

Abstract
Objectives
Recent clinical data suggest that severe kala-azar (or visceral leishmaniasis) is an exaggerated innate immune response mediated by inflammatory cytokines, leading to a systemic inflammatory syndrome similar to what is observed in malaria, sepsis and other diseases. We tested this hypothesis by measuring serum cytokines in individuals with kala-azar.
Methods
We compared patients with severe kala-azar (i.e. hemorrhagic manifestations, n = 38) with patients without evidence of hemorrhage (n = 96). We conducted a detailed clinical and laboratory evaluation, measuring serum IL-1beta, IL-6, IL-8, IL-10, IL-12, interferon-gamma, and TNF-alpha, and markers of disseminated intravascular coagulation (DIC).
Results
Infants had higher levels of inflammatory cytokines, while HIV-infected patients had lower concentrations of IL-10 and interferon-gamma. Higher levels of IL-6, interferon-gamma, and IL-8 were found among deceased patients. IL-8 and interferon-gamma were independently associated with bleeding. Several cytokines were associated with different signs of severe clinical and laboratory manifestations, including DIC. IL-6 was highly positively and independently associated with IL-1beta, IL-8, IL-10, and negatively associated with TNF-alpha. IL-1beta and TNF-alpha were also highly independently associated with disease severity.
Conclusion
In its severe form, kala-azar, a neglected tropical disease, initiates a systemic inflammatory response that leads to DIC and other manifestations. Children may have higher risk of death due to the more intense cytokine release. The data supports the notion that IL-6 is the central cytokine that is associated with lethal disease, but interferon-gamma, IL1beta, IL-8, and TNF-alpha are also involved with disease severity. Inhibition of IL-6 is a potential target of adjuvant therapy for severe or pediatric forms of this disease.
Keywords: Cytokines, Kala-azar, Leishmania infantum, Disseminated intravascular coagulation, Systemic inflammatory response
Introduction
Kala-azar, or visceral leishmaniasis, typically leads to fever, anemia, wasting, and hepatosplenomegaly and is cured with known therapies.1 For unknown reasons, some patients progress with heavy bleeding, bacterial co-infections, dyspnea, edema (lower limbs or anasarca), jaundice, and/or emesis that lead to death, even when treated with recommended therapies.2–5 Research suggests that this is not due to different parasite species, as the disease is caused by two species of protozoa of the genus Leishmania, obligatory parasites of mononuclear phagocytes, normally found in the spleen, liver, and bone marrow. L. donovani has a limited distribution in the Indian Subcontinent and East Africa, and L. infantum is found in Central Asia, Middle-East, Mediterranean, West Africa, and South and Central America.6,7
Herein, we develop a novel biological framework involving inflammatory cytokines and disease severity to explain different disease courses. A lack of proper understanding of the pathogenesis of severe disease and, therefore, inefficient therapy could explain the difficulties of treating the endemic Leishmania in Brazil where active surveillance and free treatment is provided, but mortality continues to rise annually.8 Inflammatory cytokines have been found to be elevated in non-complicated kala-azar.9 Further, patients with severe presentation of sepsis and malaria show marked increase of certain cytokines compared with those with milder clinical presentation. Thus, we hypothesize that inflammatory cytokines could also be more elevated in lethal kala-azar and this might provide insight into why some patients with severe disease cannot be rescued by specific treatment. This builds from observations in a range of cohorts where patients that have died from kala-azar were observed to have worse anemia, hepatitis, pneumonitis, nephritis, diarrhea, neutropenia, thrombocytopenia, and disseminated intravascular coagulation (DIC), which overlap with those clinical manifestations triggered by inflammatory cytokines.1–5,10 Finally, since Leishmania organisms do not produce toxins and do not destroy parenchyma cells, it is reasonable to hypothesize that these cytokines might be strongly related, or be the cause itself, of the most severe kala-azar cases, by generating a damaging systemic inflammatory response.11 This hypothesis would explain why, like sepsis and severe malaria, some patients do not respond to antibiotics or to chemotherapy and provide a basis for further research into anti-cytokines therapy for treating the most severe forms of kala-azar, as well as other severe systemic infectious diseases.
Materials and Methods
The Research Ethics Committee of the Federal University of Piauí approved the protocol. Written informed consent was obtained from each participant or his/her legal representative. From an ongoing open cohort study at a reference hospital in the city of Teresina, northeast Brazil, of 884 in-hospital patients, including 217 with any sort of bleeding (24.6%), we randomly selected 34 patients with spontaneous and clinically apparent skin or mucosal bleeding as manifested by petequiae, echimoses, hematomas, epistaxis, bleeding by gums, gastrointestinal or urinary tracts or any other site, and 96 patients with kala-azar, but without any hemorrhagic complications, to compare the concentrations of cytokines. Patients with clinical or laboratorial data associated with an increased risk of death or that had hemorrhagic complications were classified as having ‘severe kala-azar’. The diagnosis was performed based on the typical symptomatology, including fever, anemia, weight loss, and hepatosplenomegaly plus a direct parasite detection or culture from bone marrow aspiration or a positive immunofluorescent test (Leishmaniose Humana, Biomanguinhos, Rio de Janeiro, Brazil) with dilutions of 1∶80 or greater. Death was attributed to kala-azar by the investigators if it occurred before or during the treatment, or after unsuccessful drug therapy. To obtain corrected organ sizes, spleen and liver sizes were divided by individuals’ body area. Edema was reported as any medical observation of generalized edema (anasarca) or local, such as facial, ascitis or in the lower limbs.
