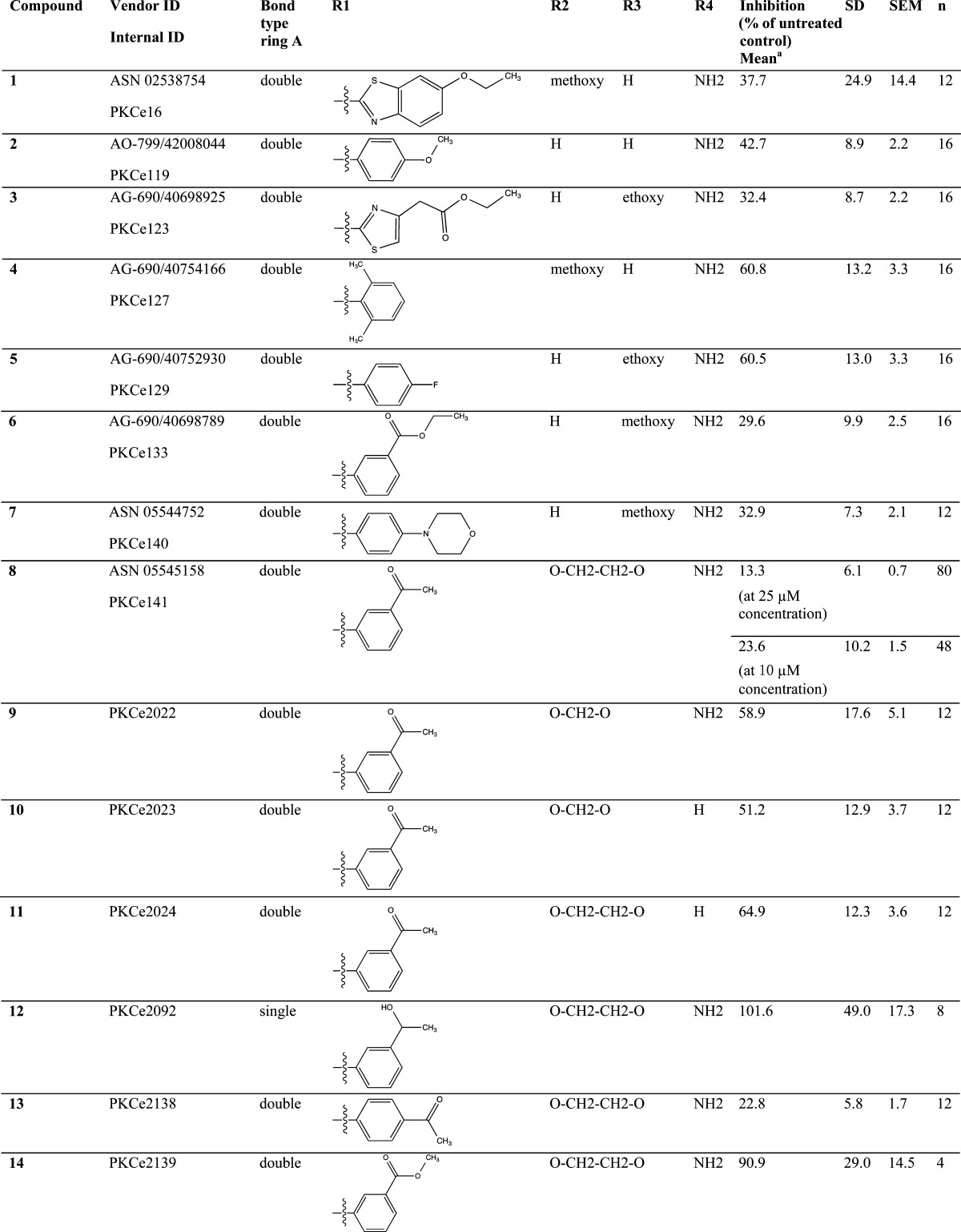
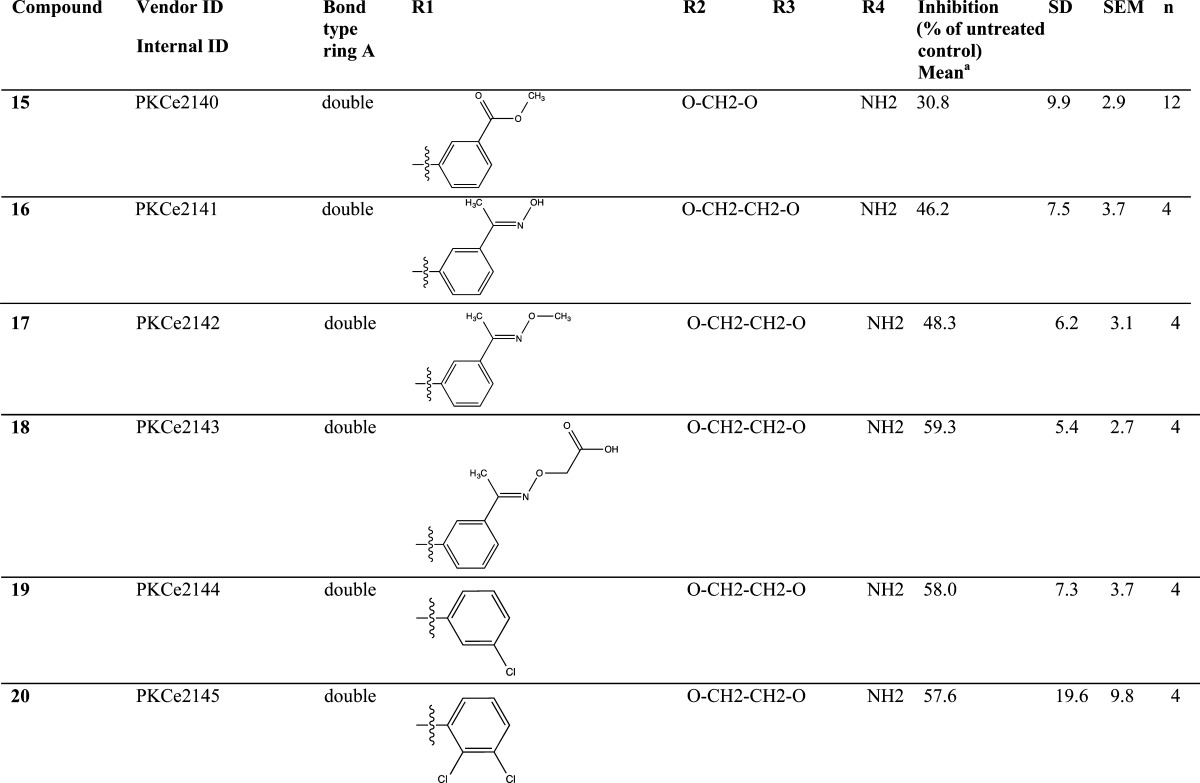
PKCε/RACK2 in vitro binding assay.
