

Abstract
Expression of the gene encoding the S100 calcium–modulated protein family member MRP-14 (also known as S100A9) is elevated in platelets from patients presenting with acute myocardial infarction (MI) compared with those from patients with stable coronary artery disease; however, a causal role for MRP-14 in acute coronary syndromes has not been established. Here, using multiple models of vascular injury, we found that time to arterial thrombotic occlusion was markedly prolonged in Mrp14–/– mice. We observed that MRP-14 and MRP-8/MRP-14 heterodimers (S100A8/A9) are expressed in and secreted by platelets from WT mice and that thrombus formation was reduced in whole blood from Mrp14–/– mice. Infusion of WT platelets, purified MRP-14, or purified MRP-8/MRP-14 heterodimers into Mrp14–/– mice decreased the time to carotid artery occlusion after injury, indicating that platelet-derived MRP-14 directly regulates thrombosis. In contrast, infusion of purified MRP-14 into mice deficient for both MRP-14 and CD36 failed to reduce carotid occlusion times, indicating that CD36 is required for MRP-14–dependent thrombosis. Our data identify a molecular pathway of thrombosis that involves platelet MRP-14 and CD36 and suggest that targeting MRP-14 has potential for treating atherothrombotic disorders, including MI and stroke.
Introduction
Acute myocardial infarction (MI) commonly results from the atherosclerotic plaque disruption (1) and thrombosis that cause coronary artery occlusion (2). Angioscopic (3) and pathological (1) observations indicate that platelets constitute a major component of such thrombi, yet the precise platelet-related molecular events that immediately precede acute MI remain uncertain. Our previous study used a transcriptional profiling strategy to identify novel regulators of vascular inflammation and atherothrombosis by examining platelet mRNA transcripts that are differentially expressed in patients with ST-segment elevation myocardial infarction (STEMI) compared with those with stable coronary artery disease (4). Myeloid-related protein-14 (MRP-14, also referred to as S100A9) was one of the strongest predictors of STEMI that arose from the transcriptional profiling analysis. MRP-14, a member of the S100 family of calcium-modulated proteins, complexes with MRP-8 (S100A8), and together the MRP-8/14 heterodimer regulates myeloid cell function by modulating calcium signaling (5) and cytoskeletal reorganization (6), by operating as a chemoattractant (7), and by binding to cell surface receptors, including CD36 (8), toll-like receptor 4 (TLR4) (9), and receptor for advanced glycation end products (RAGE) (10). In two prospective, nested case-control studies, one in apparently healthy postmenopausal women (4) and the other in patients presenting with acute coronary syndromes (11), elevated plasma levels of MRP-8/14 predicted the risk of future cardiovascular events, independent of traditional cardiovascular risk factors and high-sensitivity C-reactive protein (hs-CRP). Elevated plasma levels of MRP-8/14 also serve as an early and sensitive marker of myocardial necrosis in the setting of chest pain (12).
Despite the presence of Mrp8 mRNA transcripts, Mrp14–/– mice lack both MRP-8 and MRP-14 protein, possibly due to the instability of MRP-8 protein in the absence of MRP-14 (5, 13). In vitro studies with Mrp14–/– neutrophils showed markedly diminished migration through endothelial monolayers and attenuated chemokinesis in a three-dimensional collagen matrix (13). An essential role for MRP-8/14 in leukocyte recruitment in vivo is supported by evidence that Mrp14–/– mice have diminished granulocyte recruitment during tissue wound healing (6) and acute pancreatitis (14). Recent studies using Mrp14–/– mice have also demonstrated that MRP-8 and MRP-14 play a regulatory role in endotoxin-induced phagocyte function by binding to TLR4 and promoting myeloid MyD88–dependent activation of NF-κB (9). Last, we have reported that MRP-8/14 broadly regulates vascular inflammation and contributes to the biological response to vascular injury in murine models of atherosclerosis, vasculitis, and restenosis by promoting leukocyte recruitment (15).