Sera and plasma were collected at admission. Immediately after collection in a flask with citrate (with the exception for fibrin degradation products that came with its own anticoagulant in the dosage kit), plasma samples were frozen at −80°C, and use up to 6 months after collection. Sera were stored at −20°C. All patients were tested for HIV. Other tests were fibrinopeptide-A (Imunoclone FPA ELISA; American Diagnostica Inc., Stamford, CT, USA), fibrinogen degradation products (ActiScreenTM XL-FDP; American Diagnostica Inc.), D-dimer (Imunoclone D-Dimer ELISA; American Diagnostica Inc.), fibrinogen (Human Fibrinogen ELISA Quantitation Kit; Gen Way Biotech Inc., San Diego, CA, USA), and C-reactive protein (Array 360 System Beckman; Abbott Laboratories, North Chicago, IL, USA). Serum levels of the cytokines IL-1beta, IL-2, IL-6, IL-10, IL-12p70, IL-8, TNF-alpha, and IFN-gamma were analyzed by flow cytometry from (Cytometric Bead Array Human Inflammatory Cytokines Kit Assay; BD Biosciences, Rockville, MD, USA, and Cytometric Bead Array Human IFN-γ Flex Set; BD Biosciences). The flow cytometric assay was performed and analyzed by a single operator, and standard curves were derived from cytokine standards.
Statistical analysis was performed with the Stata/SE® 10.0 (StataCorp. LP, College Station, TX, USA) statistical software. The Spearman analysis was applied to detect correlation between continuous variables with sparse data. Wilcoxon and the Kruskal–Wallis tests were used to compare medians of variables with a non-parametric distribution. To determine which of the variables had independent associations, linear multivariate regression analysis (MRA) and logistic MRA with a backward elimination procedure were performed. The normality of distributions was examined and confirmed with kurtosis and skewness tests.
Results
The distribution of age and gender was similar between patients who had evidence of bleeding disorders and those without. Ninety-five percent of those with hemorrhage were already bleeding when they were admitted at the hospital. Among the patients bleeding 6/38 (15.8%) died, while only 2/96 (2.1%) died among those without evidence of hemorrhage. Twelve patients were infected with HIV, but there was no difference among these patients with respect to the bleeding status. The presence of bleeding was irrespective of the length of symptoms (Table 1).
Table 1. Characteristics of the study population.
| Characteristic | Hemorrhage (n = 38), n (%) | No hemorrhage (n = 96), n (%) | P value |
| Age (years) | 0.13 | ||
| <1 | 15 (39.5) | 22 (22.9) | |
| 1<5 | 6 (15.8) | 23 (23.9) | |
| 5<20 | 2 (5.3) | 18 (18.8) | |
| 20–40 | 10 (26.3) | 21 (21.9) | |
| >40 | 5 (13.1) | 12 (12.5) | |
| Gender | 0.76 | ||
| Male | 26 (68.4) | 63 (65.6) | |
| HIV infection | 0.71 | ||
| Yes* | 4 (11.4) | 8 (9.2) | |
| Outcome | 0.003 | ||
| Death | 6 (15.8) | 2 (2.1) | |
| Duration of symptoms days (95% IC) | 73.3 (48.2–98.4) | 79.6 (51.7–107.5) | 0.78 |
Note: *Only 126 tested for HIV.
Association of cytokines with death, age, and HIV infection
Higher serum levels of cytokines among patients who died as compared to survivors were found for IL-6, interferon-gamma, and IL-8 (Fig. 1). Children 2 years old or less had higher concentration of IL-6 (62.6 versus 20.7; P = 0.01) and of interferon-gamma (46.1 versus 24.8; P = 0.05). HIV-infected patients had lower concentration of IL-10 (15.0 versus 38.1; P = 0.01) and of interferon-gamma (3.5 versus 32.1; P = 0.05).
Figure 1.

Box plots of the serum concentration of cytokines in patients with kala-azar according to the occurrence of death.
Correlations of cytokines with clinical and laboratorial findings
The following signs and symptoms were found to be associated with mortality after a logistic MRA: higher body temperature, presence of bleeding, presence of sepsis, vomiting, diarrhea, edema, dyspnea, jaundice, liver, and spleen size. However, among them, only bleeding, edema, and vomiting were associated to serum cytokine concentrations. Patients with hemorrhage had higher concentrations of IL-1beta, IL-6, IL-8, and interferon-gamma, but the logistic MRA showed that only higher levels of IL-8 (P = 0.004) and interferon-gamma (P = 0.006) were independently associated with bleeding. Patients with edema had higher concentration of IL-6 and IL-8 and patients with vomiting had higher concentration of IL-6 (P = 0.02) at the uni- and MRA (Fig. 2).
Figure 2.

Box plots of the serum concentration of cytokines in patients with kala-azar according to the presence of bleeding, edema, and vomiting. Only statistically significant association results are shown.