A pharmacophore-based alignment of 8 with the peptide itself might allow the formulation of a binding mode hypothesis. Compound 8 may cover the pharmacophore features of the solvent-exposed side chains of EAVSLKPT. However, these considerations remain highly speculative, and hopefully more data on structure–activity relationships will help to shed some light on the potential molecular binding mode of 8 in the future.
Compound 8 Prevents PKCε/RACK2 Interaction in Vitro
Compound 8 led to a dose-dependent inhibition of the PKCε/RACK2 interaction (Figure 2A) and exhibited an IC50 of 5.9 μM. The peptide EAVSLKPT-RRRRRRR was used as a control. Seven arginines were added to EAVSLKPT to increase internalization of the peptide for experiments in intact cells. IC50 for the inhibition of the PKCε/RACK2 interaction by EAVSLKPT-RRRRRRR in this assay was 1.02 μM (Supporting Information Figure 1). It has been shown previously that PKCβII interacts with the adaptor protein RACK1.22 Therefore, we investigated whether 8 can also prevent the PKCβII/RACK1 interaction. As shown in Figure 2B, compound 8 does not prevent the PKCβII/RACK1 interaction, indicating specificity of 8 for the PKCε/RACK2 interaction.
Figure 2.

Effects of 8 in vitro. (A) Compound 8 prevents the in vitro interaction of PKCε with RACK2 in a dose-dependent manner. (B) Compound 8 does not prevent the in vitro interaction between PKCβII and RACK1. In (A) and (B), the mean values (±SD) of four independent experiments in which four samples were taken within each experiment are shown. Because the color development varies from experiment to experiment, the data were normalized to the control (100%). Therefore, the standard deviation for the control is 0.
Effect of Compound 8 on PKCε Translocation
Upon activation, PKCε associates with RACK2 and is translocated from the cytosolic to the membrane fraction.23,24 We investigated whether 8 also inhibits the translocation of PKCε to the membrane. For these experiments PC-3 prostate adenocarcinoma cells were employed because they express relatively high levels of endogenous PKCε (Figure 3). In PC-3 cells, treatment with 12-O-tetradecanoylphorbol 13-acetate (TPA) led to an increase of PKCε in the membrane fraction. Accordingly, the amount of PKCε in the cytosol decreased (Figure 3). Furthermore, activated PKCε is phosphorylated at Ser729. TPA increased the phosphorylated form of PKCε. Compound 8 does not inhibit PKCε in vitro (Supporting Information Figure 2). Treatment of cells with 8 similarly did not reduce Ser729-phosphorylated PKCε at the membrane (Figure 3). This result illustrates that PKCε is phosphorylated following treatment with TPA and 8 does not prevent this phosphorylation. However, compound 8 partially inhibits the cellular translocation of activated and phosphorylated PKCε to the membrane fraction. PKCδ shows a high degree of homology with PKCε. Therefore, we investigated the influence of 8 on PKCδ. As shown in Figure 3, compound 8 did not decrease PKCδ in the membrane. A similar result was obtained with immunocytochemistry (Figure 4). Short-term treatment with TPA led to an increase of PKCε in the plasma membrane. Compound 8 prevented the TPA-induced PKCε translocation to the plasma membrane (Figure 4).
Figure 3.

Effect of 8 on PKCε and PKCδ translocation. IGFI-βR and GAPDH were used as loading controls for the membrane and the cytosolic fraction, respectively. In the densitometric analysis, four independent experiments were scanned and the mean values (±SD) are shown: (∗) p = 0.02. Depending on the exposure time of the membrane to the film, the bands vary from experiment to experiment. Therefore, the data were normalized to the control (100%) and no standard deviation can be reported.
Figure 4.

Effect of 8 on PKCε translocation by immunofluorescence. PC-3 cells were employed for these experiments. TPA induces translocation of PKCε to the plasma membrane. Compound 8 inhibits this TPA-induced translocation of PKCε. Experiments 1, 2, and 3 are three independent experiments.