Despite our identification of MRP14 transcripts in platelets, freshly isolated human bone marrow megakaryocytes, and megakaryocytes generated in vitro by differentiation of human CD34-positive cells (4), the role of MRP-8/14 in thrombosis and hemostasis is unknown. In this study, we provide evidence that platelet-derived MRP-14 directly modulates platelet function and thrombosis without influence on tail bleeding time or other hemostatic parameters.
Results
Photochemical injury–induced arterial thrombosis is delayed in Mrp14–/– mice.
To elucidate the effect of MRP-8/14 on the development of arterial thrombosis in real time, carotid arteries of WT and Mrp14–/– mice were subjected to the Rose Bengal model of thrombosis, an endothelial cell photochemical injury model caused by local free-radical release (16, 17). We then continuously monitored carotid artery blood flow with a vascular flow probe. Mean time to occlusive thrombus formation in WT mice was 27.1 ± 5.9 minutes and was significantly prolonged in Mrp14–/– mice to 46.6 ± 22.9 minutes (n = 16 per group, P = 0.004) (Figure 1A). We harvested carotid arteries 25 minutes after photochemical injury for histological analysis. Both perfusion and nonperfusion fixation techniques were used in order to visualize thrombus in situ. In nonperfused animals, a fibrin-platelet–rich thrombus with some red blood cells was evident in the lumen of WT arteries (Figure 1B). In contrast, the lumen of Mrp14–/– arteries was filled with blood at this 25-minute time point when the flow probe indicated that the vessel was widely patent. Nonocclusive thrombus (arrow) was visible along the wall of vessel. With perfusion fixation, we found that the occlusive thrombus within the lumen of injured WT arteries was still visible, whereas the lumen of Mrp14–/– arteries was devoid of blood elements, indicating delayed thrombus formation and the instability of nonocclusive thrombus formed in Mrp14–/– mice. Immunohistochemical analysis of serial sections of injured arteries from WT and Mrp14–/– mice with anti–MRP-14 and the platelet-specific anti-GPIIb antibodies showed positive MRP-14 staining that colocalized with platelets in WT arteries (Figure 1C). We observed no staining for MRP-14 in Mrp14–/– arteries.
Figure 1. MRP-14 deficiency prolongs thrombotic occlusion time.

(A) Occlusion times following photochemical injury of the carotid artery in WT and Mrp14–/– mice (mean ± SD, n = 16 per group). (B) Histologic sections of nonperfused and perfusion-fixed carotid arteries 25 minutes after injury demonstrating occlusive thrombus in WT arteries and patency in Mrp14–/–arteries. (C) Immunohistochemistry of MRP-14 and the platelet-specific marker GPIIb after carotid artery photochemical injury. (D) Thrombus formation in vivo after laser-induced injury to the arteriolar wall in the cremaster microcirculation of Mrp14–/– mice was compared with that of WT mice using intravital microscopy (see Supplemental Videos 1 and 2). Platelets were labeled in vivo using an FITC-conjugated rat anti-mouse CD41 antibody. Representative intravital images at indicated times following laser pulse. Scale bars: 50 μm (B) and 20 μm (C and D). (E) Continuous, real-time thrombosis profiles of one representative experiment (n = 10–15 arterioles per group).
Impaired thrombus formation after laser-induced injury of the cremaster microvasculature in Mrp14–/– mice.
We used intravital microscopy to compare thrombus formation in vivo after laser-induced injury to the arteriolar wall in the cremaster microcirculation of Mrp14–/– mice with that of WT mice (18). In WT mice, platelet accumulation in arterioles was evident within 30 seconds of laser injury (Figure 1, D and E, and Supplemental Videos 1 and 2; supplemental material available online with this article; doi: 10.1172/JCI70966DS1). In contrast, platelet accumulation was markedly attenuated in Mrp14–/– mice (mean percentage of inhibition over time = 85.0% ± 5.1%, n = 10–15 arterioles per group). Initial platelet adhesion and small platelet aggregates were observed, but developing thrombi were unstable and embolized frequently.