Many hematological findings, liver enzymes, acute phase proteins, and kidney function abnormalities were associated with the risk of death (data not shown), but not all were correlated with serum cytokine concentrations. Figure 3 shows the significant correlations of laboratorial data with cytokines. Anemia, one of the most common signs of the disease, as measured by the hemoglobin concentration, had significant negative correlation with IL-1beta, IL-6, IL-8, and IL-10, but only IL-1beta was associated at the MRA (P = 0.049; F-test = 0.02; R2 = 0.12). Neutrophil count was negatively correlated to IL-10, interferon-gamma, and TNF-alpha at the univariate analysis; IL-6 was the sole cytokine correlated to neutrophil count (P = 0.044; F-test = 0.11; R2 = 0.03). Lymphocyte count displayed two patterns of association with cytokines: at 2000/μl and above, it was negatively correlated with IL-1beta, IL-8, and TNF-alpha, but bellow 2000/μl, it was positively correlated with TNF-alpha; however, there was no association found with the MRA. The liver enzyme aspartate aminotransferase (AST) had a positive correlation with IL-1beta, IL-6, and IL-8, while, in the MRA, IL-6 was the one significantly correlated to liver damage as measured by AST (P = 0.006; F-test = 0.049; R2 = 0.11). Finally, considering the acute phase proteins: C-reactive protein was positively correlated with IL-6 and interferon-gamma (but only with IL-6 in the MRA, P<0.001; F-test <0.001; R2 = 0.43), while fibrinogen was correlated with IL-6 and interferon-gamma (not shown). Alkaline phosphatase, serum albumin, and glomerular filtration were not correlated to any inflammatory cytokine in this data set.
Figure 3.

Correlation of serum cytokines concentration with the results of laboratory tests in patients with kala-azar.
Correlations of cytokines with DIC
Platelet count was negatively correlated with IL-1beta, IL-6, IL-8, and interferon-gamma (Fig. 4). The observations that patients with any kind of bleeding had a lower platelet count (P<0.0001), higher concentration of plasma D-dimer (P = 0.0001), and presence of fibrin degradation products (P = 0.009) highlight the relationship of platelet count with markers of DIC in patients with severe kala-azar, is reinforced by the strong negative correlation between platelet count and D-dimer (data not shown) (P<0.0001). D-dimer was also correlated with inflammatory cytokines, as seen also in Fig. 4. Moreover, patients with detected fibrin degradation products had higher concentration of IL-6 and interferon-gamma, by uni- and MRA (Fig. 5).
Figure 4.

Correlation of serum cytokines concentration with platelet count and D-dimer levels.
Figure 5.

Box plots of the concentration of serum cytokines in patients with kala-azar according to the detection of fibrin degradation products.
Correlation among cytokines
Figure 6 shows the mutual correlations among cytokines. All were strongly positively associated among themselves, but some particularly strong correlations were observed. These include IL-1beta and TNF-alpha (rho = 0.75), IL-1beta and IL-6 (rho = 0.66), IL-6 and IL-8 (rho = 0.66), IL-1beta and IL-8 (rho = 0.62), and IL-12 and TNF-alpha (rho = 0.62). In the MRA of the natural logarithm of each cytokine as an explanatory variable to the others, all linear models were robust, with F-tests <0.0001 in all, and R2 varying of 0.19 for interferon-gamma; 0.37 for IL-10; 0.38 for IL-12; 0.49 for IL-8; 0.65 for IL-6; 0.74 for TNF-alpha; and 0.82 for IL-1beta.
Figure 6.
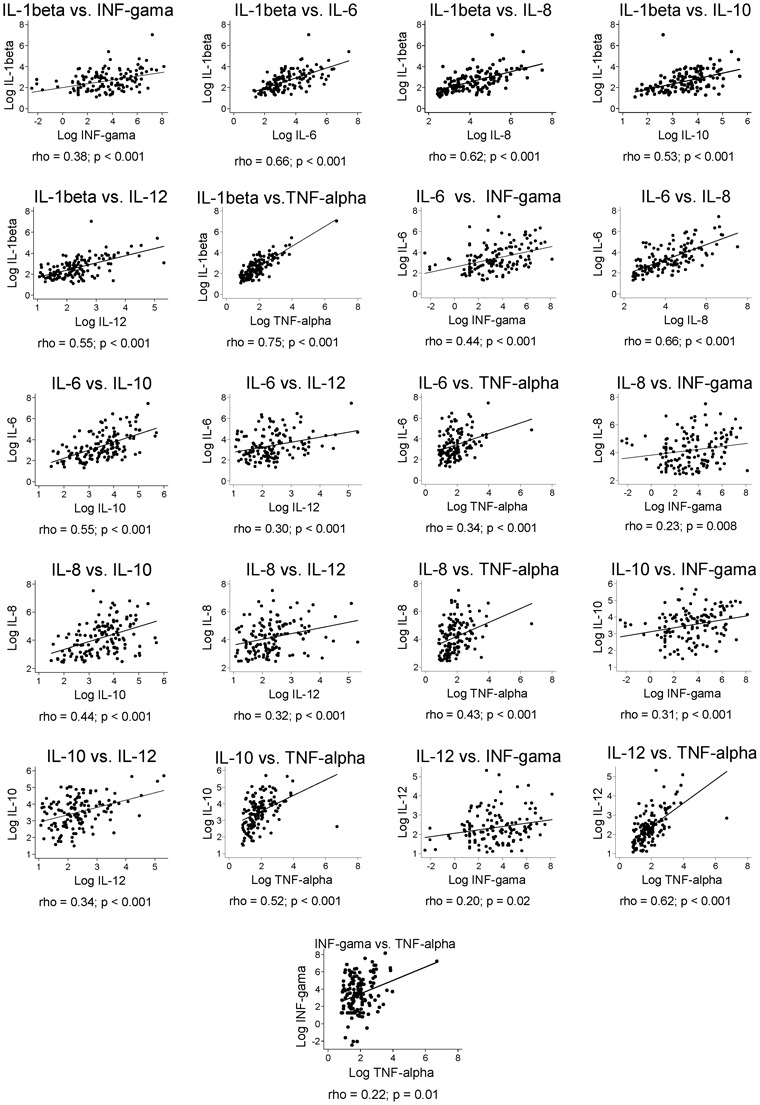
Mutual correlation of serum cytokines in patients with kala-azar.