Compound 8 Inhibits Elk-1 and MARCKS Phosphorylation in Vitro
PKCε is situated in the signal transduction cascade upstream of Raf-1.25 In a HeLa cell line containing a doxycycline-inducible constitutively active PKCε, active PKCε leads to phosphorylation of the transcription factor Elk-126 (Figure 5A, Figure 5B). To obtain information on whether 8 is able to prevent the PKCε/RACK2 interaction in intact cells, we tested the compound for inhibition of Elk-1 phosphorylation in a PathDetect HeLa luciferase (HLR) trans-reporting HeLa cell line. In these cells, activation of PKC by TPA leads to the expression of luciferase. As shown in Figure 5C, compound 8 inhibited the phosphorylation of Elk-1 in a dose-dependent manner with an IC50 of 11.2 μM in intact cells.
Figure 5.

PKCε induces phosphorylation of Elk-1. (A) In HeLa cells, a doxycycline-inducible constitutively active PKCε27 leads to phosphorylation of Elk-1. The cells were left untreated or induced with doxycycline (2 μg/mL) for 24 h. Additional stimulation with TPA (50 nM) was performed for 10 min. Phosphorylation of Elk-1 was detected with a phospho-specific antibody against the Ser383 residue of Elk-1. GAPDH was used as loading control. (B) Densitometric analysis of Western blots. Elk-1 phosphorylation was normalized to the GAPDH loading control, and data are expressed relative to untreated cells (control). Bar graphs represent quantitation of three independent experiments (±SD). Dox = doxycycline. Fold of control is calculated from Dox and TPA. (C) Compound 8 inhibits the phosphorylation of Elk-1 in PathDetect HeLa-HLR cells. Luciferase activity following activation of Elk-1 is shown. Data shown are the mean values (±SD) of three independent experiments. Because the relative light units vary from experiment to experiment, the data were normalized to the controls. Therefore, the standard deviation for the control is 0.
In cells, PKCε phosphorylates myristoylated alanine-rich protein kinase C substrate (MARCKS).26 As shown in Figure 6A, compound 8 inhibited the phosphorylation of MARCKS in HeLa cells. These results show that 8 exhibits the features of an inhibitor of PKCε signaling in vitro and also in intact cells.
Figure 6.

Compound 8 inhibits MARCKS phosphorylation, cell proliferation, and invasion following treatment with 8. (A) HeLa cells were starved for 16 h in medium without fetal calf serum. Subsequently, they were treated with 50 μM solution of compound 8 for 30 min and 100 nM TPA for the last 10 min. An amount of 150 μg of protein from lysed cells was loaded onto SDS 8–16% gels. Tubulin was used as loading control. Below a representative blot, a bar graph corresponding to quantitative scans of three independent experiments (±SD) is shown. (B) Cell proliferation following treatment of HeLa and PC-3 cells with 8. The mean values (±SD) of three independent experiments, in which three samples were taken within each experiment, are shown. (C) Invasion of cells through a membrane. Cell invasion was determined as described in Experimental Section. In HeLa cells with a doxycycline-inducible PKCε the expression of PKCε was induced with doxycycline (control + Dox). The mean of three experiments (±SD) is shown: (∗) p = 0.03. The data are normalized to the control (100%). Therefore, the standard deviation for the control is 0.
Effects of Compound 8 on Cell Proliferation, Migration, Invasion, and Angiogenesis
A major question is whether such an inhibitor is toxic or in other words whether the observed effects are due to inhibition of cell proliferation rather than of Elk-1 mediated signaling. Therefore, we tested the compound for inhibition of cell proliferation in HeLa-HLR and human PC-3 prostate adenocarcinoma cells. These cell lines were used because HeLa-HLR cells were employed for Elk-1 phosphorylation and PC-3 cells for the PKCε translocation experiments described above. As shown in Figure 6B, in both of the cell lines even 50 μM 8 did not show any inhibition of cell proliferation. It has been shown previously that PKCε does not increase cell proliferation. However, it increases cell migration8,26 and is associated with metastatic spread and invasiveness of human cancer cells.27 Therefore, we investigated whether 8 inhibits PKCε-induced cell migration or invasion. Invasion of cells through an extracellular matrix was indeed inhibited by 8 (Figure 6C). As shown in Figure 7, untreated HeLa cells containing a constitutively active doxycycline-inducible PKCε showed only low migration into a scratch made with a pipet tip on a tissue culture dish. If the scratch was made into the monolayer cells and the expression of constitutively active PKCε was induced by doxycycline, after 24 h a significant part of the scratch was covered with cells. PKCε increased migration of cells into a gap, and 8 indeed inhibited this PKCε-induced migration (Figure 7). This result is interesting for cancer research. Another question in the cancer area is whether the compound inhibits angiogenesis. As shown in Figure 8, in a chicken egg assay the induction of angiogenesis by vascular endothelial growth factor is indeed inhibited.
Figure 7.

PKCε-induced migration of HeLa cells into a gap. The expression of constitutively active PKCε was induced by doxycycline for 24 h after a scratch was made into monolayer cells with a pipet tip.
Figure 8.

Inhibition of angiogenesis by 8. Angiogenesis was observed in a chicken egg assay with and without treatment with 8. Arrows indicate blood vessels.
Inhibition of Kinases
Earlier, we found that the barbituric acid derivative BAS 02104951 prevented PKCε/RACK2 interaction (IC50 = 28.5 μM). In addition, this compound also inhibited PKCη and PKCε directly.28 Therefore, we tested 8 for its inhibition of PKC isozymes. As shown in Supporting Information Figure 2, 50 μM compound inhibited PKCs α, βI, and η to approximately 80%. If compared to the inhibition of the PKCε/RACK2 interaction (IC50 = 5.90 μM), this is not a major effect. However, these are significant inhibitions, in particular for PKCη. This has to be considered in further investigations into this compound.