Platelet count and coagulation assays are similar in WT and Mrp14–/– mice.
Having observed delayed thrombosis in Mrp14–/– mice, we set out to determine the mechanism by first performing screening platelet and coagulation assays in WT and Mrp14–/– mice (Supplemental Figure 1). The platelet count was similar in WT (736,000 ± 307,000 platelets/μl) and Mrp14–/– (753,000 ± 241,000 platelets/μl, P = 0.90) mice. We assessed the coagulation activity of plasma using the activated partial thromboplastin time (aPTT) and a thrombin generation assay. The aPTT was not prolonged in Mrp14–/–mice (WT: 66 ± 28 seconds versus Mrp14–/–: 55 ± 16 seconds, P = 0.54). Tissue factor–induced total thrombin generation was similar in WT and Mrp14–/– plasma (WT: 21,055 ± 407 versus Mrp14–/–: 21,001 ± 4,041 arbitrary fluorescent units, P = 0.54). Moreover, multiple parameters of thrombin generation, including the lag time of thrombin generation, the maximum rate of thrombin generation, and the time to reach maximal thrombin activity, were comparable in WT and Mrp14–/– mice (data not shown). Taken together, these data indicate that neither platelet count nor coagulation parameters likely account for delayed thrombosis in Mrp14–/– mice.
Platelets express MRP-8 and MRP-14 proteins.
Although we have detected MRP14 transcripts in human platelets, freshly-isolated human bone marrow megakaryocytes, and megakaryocytes generated in vitro by differentiation of human CD34–positive cells (4), we turned our attention to evaluating the expression of MRP-8 and MRP-14 protein in mouse platelets by Western blot analysis. Both MRP-8 and MRP-14 were detected in gel-filtered and washed platelets from WT, but not Mrp14–/–, mice (Figure 2A). To exclude the possibility that contaminating leukocytes were responsible for these S100 proteins, we also investigated MRP-8/14 expression using immunofluorescence and costaining for the platelet-specific marker glycoprotein IX (GPIX). Robust MRP-8/14 staining was evident in GPIX-positive human platelets (Figure 2B). Finally, to further examine platelet MRP-8/14 expression, we performed 2-color flow cytometry on gel-filtered human platelets using platelet (anti-GPIIb/IIIa), leukocyte (anti-CD45), and anti–MRP-8/14 antibodies (Supplemental Figure 2) and observed abundant double-positive (GPIIb/IIIa-positive/MRP-8/14–positive) platelets (Supplemental Figure 2B). Importantly, this staining was not accounted for on the basis of contaminating leukocytes, because MRP-8/14–positive cells were CD45 negative (Supplemental Figure 2C).
Figure 2. Platelets express MRP-8 and MRP-14.

(A) MRP-8 (8 kDa) and MRP-14 (14 kDa) protein expression was examined in gel-filtered (lanes 1 and 2) and washed (lanes 3 and 4) WT and Mrp14–/– platelets. Platelets were lysed with SDS-PAGE–reduced sample buffer and then immunoblotted sequentially with anti–MRP-8, anti–MRP-14, and anti-tubulin antibodies. (B) Expression of MRP-8/14 in human platelets adherent to fibrinogen-coated coverslips was investigated using combined immunofluorescence with one antibody that recognizes the MRP-8/14 complex and another the platelet-specific marker GPIX. Robust MRP-8/14 staining was evident in GPIX-positive human platelets. Scale bars: 10 μm. (C) Platelet surface expression of MRP-14 in resting and thrombin-stimulated (FIIa-stimulated) platelets was determined by flow cytometry (mean fluorescence intensity; n = 4 per group). (D) MRP-8/14 and CD40L protein content in gel-filtered human platelets (mean ± SD, n = 5 normal human donors). (E) Immunoblot of plasma (18 μl per lane) from noninjured animals and from animals 10 minutes after photochemical carotid artery injury using anti–MRP-14 antibody. Equal loading was verified by IgG light chain. (F) Paired plasma and serum levels of MRP-8/14 (μg/ml) from 5 normal human donors.