Discussion
The associations between these serum cytokine concentrations with certain clinical presentations of kala-azar of severe disease provides insights into the involvement of these proteins in lethal kala-azar, particularly IL-6, interferon-gamma, and IL-8, which are those cytokines independently directly associated with death, particularly among the young. These findings also suggest that the higher mortality found among infants is due to the higher concentration of IL-6 and interferon-gamma as seen in other conditions.12,13 IL-6 is a pleiotropic cytokine that is both produced and targeted by a variety of cell types, and thus has the capacity to induce several different intracellular signaling pathways. For example, IL-6 has a role on the pathogenesis of sepsis and rheumatoid arthritis and its main actions are promoting acute phase response together with IL-1 and on tissue factor expression, which, initiates the coagulation cascade.14,15 interferon-gamma is produced by T- and B-cells, natural killer cells and antigen presenting cells and orchestrates many distinct cellular programs through transcriptional control over large numbers of genes.16 Also, interferon-gamma enhances immune reactivity by increasing antigen presentation to lymphocytes via induction of HLA-DR that improves the outcome of patients with multiple organ failure following severe trauma.17,18 The other disease inducing cytokines are IL-8, IL-1beta, and TNF-alpha. IL-8 is a chemokine19 mostly produced by monocytes and macrophages, with the important role of recruiting leukocytes. IL-1beta is normally produced in response to infection, injury, or immunologic challenge by blood monocytes; at minimal concentrations, it causes fever, hypotension, and production of additional pro-inflammatory cytokines, such as IL-6.20 TNF-alpha is secreted by macrophages, monocytes, neutrophils, natural killer cells, and T cells and is known to affect the growth, differentiation, survival and physiological function of a variety of different cells including macrophage-mediated cytotoxicity due to the proapoptotic effects of TNF-alpha. These cytokines can directly lead to bleeding either by inducing coagulopathy, by directly provoking thrombocytopenia or by damaging the liver of those infected. They are involved with severity not only of kala-azar but of other infectious diseases, like sepsis and Plamodium falciparum malaria.21 Besides acting on kala-azar the inflammatory cytokines play a critical role on the pathogenesis of sepsis and the systemic inflammatory response syndrome) via innate immunity.22
Bleeding and edema were associated with high concentrations of serum cytokines and are linked to endothelium and mononuclear cells in the microvasculature.23 Edema in severe kala-azar seems to be a consequence of both low serum albumin and capillary leakage11 while DIC leads to bleeding.24–27 This study shows that edema is strongly related to IL-6 and to IL-8. We hypothesize that the release of IL-6 and IL-8 by macrophages and neutrophils could induce the expression of adhesion molecules in the endothelium and could recruit leucocytes, which are precursors of vascular damage19,28 and therefore edema.29 Additionally, IL-6 is known to inhibit the production of albumin in the liver,30 which might exacerbate edema.
Besides inducing edema, IL-8, together with interferon-gamma, IL-6 and IL-1beta, up-regulates the expression of tissue factor by endothelial cells initiating the coagulation cascade that leads to DIC.31 DIC is the primary pathway to hemorrhage, indicated by the robust association of markers of DIC such as D-dimer and fibrinogen degradation products with IL-6 and interferon-gamma. Coagulation has also been found to induce IL-6 secretion and inflammation.22 DIC may not be the only cause of bleeding. Indeed, liver injury indicated by alterations of a hepatic enzyme, which is common in severe kala-azar,24,32 was independently related to IL-6 and likely is a complementary cause of bleeding. Therefore, while these proteins are up-regulated by IL-6,33 fibrinogen appears to be down-regulated.34 We have previously shown the association of genetic polymorphisms of another acute phase protein, mannan-binding lectin, with severe kala-azar,35 and since this lectin activates coagulation36,37 it can lead to DIC via MBL-dependent complement activation.38
Neutropenia, thrombocytopenia and anemia are not a consequence of depletion of bone marrow cellularity by Leishmania infiltration.39 Rather, our data suggest that they are a consequence of systemic inflammation. For example, IL-6 was shown to be the main cytokine inducing neutropenia, yet this is not peculiar to kala-azar, since it does not occur in bacteremic neutropenic patients.39 Thrombocytopenia appears to be potentially induced by DIC suggested by its association with the markers of DIC. Platelet count exhibited a strong association with a range of different cytokines. Anemia, the commonest sign of kala-azar, indicated by hemoglobin concentration, was associated independently with IL-1beta, IL-6, IL-8, and IL-10. This result suggests that a pathway via hepcidin exists.40 Hepcidin is an acute phase protein released by the liver, which leads to iron-deficient anemia,41 typical of the anemia observed in patients with kala-azar.39 Finally, lymphocyte count in kala-azar was shown to be possibly dependent on TNF-alpha, IL-1beta, and IL-8, although not among patients with cancer.42 A likely role of CD4 cells in the secretion of IL-10 and interferon-gamma is indicated by its lower concentration in L. infantum-HIV-infected patients.