A screen of 109 kinases showed that many kinases are not or only slightly affected by 8 (Table 2). Among the kinases inhibited by 8 are ERK1, NUAK1 PIM3, BTK, and RSK2. However, compound 8 affected these kinases less than the PKCε/RACK2 interaction. As shown in Figure 2A, 25 μM solution of 8 inhibited the PKCε/RACK2 interaction to approximately 10% of untreated controls, whereas 25 μM solution of 8 inhibited the most affected kinase RSK2 to 27% (Table 2). All other kinases were less affected.
Table 2. Profile of Kinase Inhibition by 25 μM Solution of 8a.
| % of control | SD, % | |
|---|---|---|
| 25 μM Solution of 8 | ||
| control | 100 | 0 |
| ERK1 | 55 | 5 |
| RSK2 | 27 | 1 |
| NUAK1 | 49 | 6 |
| PIM3 | 46 | 4 |
| BTK | 42 | 1 |
109 different protein kinases were tested for their inhibition by 8. The five kinases that were inhibited most strongly are reported in this table. All other kinases were affected less. Most of them are not affected at all. Screening was performed by the National Centre for Protein Kinase Profiling, Division of Signal Transduction Therapy, University of Dundee. The data are portrayed as mean % activity and standard deviation of assay duplicates.
Discussion
PKC isozymes seem to play important roles in several signal transduction pathways. The exact functions of the different PKCs in the cells are still being actively investigated. Isozyme-selective inhibitors would contribute to explain their functions. Kinase inhibitors usually bind to the ATP binding site. The corresponding sequences, in particular in PKC isozymes, exhibit strong structural similarities. Therefore, kinase inhibitors frequently are not selective for a particular kinase. In order to identify a selective inhibitor of PKCε signaling, we sought to prevent the PKCε/RACK2 interaction. A pharmacophore-based approach utilizing the crystal structure of the C2 domain of PKCε was employed to identify a candidate molecule (1, Figure 1C), which is based on a thienoquinoline core. Further thienoquinolines related to the structure of 1 were explored, leading to the discovery of 8, which prevents this interaction with a half-maximal inhibitory concentration of 5.9 μM. Although 8 inhibits other kinases to a certain extent (Figure 2A, Table 2), this level of selectivity is acceptable for an inhibitor lead. In addition, detergent-sensitive aggregation effects may also be involved at such high concentrations of inhibitors.29 Medicinal chemistry-driven optimization of this lead structure is expected to lead to a substantial improvement of inhibitor specificity.
The aim of developing an inhibitor of signal transduction is ultimately to use it for therapeutic purposes. Many enzyme inhibitors used as drugs exhibit an IC50 in the nanomolar range. However, because of their particular physicochemical properties (extensive, hydrophobic, surface-exposed interfaces), protein–protein interfaces are more difficult to target than classical ligand-binding sites. Attempts to tackle protein–protein interactions have been reported in recent years, but experience is still quite limited and experimental aspects are challenging as well.30 The EAVSLKPT peptide, derived from the binding site of PKCε to RACK2 and used for molecular modeling, inhibits this interaction with an IC50 of 1.02 μM. Therefore, a peptidomimetic of EAVSLKPT with an IC50 of 5.9 μM may be approaching the maximum achievable inhibitory activity, considering the size of this molecule. Also aurothiomalate, which prevents the interaction of PKCι and its adaptor protein Par6, is active in the range of 10 μM.19 For clinical application a compound with an IC50 in the nanomolar range would be preferable. However, this is not essential. For example, the ribonucleotide reductase inhibitor hydroxyurea is used in the clinic as an anticancer agent. It inhibits ribonucleotide reductase with an IC50 of 37.2 μM.31 Furthermore, many clinically relevant kinase inhibitors that are active at nanomolar concentrations in vitro have cellular IC50 values closer to the micromolar range because of the higher physiological concentrations of ATP relative to those typically used for in vitro assays. In particular for compounds with high IC50 values solubility might be a problem. However, for 8 this seems solvable.
It has been reported previously that PKCε appears to have oncogenic properties.1 Therefore, an inhibitor of the PKCε signal transduction might exhibit antitumor activity. However, as shown in Figure 6B, the proliferation of HeLa and PC-3 tumor cells was not inhibited by 8, which illustrates that the compound seems not to exhibit general antitumor activity, although mechanism-based anticancer applications cannot be ruled out in tumors in which PKCε activation is associated with the driving mutation. The data also indicate that the compound does not exhibit toxicity, a particularly favorable characteristic for a potential application as an inhibitor of PKCε signaling to treat diseases such as myocardial hypertrophy,32,33 diabetes,6 stroke, or pain.8,9 Our results are in agreement with reports27,34 showing that PKCε does not increase cell proliferation (Figure 6B) but is involved in cell migration and invasion (Figures 6C and 7). This is an indication that 8 might be an inhibitor of metastasis. The potential use of the compound for pharmaceutical purposes has to be examined in additional projects.
Conclusions
Sequence similarities within the 10 members of the PKC family render the development of isozyme-selective small molecule inhibitors of PKCε a highly challenging task. In this study, our aim was to develop a molecule blocking the activity of PKCε by preventing it from binding to its adaptor protein, RACK2. We successfully identified a cluster of active molecules based on a thienoquinoline ligand core. The most active molecule, compound 8, interferes with the PKCε/RACK2 interaction with an activity comparable to that of a known peptidic inhibitor of this interface (EAVSLKPT). Compound 8 inhibits the phosphorylation of the PKCε-downstream target Elk-1, the phosphorylation of MARCKS, and the TPA-induced translocation of PKCε (but not that of its nearest relative PKCδ) from the cytosol to the membrane. It reduces cell migration into a gap and invasion through a reconstituted basement membrane matrix and inhibits angiogenesis in a chicken egg assay. We believe that the herein reported disruptor will be useful for investigating the function of PKCε and may serve as an interesting starting point for the development of drug molecules targeting this protein with adequate specificity.