Next, we evaluated whether agonist stimulation is associated with increased surface expression and secretion of MRP-14. Thrombin stimulation of platelets was accompanied by increased platelet surface expression of MRP-14 (Figure 2C). To verify secretion of MRP-14, gel-filtered human platelets were stimulated with thrombin, pelleted, and the supernatant was harvested for determination of MRP-8/14 concentration by ELISA. The concentration of MRP-8/14 in 1 ml of supernatant from 400 million thrombin-stimulated platelets was 1.03 ± 0.56 μg/ml.
To determine the relative abundance of MRP-8/14 compared with other intracellular platelet proteins/agonists, we assayed the protein content of human platelet MRP-8/14 compared with that of platelet CD40L, a platelet α-granule agonist that binds to GPIIb/IIIa and promotes thrombosis in an autocrine manner (19). We found that human platelet MRP-8/14 protein was more abundant than human platelet CD40L (Figure 2D). To verify that platelet activation and thrombosis are associated with secretion of MRP-14, we hypothesized that MRP-14 levels would increase after carotid artery photochemical injury. Although the source of MRP-14 is uncertain (i.e., platelet, leukocyte, or endothelial cell derived), plasma MRP-14 levels increased 10 minutes after injury compared with levels in noninjured animals (Figure 2E). In addition, when plasma and serum levels of MRP-8/14 in paired samples from 5 normal human donors were compared, we found that MRP-8/14 levels were significantly higher in serum than in plasma (Figure 2F). Taken together, these observations indicate that platelets express both MRP-8 and MRP-14 protein and are capable of secreting MRP-8/14 after agonist stimulation and thrombus formation.
Deficiency of MRP-14 attenuates platelet thrombus formation under flow ex vivo.
Having demonstrated that thrombus formation is attenuated in Mrp14–/– mice and that platelets express MRP-8/14, we next determined whether platelet MRP-14 itself regulates platelet function. We examined the role of MRP-14 in platelet thrombus formation under physiologic arterial shear conditions using a highly automated dynamic flow system (20, 21). Thrombus formation was achieved by perfusion of anticoagulated blood labeled with rhodamine 6G through collagen-coated rectangular capillaries at an arterial shear rate of 625 s–1. We quantified the thrombus area and volume in real time using computer-assisted imaging analysis (Figure 3A). Perfusion of blood resulted in the rapid formation of platelet thrombi that were significantly reduced in Mrp14–/– compared with WT mice (percentage of inhibition of thrombus area = 49, P < 0.001; percentage of inhibition of thrombus volume = 60, P < 0.001) (Figure 3, B and C).
Figure 3. MRP-14 deficiency attenuates thrombus formation under flow and is associated with defects in collagen-induced platelet activation.

(A) Platelet thrombi on collagen-coated capillaries following perfusion of rhodamine 6G–labeled blood from WT and Mrp14–/– mice at an arterial shear rate of 625 s–1. Original magnification, ×40; observation area, 360 × 270 mm. Thrombus formation (B, area; C, volume) was quantified using computer-assisted imaging analysis (n = 3–5 per group). Flow cytometric analysis of P-selectin expression (D) and assessment of GPIIb/IIIa activation using staining with the JON/A antibody (E) following stimulation of washed platelets from WT (black bars) and Mrp14–/– (white bars) mice with 0 to 10 μg/ml collagen (n = 5 per group).
Given the defect in platelet thrombus formation on collagen under flow, we assessed platelet activation by monitoring the expression of P-selectin and activated GPIIb/IIIa (JON/A-positive staining) in response to agonist stimulation. Washed platelets from WT and Mrp14–/– mice were stimulated with collagen, thrombin, arachidonic acid, or ionomycin. P-selectin expression was significantly decreased in Mrp14–/– compared with that in WT platelets following stimulation with collagen at 5 and 10 μg/ml (P = 0.005 and P = 0.048, respectively; Figure 3D) and with 800 μM arachidonic acid (P = 0.027) (Supplemental Figure 3), but not with thrombin or ionomycin (Supplemental Figure 3). Collagen-induced activation of GPIIb/IIIa was also significantly reduced (P = 0.010) in Mrp14–/– versus WT platelets (Figure 3E). Importantly, we verified that the expression levels of the platelet receptors GPIbα, GPVI, and α2β1 were comparable on WT and Mrp14–/– platelets (Supplemental Figure 4A).