These correlations between cytokines permit speculations about a dynamic network among cytokines in severe kala-azar. The regression analyses allowed identifying the dependency of each cytokine on another, which opened the window for deeper inferences on their relationships. For instance, IL-12 seems to be a strongly linked to TNF-alpha, while TNF-alpha and IL-1beta show a strong relationship. We hypothesize that they are secreted by the same type of cells, possibly parasitized macrophages, stimulated by the same pathogen-associated molecular patterns,43–45 very likely in the spleen, bone marrow, and liver. IL-1beta is strongly controlled by IL-6 in kala-azar, leading to anemia, one of the commonest sign of kala-azar and a predictor of death. Together with IL-6, IL-1beta controls the secretion of IL-8. The tight correlation between IL-1beta, IL-6, and IL-8 also suggests a particular interplay between them leading to death with bleeding and edema. A possible pathway would be the production of IL-1beta and IL-6 by macrophages, inducing the secretion of IL-8,46 and hence, endothelial activation with further secretion of IL-1beta and IL-6. Both IL-6 and IL-8, albeit mostly the former, were linked to interferon-gamma. Interferon-gamma is an important disease causing factor as seen by its independent association with death, bleeding, DIC, liver involvement, thrombocytopenia, neutropenia, and the acute phase response indicated by C-reactive protein secretion, nevertheless its protective effect for Leishmania, as a component of acquired immunity. This dual action of interferon-gamma suggests that the molecules identified in the sera are rather part of the harmful excessive innate response secreted by neutrophils, eosinophils, and natural killer T-cells.9 IL-6 appears to play a central role in the pathogenesis of severe kala-azar, not only because it is directly and independently associated with a range of pathological manifestations such as edema, DIC, neutropenia, liver damage (as seen by AST), and acute phase response, but also due to its induction of several other disease causing cytokines such as IL-1beta, interferon-gamma, and IL-8. Besides possibly down-regulating TNF-alpha, it is strongly related to IL-10 secretion, therefore establishing a link with the innate response with the acquired immunity in kala-azar patients, via the T-regulatory response. Indeed, IL-10 production by splenic CD4+CD25−Foxp3− T cells seems to lead to the immunoparalysis that allow parasite proliferation and infection success.43–45,47,48 One hypothesis is that an initial localized parasite proliferation initiates an innate response inhibiting the effective Th1 response via IL-10 control of IL-12,49 and by the inhibition of an effective innate response by TNF-alpha via IL-6. These mechanisms may be the cause of severe disease when a high parasite burden is progressively reached, leading to an exaggerated release of pathogen-associated molecular pattern that triggers the lethal innate immune response of kala-azar. Progression to severity appears to occur as indicated by the known effect of time on the chance of death. However, it seems to be a very slow process, since we found no effect of time on bleeding. Moreover, the effect of time on mortality is very late. Taken together, these data suggest that most patients reach an early plateau of higher risk; thereafter, the increase in the chance of death would be due only to the accumulation of daily repeated chances of complications and death.
Limitations
Some points related to the study design must be addressed. While a larger sample would have been attractive, it is uncommon to study such a number of severe kala-azar patients. Since almost all the patients with hemorrhage were already bleeding when they entered in the hospital (and the same is true for other symptoms), it was not possible to develop a longitudinal study to clarify a cause–effect relationship. While scientifically attractive, it would be unethical to admit patients without bleeding and then to wait the development of hemorrhage just to observe a causal effect of cytokine sera concentration.
Regarding causality, the only possible detectable temporal relationship with cytokines and disease was death, and HIV infection, and therefore, all the other associations shall not be taken as causal. Consequently, while IL-6, IL-8, and interferon-gamma can be inferred as being part of a causal pathway to death, or, at least, as predictors of mortality, the other associations of cytokines with symptoms and signs should be taken only as markers of severity. Which of the clinical and pathogenic alterations are the immediate causes of death cannot be ascertained in this study. However, since with the exception of shock, the manifestations of severe kala-azar are similar to those of sepsis and systemic infection where inflammatory cytokines play a critical role, multiple organ failure and bleeding are the main consequences of inflammation that lead to death.22,50,51 Finally, an interesting future analysis would expand upon the limitation of being able to stratify patients based on ‘severity’, perhaps by using metrics such as parasite load.
Conclusion
Serum cytokines are part of a complex host response in kala-azar, which is not well understood. Experimental models of severe disease, systems studies, and cohort designs followed by pathological observations of the deceased patients represent the limit of ethical scientific investigation for this area. In this context, the present findings suggest that the late stage of severe kala-azar, like other severe acute infectious diseases such as sepsis, malaria, dengue, leptospirosis, and viral hemorrhagic fevers, should be interpreted as a consequence of a systemic inflammatory response.22,52–55
Only a limited number of proteins and pathways were studied, while many others need to be observed. As examples, it has been shown by our group that polymorphisms in the MBL gene alleles and the TGF-alpha gene –509 allele T are associated with more severe kala-azar.35,56 The identification of parasite and host molecules that participate in the development of severe kala-azar will help to design better adjuvant therapy. Clinical experience has shown that after patients start to bleed or if they have other complications, it is much harder to save their lives, even when using first line drugs like liposomal amphotericin B. Targeting critical molecules that are part of the host response with biopharmaceuticals may eventually save lives as it has for acute critical diseases,57–59 or may help to assemble data for future analysis.60 From our findings, the first and most promising pharmaceutical targets are the antagonists of IL-6.