Experimental Section
Compounds
For in vitro screening purposes, all 19 initially selected compounds (including compound 1, reported in Supporting Information Table 1) were purchased from Asinex Ltd., Moscow, Russia, together with compounds 7 and 8. Compounds 2–6 were purchased from Specs, Delft, The Netherlands. Compounds 9–20 were synthesized in house. The 25 mM stock solutions were prepared in DMSO or in DMF. This stock was incubated at 56 °C for 1 h to dissolve the compound completely.
Characterization of Compound 8
For biological characterization of compound 8 a fully characterized batch synthesized in house and checked for identity and purity was used. Mp >250 °C (dec) (crystallized from DMF–MeOH). 1H NMR (DMSO-d6): δ 2.58 (s,3H), 4.42 (d, J = 6 Hz, 4H), 7.45–7.49 (m,3H), 7.55 (s,2H), 7.67 (d, J = 6 Hz, 1H), 8.01 (d, J = 6 Hz, 1H), 8.34 (s, 1H), 8.89 (s, 1H), 9.58 (s, 1H). HRMS-ESI calcd (C22H17N3O4S + H), 420.101 82; found, 420.1043 (M + H)+; 442.0866 (M + Na)+; 861.1839 (2M + Na)+. HPLC, 95.05% (H2O + H3PO4, 0.01 M, pH 2.6/MeCN). Identity and purity (>95%) of 8 was determined using thin-layer chromatography and liquid chromatography–mass spectrometry. 1H NMR, ESI-MS, and HPLC analyses of 8 are shown in the Supporting Information Figures 3–6.
Computational Approaches
PDB entry 1GMI was used as a basis for the definition of structure-based pharmacophores of the PKCε protein fragment EAVSLKPT. Catalyst (version 4.11, Accelrys Inc., San Diego, CA, USA) was employed for pharmacophore modeling and screening. Multiconformational databases of the Asinex Gold and Platinum libraries (Asinex Ltd., Moscow, Russia) were generated applying the FAST conformational search algorithm with a maximum of 250 conformers created per molecule. The two databases were screened using the Catalyst FAST algorithm.
Cells and Cell Proliferation
HeLa cells containing a tetracycline/doxycycline-inducible constitutively active PKCε were described previously.26 A PathDetect HeLa HLR cell line was obtained from Agilent, Technologies, Wokingham, U.K. PC-3 prostate adenocarcinoma cells were obtained from Dr. Helmut Klocker, Department of Urology, Innsbruck Medical University. For cell proliferation HeLa HLR and PC-3 cells were seeded at ∼10 000 cells per well in 96-well plates. After 4 h various concentrations of 8 were added and left for 72 h. Cell proliferation was determined by the SRB assay.35
PKCε/RACK2 in Vitro Binding Assay
Recombinant RACK2 tagged with maltose-binding protein (RACK2-MBP) was purified on columns with amylose resin (New England Biolabs, Ipswich, MA) as described by the manufacturer. The recombinant protein was analyzed by Coomassie Blue staining and Western blotting after SDS–PAGE. Aliquots were stored in liquid nitrogen. The interaction between PKCε and RACK2 was measured using an ELISA-based assay. The 96-well EIA/RIA high binding plates (Costar) were coated with 100 ng of recombinant PKCε (ProQinase, Freiburg, Germany) in buffer A (20 mM Tris-HCl/100 mM NaCl, pH 7.5) at 4 °C on a shaker with gentle agitation overnight. The plate was washed twice with 225 μL/well buffer A. After blocking of unspecific binding sites with 225 μL of sterile-filtered 3% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) in buffer A at room temperature for 3 h, the plate was washed twice with 225 μL of this buffer. PKCε was left untreated or activated by addition of 60 μg/mL phosphatidylserine (Sigma-Aldrich) and 100 nM TPA (Sigma-Aldrich) in a volume of 50 μL of buffer A for 10 min at 30 °C. Recombinant purified RACK2-MBP (obtained from Prof. Dr. Daria Mochly-Rosen, Stanford University, USA, as described in ref (15)) was either left untreated or incubated with EAVSLKPT-R7 or 8 at room temperature for 30 min in a final volume of 50 μL of buffer A. Then 500 ng RACK2-MBP was added to untreated or activated PKCε for 1 h at room temperature for binding. The plate was washed twice with 225 μL of buffer A. Then 100 μL of RACK2-specific rabbit anti-RACK2 polyclonal antibody (obtained from Prof. F. Wieland, University of Heidelberg, Germany) diluted 1:20 000 in 3% BSA/buffer A was added for 1 h at room temperature. The plate was subsequently washed three times with 225 μL of buffer A, and a goat anti-rabbit HRP-conjugated IgG (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:20 000 in 100 μL of 3% BSA/buffer A was added for 1 h at room temperature. After three washes with 225 μL of buffer A, 100 μL of ABTS substrate (0.5 mg/mL) diluted in ABTS buffer (Roche, Vienna, Austria) was added and the plate was incubated in the dark for 30–180 min. Color development was measured on a plate reader at a wavelength of 420 nm.