We also performed platelet secretion and aggregation studies to further characterize the nature of the Mrp14–/– platelet defect. We assessed dense granule secretion by measuring agonist-induced secretion of [14C] 5-hydroxytryptamine and observed no significant difference in the uptake of [14C] 5-hydroxytryptamine between WT and Mrp14–/– platelets (44% and 47%, respectively). After collagen stimulation, Mrp14–/– platelets secreted 45% ± 19% of [14C] 5-hydroxytryptamine compared with 33% ± 3% for WT platelets (P = 0.32; Supplemental Figure 4E). Similarly, there was no difference in α-thrombin–induced platelet secretion (Mrp14–/– platelets secreted 98% ± 1% of [14C] 5-hydroxytryptamine versus 95% ± 4% for WT platelets, P = 0.28; Supplemental Figure 4E). We monitored platelet aggregation during secretion experiments and found no difference in collagen- or thrombin-induced platelet aggregation between WT and Mrp14–/– platelets (Supplemental Figure 4F). Finally, to determine whether MRP-14 modulates the platelet aggregation threshold, we evaluated ADP-induced fibrinogen binding by flow cytometry over a range of ADP concentrations and found that MRP-14 deficiency had no effect on ADP-stimulated fibrinogen binding (Supplemental Figure 4G).
Hemostasis as determined by tail vein bleeding time is unimpaired in Mrp14–/– mice.
To assess the role of MRP-14 in hemostasis, we examined tail vein bleeding times. There was no difference in tail bleeding times between WT and Mrp14–/–mice using complete cessation of bleeding for either 3 minutes or for 30 seconds as the criterion for bleeding time determination. Mean bleeding time for WT mice was 398 ± 200 seconds compared with 384 ± 218 seconds for Mrp14–/– mice (n = 16 per group, P = 0.71) when complete absence of bleeding for 3 minutes was required (Figure 4A). With the shorter bleeding cessation period of 30 seconds, the bleeding time in Mrp14–/– mice was similar to that in WT mice (74 ± 26 seconds versus 79 ± 32 seconds, respectively, P = 0.64; Figure 4B).
Figure 4. MRP-14 deficiency has no effect on tail bleeding time.

To assess the role of MRP-14 in hemostasis, tail bleeding times were assessed in WT and Mrp14–/– mice using complete cessation of bleeding either for 3 minutes (A) or 30 seconds (B) as the criterion for determination of bleeding time (mean ± SD, n = 16 per group).
We examined additional parameters of the primary hemostatic response, including platelet adhesion to the collagen peptide that binds α2β1 integrin GFOGER and platelet adhesion to and spreading on vWF. Platelet adhesion and spreading were similar in WT and Mrp14–/– platelets (Supplemental Figure 4, B–D).
Transfusion of WT platelets shortens the prolonged time to carotid artery occlusion in Mrp14–/– mice.
Although we have provided evidence here that MRP-14 deficiency is associated with prolonged time to carotid artery occlusion after photochemical injury and with defects in collagen-stimulated platelet activation and shear-induced thrombus formation in collagen-coated capillaries, we used mice with global, rather than tissue-specific, deficiency of MRP-14, thereby limiting our ability to definitively conclude whether platelet-derived MRP-14 is critical for thrombus formation. To address this issue, we performed adoptive transfer or transfusion of WT and Mrp14–/– donor platelets into Mrp14–/– recipient mice prior to photochemical injury. Mrp14–/– recipient mice that received Mrp14–/– donor platelets formed occlusive thrombi within 44.0 ± 10.3 minutes (Figure 5A). Strikingly, the time to occlusive thrombus formation was significantly shortened in Mrp14–/–recipient mice receiving WT donor platelets to 30.3 ± 5.2 minutes (P = 0.02), nearly restoring the occlusion time to that of WT mice (27.1 ± 5.9 minutes, Figure 1A).