To briefly summarize in full, in this study, we have shown that higher concentrations of IL-6, IL-8, and interferon-gamma precede death in patients with kala-azar, but not other inflammatory cytokines. Also, IL-1beta, IL-6, IL-8, interferon-gamma, and TNF-alpha are correlated with an array of clinical and laboratorial data typically found in severe kala-azar. IL-6 and interferon-gamma were strongly associated with markers of DIC. Finally, a pattern of relationships among cytokines was identified in kala-azar patients, in which IL-6 seems to play a central role in triggering symptoms and controlling other cytokines. The present data indicates that lethal kala-azar is very likely a result of an exaggerated inflammatory response and that anti-cytokine therapy, particularly anti-IL-6, should be assayed as an adjuvant therapy for the most severe forms of the disease in animal models.
Financial disclosure
The research was funded by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant no. 306503/2008-5). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. DLC is supported by CNPq. ALN and RMC are supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior. CHNC, AB, and MBN are investigators from CNPq.
Competing interests
The authors have declared that no competing interests exist. This article is part of the DSc thesis of Dorcas L. Costa ‘Fatores de prognóstico na leishmaniose visceral: Alterações clínicas e laboratoriais associadas à resposta imune, aos distúrbios da coagulação e à morte’. Graduate Program in Health Sciences: Infectious Diseases and Tropical Medicine. Belo Horizonte, MG, Federal University of Minas Gerais, August 2009.
Acknowledgments
We thank José Fontes for his indispensable clinical pathology assistance. We are also very thankful to Fernando O. Silva, Alexandre P. Silva, Juqueline Rocha Cristal, and Adorielze Leite for their essential technical and logistic support. We are also pleased with Dr Vivianny Vasconcelos and Dr Daniela Moura for their help on the registration of clinical data. Finally, we thank Dr Isabel Santos for her precious comments.
References
- 1.Herwaldt BL. Leishmaniasis. Lancet. 1999;354(9185):1191–9. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 2.Seaman J, Mercer AJ, Sondorp HE, Herwaldt BL. Epidemic visceral leishmaniasis in southern Sudan: treatment of severely debilitated patients under wartime conditions and with limited resources. Ann Intern Med. 1996;124(7):664–72. doi: 10.7326/0003-4819-124-7-199604010-00007. [DOI] [PubMed] [Google Scholar]
- 3.Werneck GL, Batista MS, Gomes JR, Costa DL, Costa CH. Prognostic factors for death from visceral leishmaniasis in Teresina, Brazil. Infection. 2003;31(3):174–7. doi: 10.1007/s15010-003-3139-9. [DOI] [PubMed] [Google Scholar]
- 4.Abdelmoula MS, M’Hamdi Z, Amri F, Tebib N, Ben Turkia H, Ben Dridi MF. [Visceral leishmaniasis in children: prognostic factors]. Tunis Med. 2003;81(8):535–9. French. [PubMed] [Google Scholar]
- 5.Collin SDR, Ritmeijer K, Keus K, Melaku Y, Kipngetich S, Davies C. Conflict and kala-azar: determinants of adverse outcomes of kala-azar among patients in southern Sudan. Clin Infect Dis. 2004;38(5):612–9. doi: 10.1086/381203. [DOI] [PubMed] [Google Scholar]
- 6.WHO. Control of the leishmaniasis. Technical Report Series 793. Geneve: World Health Organization; 1990. [Google Scholar]
- 7.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27(5):305–18. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Harhay MO, Olliaro PL, Costa DL, Costa CH. Urban parasitology: visceral leishmaniasis in Brazil. Trends Parasitol. 2011;27(9):403–9. doi: 10.1016/j.pt.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Peruhype-Magalhaes V, Martins-Filho OA, Prata A, Silva Lde A, Rabello A, Teixeira-Carvalho A, et al. Mixed inflammatory/regulatory cytokine profile marked by simultaneous raise of interferon-gamma and interleukin-10 and low frequency of tumour necrosis factor-alpha+ monocytes are hallmarks of active human visceral Leishmaniasis due to Leishmania chagasi infection. Clin Exp Immunol. 2006;146(1):124–32. doi: 10.1111/j.1365-2249.2006.03171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampaio MJ, Cavalcanti NV, Alves JG, Filho MJ, Correia JB. Risk factors for death in children with visceral leishmaniasis. PLoS Negl Trop Dis. 2010;4(11):e877. doi: 10.1371/journal.pntd.0000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa CH, Werneck GL, Costa DL, Holanda TA, Aguiar GB, Carvalho AS, et al. Is severe visceral leishmaniasis a systemic inflammatory response syndrome? A case control study. Rev Soc Bras Med Trop. 2010;43(4):386–92. doi: 10.1590/s0037-86822010000400010. [DOI] [PubMed] [Google Scholar]