PKCβII/RACK1 in Vitro Binding Assay
This assay was similar to the PKCε/RACK2 binding assay described above. 6-his-tagged RACK1 was cloned into a pET-30a(+) vector (Novagen, Merck KGaA, Darmstadt, Germany) and purified with Ni-NTA agarose (Qiagen, Hilden, Germany). Final elution was performed with 500 nM imidazole in elution buffer (20 mM Tris-HCl/300 mM NaCl, 20% glycerol, pH 7.5). The integrity of purified recombinant protein was analyzed by Coomassie Blue staining and Western blotting. Then 200 ng RACK1 was coated onto 96-well EIA/RIA high binding plates (Costar, Corning, New York, NY) at 4 °C on a shaker with gentle agitation overnight. Compound 8 was added at room temperature for 30 min. Then 500 ng of recombinant GST-tagged PKCβII (ProQinase) was activated with CaCl2, phosphatidylserine, and TPA for 10 min. The RACK1-coated plates with and without 8 were incubated for 1 h with activated PKCβII. The interaction was determined with a primary rabbit anti-GST antibody (Santa Cruz) and corresponding HRP-conjugated goat anti-rabbit HRP conjugated secondary antibody (Santa Cruz) as described above for the PKCε/RACK2 interaction.
Elk-1 Phosphorylation
Elk-1 phosphorylation was determined with the PathDetect system (Agilent, Santa Clara, CA). PathDetect HeLa HLR-Elk-1 cells contain a luciferase reporter cassette and express a unique, stably integrated, trans-acting fusion protein. The fusion protein consists of the activation domain of the Elk-1 transcriptional activator36−38 that is fused to the yeast GAL4 DBD (residues1–147). The transcriptional activator domain of Elk-1 is activated. An amount of 200 000 PathDetect HeLa HLR-Elk-1 cells per well was seeded in a six-well plate and grown for 24 h. Cells were washed with phosphate buffered saline and starved for 16 h in starvation medium (DMEM-containing 0.5% fetal bovine serum and 1% glutamine). Compounds were added in DMEM for 30 min. Following treatment with 50 nM TPA for 5 min the cells were washed twice with phosphate buffered saline and incubated for 4 h in starvation medium and compound 8. Then 200 μL lysis buffer as described by the manufacturer was added. The plates were shaken intensively at 4 °C for 20 min. Lysates were collected and centrifuged at 11000g at 4 °C for 2 min and used immediately for luciferase activity measurement. Protein concentration was determined according to Bradford,39 and 20 μg of each sample was transferred to a white, opaque 96-well plate. An amount of 150 μL of luciferase assay buffer as described by the manufacturer was injected, and light emission from the reaction was measured for 3 s after a delay time of 2 s. Relative light units were measured with a 1450 Microbeta Wallac Jet luminometer (Perkin-Elmer, Waltham, MA).
Cell Fractionation and Western Blotting
For PKCε translocation PC-3 cells were starved for 16 h, treated with 8 for 30 min, stimulated with 50 nM TPA for 5 min, lysed, and fractionated with a CNMCS/CNM compartmental protein extraction kit (BioChain Institute, Newark, CA). Western blotting was performed by a standard procedure as described by Garczarczyk et al.27,40 Cytosolic and membrane fractions were loaded onto SDS gels and transferred to an Immobilon membrane (Millipore, Billerica, MA). The membranes were incubated with rabbit polyclonal IgG antibodies for detection of PKCε (Santa Cruz Biotechnology; dilution 1:2000), for PKCε phosphoSer729 (Millipore; dilution 1:1000), for PKCδ (Santa Cruz Biotechnology; dilution 1:1000), and for MARCKS phosphoSer152/156 (Cell Signaling Technology, Danvers, MA; dilution 1:1000). For the loading control tubulin (Santa Cruz Biotechnology; dilution 1:1000), and as marker for the membrane fraction, an IGFI-βR rabbit polyclonal IgG antibody (Santa Cruz; dilution 1:1000) and, as secondary antibodies, peroxidase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA; dilution 1:20000) were used. GAPDH (Chemicon, Millipore, Billerica, MA; dilution 1:10000) was used as loading control for the cytosolic fraction. A peroxidase-conjugated secondary antibody (AffiniPure goat anti-mouse IgG; Jackson ImmunoResearch Laboratories; dilution 1:20000) was employed for detection.
Immunofluorescence
PC-3 cells were grown on glass coverslips coated with poly-l-lysine (Sigma-Aldrich). After treatment with 8 for 30 min and with 100 nM TPA for 5 min, the cells were rinsed twice with phosphate buffered saline (PBS) and fixed with filter sterilized 4% (w/v) paraformaldehyde/4% sucrose (w/v) (both from Sigma-Aldrich) in PBS at room temperature for 10 min. Subsequently the cells were washed three times with PBS and permeabilized with 0.2% Triton X-100/0.2% IgG-free BSA in PBS at room temperature for 10 min. After blocking with 5% normal goat serum diluted in PBS as described above for 30 min, cells were incubated with the primary antibodies for PKCε (Santa Cruz; diluted 1:500) in 0.2% Triton X-100/0.2% IgG-free BSA in PBS at 4 °C overnight. Subsequently, the cells were washed three times with the same buffer and incubated with the labeled secondary antibody (Alexa Fluor, Invitrogen, Life Technologies, Carlsbad, CA; 1:4000) at room temperature for 1 h. After three more washes with 0.2% Triton X-100/0.2% IgG-free BSA in PBS, cells were mounted with Mowiol (Sigma-Aldrich) and images were taken with an Olympus BX 50 optical microscope (Olympus Corporation, Tokyo, Japan).