- 12.Ambalavanan N, Carlo WA, D’Angio CT, McDonald SA, Das A, Schendel D, et al. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics. 2009;123(4):1132–41. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavare J, Grope I, Kalnins I, Gardovska D. High-mobility group box-1 protein, lipopolysaccharide-binding protein, interleukin-6 and C-reactive protein in children with community acquired infections and bacteraemia: a prospective study. BMC Infect Dis. 2010;10:28. doi: 10.1186/1471-2334-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carson JA, Baltgalvis KA. Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev. 2010;38(4):168–76. doi: 10.1097/JES.0b013e3181f44f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Sem Thromb Hemost. 2012;38(5):506–14. doi: 10.1055/s-0032-1305782. [DOI] [PubMed] [Google Scholar]
- 16.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 17.Rhodes J, Jones DH, Bleehen NM. Increased expression of human monocyte HLA-DR antigens and Fc gamma receptors in response to human interferon in vivo. Clin Exp Immunol. 1983;53(3):739–43. [PMC free article] [PubMed] [Google Scholar]
- 18.Spruijt NE, Visser T, Leenen LP. A systematic review of randomized controlled trials exploring the effect of immunomodulative interventions on infection, organ failure, and mortality in trauma patients. Crit Care. 2010;14(4):R150. doi: 10.1186/cc9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Remick DG. Interleukin-8. Crit Care Med. 2005;33(12 Suppl):S466–7. doi: 10.1097/01.ccm.0000186783.34908.18. [DOI] [PubMed] [Google Scholar]
- 20.Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. 2008;4(1):34–42. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- 21.Prakash D, Fesel C, Jain R, Cazenave PA, Mishra GC, Pied S. Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J Infect Dis. 2006;194(2):198–207. doi: 10.1086/504720. [DOI] [PubMed] [Google Scholar]
- 22.de Jong HK, van der Poll T, Wiersinga WJ. The systemic pro-inflammatory response in sepsis. J Innate Immun. 2010;2(5):422–30. doi: 10.1159/000316286. [DOI] [PubMed] [Google Scholar]
- 23.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91(10):3527–61. [PubMed] [Google Scholar]
- 24.Costa DL. Fatores de prognóstico na leishmaniose visceral: alterações clínicas e laboratoriais associadas à resposta imune, aos distúrbios da coagulação e à morte. [Doutorado] Belo Horizonte, MG: Universidade Federal de Minas Gerais; 2009. [Google Scholar]
- 25.al-Jurayyan NA, al-Nasser MN, al-Fawaz IM, al Ayed IH, al Herbish AS, al-Mazrou AM, et al. The haematological manifestations of visceral leishmaniasis in infancy and childhood. J Trop Pediatr. 1995;41(3):143–8. doi: 10.1093/tropej/41.3.143. [DOI] [PubMed] [Google Scholar]
- 26.Lomtadze ML, Khochava MA, Shalamberidze IA, Shilakadze MA, Dzhokhtaberidze TG. [Functional status of haemostasis system in patients with visceral leishmaniasis]. Georgian Med News. 2005;(128):59–62. Russian. [PubMed] [Google Scholar]
- 27.Lomtadze ML, Khochava MA, Shalamberidze IA, Kharaishvili VI, Vorob’eva EO. [Study of intravascular coagulation activation markers in patients with visceral leishmaniasis]. Georgian Med News. 2005;(124–125):47–50. Russian. [PubMed] [Google Scholar]
- 28.Zhang J, Alcaide P, Liu L, Sun J, He A, Luscinskas FW, et al. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE. 2011;6(1):e14525. doi: 10.1371/journal.pone.0014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin WC, Lin CF, Chen CL, Chen CW, Lin YS. Prediction of outcome in patients with acute respiratory distress syndrome by bronchoalveolar lavage inflammatory mediators. Exp Biol Med (Maywood). 2010;235(1):57–65. doi: 10.1258/ebm.2009.009256. [DOI] [PubMed] [Google Scholar]
- 30.Suffredini AF, Fantuzzi G, Badolato R, Oppenheim JJ, O’Grady NP. New insights into the biology of the acute phase response. J Clin Immunol. 1999;19(4):203–14. doi: 10.1023/a:1020563913045. [DOI] [PubMed] [Google Scholar]
- 31.Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. 2000;85(9):967–72. [PubMed] [Google Scholar]
- 32.Duarte MI, Corbett CE. Histopathological patterns of the liver involvement in visceral leishmaniasis. Rev Inst Med Trop Sao Paulo. 1987;29(3):131–6. doi: 10.1590/s0036-46651987000300003. [DOI] [PubMed] [Google Scholar]
- 33.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levi M, ten Cate H, van der Poll T. Endothelium: interface between coagulation and inflammation. Crit Care Med. 2002;30(5 Suppl):S220–4. doi: 10.1097/00003246-200205001-00008. [DOI] [PubMed] [Google Scholar]
- 35.Alonso DP, Ferreira AF, Ribolla PE, de Miranda Santos IK, do Socorro Pires e Cruz M, Aecio de Carvalho F, et al. Genotypes of the mannan-binding lectin gene and susceptibility to visceral leishmaniasis and clinical complications. J Infect Dis. 2007;195(8):1212–7. doi: 10.1086/512683. [DOI] [PubMed] [Google Scholar]
- 36.Endo Y, Nakazawa N, Iwaki D, Takahashi M, Matsushita M, Fujita T. Interactions of ficolin and mannose-binding lectin with fibrinogen/fibrin augment the lectin complement pathway. J Innate Immun. 2010;2(1):33–42. doi: 10.1159/000227805. [DOI] [PubMed] [Google Scholar]
- 37.Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, et al. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129(4):482–95. doi: 10.1111/j.1365-2567.2009.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sprong T, Mollnes TE, Neeleman C, Swinkels D, Netea MG, van der Meer JW, et al. Mannose-binding lectin is a critical factor in systemic complement activation during meningococcal septic shock. Clin Infect Dis. 2009;49(9):1380–6. doi: 10.1086/606054. [DOI] [PubMed] [Google Scholar]
- 39.Kager PA, Rees PH. Haematological investigations in visceral leishmaniasis. Trop Geogr Med. 1986;38(4):371–9. [PubMed] [Google Scholar]
- 40.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–3. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 41.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakubowski AA, Casper ES, Gabrilove JL, Templeton MA, Sherwin SA, Oettgen HF. Phase I trial of intramuscularly administered tumor necrosis factor in patients with advanced cancer. J Clin Oncol. 1989;7(3):298–303. doi: 10.1200/JCO.1989.7.3.298. [DOI] [PubMed] [Google Scholar]
- 43.Verma S, Kumar R, Katara GK, Singh LC, Negi NS, Ramesh V, et al. Quantification of parasite load in clinical samples of leishmaniasis patients: IL-10 level correlates with parasite load in visceral leishmaniasis. PLoS ONE. 2010;5(4):e10107. doi: 10.1371/journal.pone.0010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gautam S, Kumar R, Maurya R, Nylen S, Ansari N, Rai M, et al. IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis. 2011;204(7):1134–7. doi: 10.1093/infdis/jir461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conti P, Kempuraj D, Kandere K, Di Gioacchino M, Barbacane RC, Castellani ML, et al. IL-10, an inflammatory/inhibitory cytokine, but not always. Immunol Lett. 2003;86(2):123–9. doi: 10.1016/s0165-2478(03)00002-6. [DOI] [PubMed] [Google Scholar]
- 46.Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation. 2003;10(3–4):335–50. doi: 10.1038/sj.mn.7800198. [DOI] [PubMed] [Google Scholar]
- 47.Nylen S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol. 2007;28(9):378–84. doi: 10.1016/j.it.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Ansari NA, Kumar R, Gautam S, Nylen S, Singh OP, Sundar S, et al. IL-27 and IL-21 are associated with T cell IL-10 responses in human visceral leishmaniasis. J Immunol. 2011;186(7):3977–85. doi: 10.4049/jimmunol.1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bacellar O, Oliveira A, Jr, Jeronimo S, Carvalho EM. IL-10 and IL-12 are the main regulatory cytokines in visceral leishmaniasis. Cytokine. 2000;12(8):1228–31. doi: 10.1006/cyto.2000.0694. [DOI] [PubMed] [Google Scholar]
- 50.Cai B, Deitch EA, Ulloa L. Novel insights for systemic inflammation in sepsis and hemorrhage. Mediators Inflamm. 2010;2010:642462. doi: 10.1155/2010/642462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med. 2008;29(4):617–25. doi: 10.1016/j.ccm.2008.06.010. viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devignot S, Sapet C, Duong V, Bergon A, Rihet P, Ong S, et al. Genome-wide expression profiling deciphers host responses altered during dengue shock syndrome and reveals the role of innate immunity in severe dengue. PLoS ONE. 2010;5(7):e11671. doi: 10.1371/journal.pone.0011671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagenaar JF, Goris MG, Sakundarno MS, Gasem MH, Mairuhu AT, de Kruif MD, et al. What role do coagulation disorders play in the pathogenesis of leptospirosis? Trop Med Int Health. 2007;12(1):111–22. doi: 10.1111/j.1365-3156.2006.01792.x. [DOI] [PubMed] [Google Scholar]
- 54.Bray M. Pathogenesis of viral hemorrhagic fever. Curr Opin Immunol. 2005;17(4):399–403. doi: 10.1016/j.coi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Clark IA, Budd AC, Alleva LM, Cowden WB. Human malarial disease: a consequence of inflammatory cytokine release. Malar J. 2006;5:85. doi: 10.1186/1475-2875-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frade AF, Oliveira LC, Costa DL, Costa CH, Aquino D, van Weyenbergh J, et al. TGFB1 and IL8 gene polymorphisms and susceptibility to visceral leishmaniasis. Infect Genet Evol. 2011;11(5):912–6. doi: 10.1016/j.meegid.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 57.Ratsimandresy RA, Rappaport J, Zagury JF. Anti-cytokine therapeutics: history and update. Curr Pharm Des. 2009;15(17):1998–2025. doi: 10.2174/138161209788453130. [DOI] [PubMed] [Google Scholar]
- 58.Webster NR, Galley HF. Immunomodulation in the critically ill. Br J Anaesth. 2009;103(1):70–81. doi: 10.1093/bja/aep128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wheeler DS, Zingarelli B, Wheeler WJ, Wong HR. Novel pharmacologic approaches to the management of sepsis: targeting the host inflammatory response. Recent Pat Inflamm Allergy Drug Discov. 2009;3(2):96–112. doi: 10.2174/187221309788489779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parker RS, Clermont G. Systems engineering medicine: engineering the inflammation response to infectious and traumatic challenges. J R Soc Interface. 2010;7(48):989–1013. doi: 10.1098/rsif.2009.0517. [DOI] [PMC free article] [PubMed] [Google Scholar]