Cell Migration and Invasion
For cell migration and motility, a scratch migration assay described by Cha et al.41 was employed. In a tissue culture dish, with logarithmically growing HeLa cells containing a doxycycline-inducible constitutively active PKCε, a migration gap of approximately 1 mm was created by introducing a “scratch” to the adherent layer of cultured cells using a sterile Gilson 200 μL pipet tip. The scratch was administered by hand with sufficient pressure to remove adherent cells from the polystyrene substrate but without causing physical damage to the polystyrene surface. The dish was washed with PBS to remove the cells and further incubated with 2 μg/mL doxycycline and 25 μM 8 for 24 h. Controls were left either untreated or were incubated with 2 μg/mL doxycycline and DMSO. Migration into the scratch was observed with an Olympus microscope. Invasion of HeLa cells through a reconstituted basement membrane matrix was determined with a cell invasion assay kit from Chemicon (no. ECM550) as described by the manufacturer.
Chicken Egg Assay
Eggs from hen were incubated at 37 °C for 3 days, opened, and incubated for further 7 days. On day 10, the growth factor VEGF and 8 were added as indicated in Figure 8. After further incubation for 5 days, angiogenesis was observed by microscope.
IC50 Determination and Statistics
IC50 values were determined with CalcuSyn (Biosoft, Cambridge, U.K.). Significance was calculated by the t test with GraphPad Prism, version 5.0.
Acknowledgments
We thank Dr. Gerold Untergasser, Institute for Internal Medicine, Medical University of Innsbruck, for performing the chicken egg assay. This work was supported by Grants P16477-B12 and P25491-B21 from the Austrian Science Fund, grant Prokinase Research from the European Commission (European Union FP6 Integrated Project LSHB-CT-2004-503467), and Grant GZ: UNI-0404/728 from the Tyrolean Science Fund.
Glossary
Abbreviations Used
- BSA
bovine serum albumin
- cpd
compound
- MBP
maltose-binding protein
- MARCKS
myristoylated alanine-rich protein kinase C substrate
- PBS
phosphate buffered saline
- PKC
protein kinase C
Supporting Information Available
All compounds selected by virtual screening (and tested in vitro); inhibition of RACK2-binding to PKCε by the peptide EAVSLKPT; effect of 8 on PKC isozymes; 1H NMR of 8 purchased from Asinex; 1H NMR, ESI-MS, and HPLC of 8 synthesized at Gause Institute of New Antibiotics. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∞ F.R., P.G., and J.K. contributed equally to this work.
The authors declare the following competing financial interest(s): Innsbruck Medical University and University of Innsbruck have filed a patent on this topic. F.R., P.G., J.K., G.H., M.B., D.G., G.L., M.N.P., T.L., and J.H. are co-assignees of the patent application.
Supplementary Material
References
- Gorin M. A.; Pan Q. Protein kinase C epsilon: an oncogene and emerging tumor biomarker. Mol. Cancer 2009, 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pass J. M.; Zheng Y.; Wead W. B.; Zhang J.; Li R. C.; Bolli R.; Ping P. PKCepsilon activation induces dichotomous cardiac phenotypes and modulates PKCepsilon-RACK interactions and RACK expression. Am. J. Physiol.: Heart Circ. Physiol. 2001, 280, H946–H955. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Gray M. O.; Chen C. H.; Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 1996, 271, 24962–24966. [DOI] [PubMed] [Google Scholar]
- Cesare P.; Dekker L. V.; Sardini A.; Parker P. J.; McNaughton P. A. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron 1999, 23, 617–624. [DOI] [PubMed] [Google Scholar]
- Castrillo A.; Pennington D. J.; Otto F.; Parker P. J.; Owen M. J.; Boscá L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. J. Exp. Med. 2001, 194, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y.; Olsen G. S.; Ziv E.; Hansen L. L.; Busch A. K.; Hansen B. F.; Shafrir E.; Mosthaf-Seedorf L. Cellular mechanism of nutritionally induced insulin resistance in Psammomys obesus: overexpression of protein kinase Cepsilon in skeletal muscle precedes the onset of hyperinsulinemia and hyperglycemia. Diabetes 2001, 50, 584–592. [DOI] [PubMed] [Google Scholar]
- Choi D. S.; Wang D.; Dadgar J.; Chang W. S.; Messing R. O. Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. J. Neurosci. 2002, 22, 9905–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita Y. Protein kinase Cepsilon: multiple roles in the function of, and signaling mediated by, the cytoskeleton. FEBS J. 2008, 275, 3995–4004. [DOI] [PubMed] [Google Scholar]
- Shirai Y.; Adachi N.; Saito N. Protein kinase Cepsilon: function in neurons. FEBS J. 2008, 275, 3988–3994. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Marsden B.; Pogacic V.; Rellos P.; Müller S.; Bullock A. N.; Schwaller J.; Sundström M.; Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20523–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S.; Chikaraishi Y.; Tsuruma K.; Shimazawa M.; Hara H. Ruboxistaurin, a PKCbeta inhibitor, inhibits retinal neovascularization via suppression of phosphorylation of ERK1/2 and Akt. Exp. Eye Res. 2010, 90, 137–145. [DOI] [PubMed] [Google Scholar]
- Chen Y. B.; LaCasce A. S. Enzastaurin. Expert Opin. Invest. Drugs 2008, 7, 939–944. [DOI] [PubMed] [Google Scholar]
- Soltoff S. P. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol. Sci. 2007, 28, 453–458. [DOI] [PubMed] [Google Scholar]
- Csukai M.; Chen C.-H.; De Matteis M. A.; Mochly-Rosen D. The coatomer protein b′COP: a selective protein (RACK) for epsilon protein kinase C. J. Biol. Chem. 1997, 272, 29200–29206. [DOI] [PubMed] [Google Scholar]
- Schechtman D.; Mochly-Rosen D. Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene 2001, 20, 6339–6347. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D.; Khaner H.; Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 3997–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M. O.; Karliner J. S.; Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997, 272, 30945–30951. [DOI] [PubMed] [Google Scholar]
- Erdogan E.; Lamark T.; Stallings-Mann M.; Jamieson L.; Pellecchia M.; Thompson E. A.; Johansen T.; Fields A. P. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J. Biol. Chem. 2006, 281, 28450–28459. [DOI] [PubMed] [Google Scholar]
- Beeley N. R. Can peptides be mimicked?. Drug Discovery Today 2000, 5, 354–363. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Wells J. A. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat. Rev. Drug Discovery 2004, 3, 301–317. [DOI] [PubMed] [Google Scholar]
- Ron D.; Jiang Z.; Yao L.; Vagts A.; Diamond I.; Gordon A. Coordinated movement of RACK1 with activated betaIIPKC. J. Biol. Chem. 1999, 274, 27039–27046. [DOI] [PubMed] [Google Scholar]
- Brodie C.; Bogi K.; Acs P.; Lazarovici P.; Petrovics G.; Anderson W. B.; Blumberg P. M. Protein kinase C plays a role in neurite outgrowth in response to epidermal growth factor and nerve growth factor in PC12 cells. Cell Growth Differ. 1999, 10, 183–191. [PubMed] [Google Scholar]
- Fenton R. A.; Komatsu S.; Ikebe M.; Shea L. G.; Dobson J. G. Jr. Adenoprotection of the heart involves phospholipase C-induced activation and translocation of PKC-epsilon to RACK2 in adult rat and mouse. Am. J. Physiol.: Heart Circ. Physiol. 2009, 297, H718–H725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan Y. T.; Guo Y.; Zhu Y.; Wang O. L.; Rokosh G.; Messing R. O.; Bolli R. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation 2005, 112, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garczarczyk D.; Toton E.; Biedermann V.; Rosivatz E.; Rechfeld F.; Rybczynska M.; Hofmann J. Signal transduction of constitutively active protein kinase C epsilon. Cell. Signalling 2009, 21, 745–752. [DOI] [PubMed] [Google Scholar]
- Stensman H.; Larsson C. Protein kinase Cepsilon is important for migration of neuroblastoma cells. BMC Cancer 2008, 8, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber P.; Rechfeld F.; Kirchmair J.; Hauser N.; Boehler M.; Garczarczyk D.; Langer T.; Hofmann J. Barbituric acid derivative BAS 02104951 inhibits PKCε, PKCη, PKCε/RACK2 interaction, Elk-1 phosphorylation in HeLa and PKCε and η translocation in PC3 cells following TPA-induction. J. Biochem. 2011, 149, 331–336. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Helfand B. T.; Feng B. Y.; Shoichet B. K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- Rechfeld F.; Gruber P.; Hofmann J.; Kirchmair J. Modulators of protein–protein interactions—novel approaches in targeting protein kinases and other pharmaceutically relevant biomolecules. Curr. Top. Med. Chem. 2011, 11, 1305–1319. [DOI] [PubMed] [Google Scholar]
- Easmon J.; Puerstinger G.; Roth T.; Fiebig H. H.; Jenny M.; Jaeger W.; Heinisch G.; Hofmann J. 2-Benzoxazolyl and 2-benzimidazolyl hydrazones derived from 2-acetylpyridine: a novel class of antitumor agents. Int. J. Cancer 2001, 94, 89–96. [DOI] [PubMed] [Google Scholar]
- Takeishi Y.; Ping P.; Bolli R.; Kirkpatrick D. L.; Hoit B. D.; Walsh R. A. Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circ. Res. 2000, 86, 1218–1823. [DOI] [PubMed] [Google Scholar]
- Ferreira J. C.; Brum P. C.; Mochly-Rosen D. βIIPKC and εPKC isozymes as potential pharmacological targets in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2010, 51, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perletti G.; Tessitore L.; Sesca E.; Pani P.; Dianzani M. U.; Piccinini F. Epsilon PKC acts like a marker of progressive malignancy in rat liver, but fails to enhance tumorigenesis in rat hepatoma cells in culture. Biochem. Biophys. Res. Commun. 1996, 221, 688–691. [DOI] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Rao V. N.; Huebner K.; Isobe M.; ar-Rushdi A.; Croce C. M.; Reddy E. S. elk, tissue-specific ets-related genes on chromosomes X and 14 near translocation breakpoints. Science 1989, 244, 66–70. [DOI] [PubMed] [Google Scholar]
- Price M. A.; Rogers A. E.; Treisman R. Comparative analysis of the ternary complex factors Elk-1, SAP-1a and SAP-2 (ERP/NET). EMBO J. 1995, 14, 2589–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais R.; Wynne J.; Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Garczarczyk D.; Szeker K.; Galfi P.; Csordas A.; Hofmann J. Protein kinase Cgamma in colon cancer cells: expression, Thr514 phosphorylation and sensitivity to butyrate-mediated upregulation as related to the degree of differentiation. Chem.-Biol. Interact. 2010, 185, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha D.; O’Brien P.; O’Toole E. A.; Woodley D. T.; Hudson L. G. Enhanced modulation of keratinocyte motility by transforming growth factor-alpha (TGF-alpha) relative to epidermal growth factor (EGF). J. Invest. Dermatol. 1996, 106, 590–597. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.