Figure 5. Transfusion of WT platelets or infusion of purified MRP-14 or MRP-8/14 shortens the prolonged time to carotid artery occlusion in Mrp14–/–mice.

(A) Transfusion of WT or Mrp14–/– donor gel-filtered platelets (1 × 108 in 250 μl) into Mrp14–/– recipient mice prior to carotid artery photochemical injury. Occlusion times following injury for Mrp14–/– recipient mice receiving WT donor or Mrp14–/– donor platelets (mean ± SD, n = 6 per group). (B) Thrombotic occlusion time with intravenous infusion of saline (n = 14), purified human MRP-8 (0.08 μg/g mouse; n = 9), MRP-14 (0.08 μg/g mouse, n = 10), or MRP-8/14 (0.08 μg/g mouse; n = 10) into Mrp14–/– recipient mice prior to carotid artery photochemical injury. (C) Exogenous MRP-8/14 enhanced platelet thrombus formation on collagen under flow. Platelet thrombi on collagen-coated capillaries following perfusion (shear rate of 625 s–1) of rhodamine 6G–labeled blood from Mrp14–/– blood that was treated with purified human MRP-8/14 (5 μg/ml) or control buffer. Original magnification, ×40; observation area, 360 × 270 mm. (D) Continuous, real-time thrombosis profiles of one representative experiment. (E) Platelet thrombus formation under flow in anticoagulated human whole blood was evaluated in the presence of the anti–MRP-14 monoclonal antibody 1H9 versus control antibody (10 μg/ml). Continuous, real-time thrombosis profiles of one representative experiment.
Role of intracellular versus extracellular MRP-8/14 in thrombosis.
Transfusion of WT gel-filtered platelets nearly corrected the thrombotic defect in Mrp14–/– mice, indicating that platelet MRP-8/14 content regulates thrombosis (Figure 5A). To determine whether extracellular MRP-8/14 action modulates thrombosis, we infused purified, recombinant human MRP-8, MRP-14, or MRP-8/14 (0.08 μg/g mouse) into Mrp14–/– mice (Figure 5B). Intravenous infusion of purified MRP-14 (30.9 ± 10.5 versus saline control infusion 46.4 ± 20.5 minutes, P = 0.025) or MRP-8/14 (25.7 ± 13.3 versus saline control 46.4 ± 20.5 minutes, P = 0.007) into Mrp14–/– mice shortened thrombotic occlusion time to that observed in WT mice (27.1 ± 5.9 minutes) (Figure 5B). In contrast, infusion of purified MRP-8 alone had no significant effect on thrombotic occlusion time (MRP-8: 41.1 ± 24.4 minutes, P = 0.625), strongly suggesting that MRP-14 is responsible for thrombotic action of the MRP-8/14 heterodimer complex.
To verify the importance of extracellular MRP-8/14 in modulating platelet function, we assessed the effect of purified MRP-8/14 on platelet thrombus formation under flow conditions. Purified MRP-8/14 enhanced thrombus formation of Mrp14–/– whole blood perfused through collagen-coated capillaries (Figure 5, C and D). Finally, to extend these findings to human platelets, we examined the effect of targeting extracellular MRP-8/14 on platelet aggregate formation by assessing the effect of anti–MRP-14 monoclonal antibody on platelet thrombus formation under flow of anticoagulated human whole blood and found that anti–MRP-14 antibody inhibited platelet thrombus formation compared with control antibody (Figure 5E).
Identification of the candidate platelet receptor for MRP-14.
Having observed that extracellular MRP-14 promotes thrombosis, we next sought to identify candidate platelet receptor(s). Putative receptors for MRP-8/14 on target cells include CD36 (8), RAGE (10), and TLR4 (9). Interestingly, both CD36 (22) and RAGE (23) signaling have been directly implicated in platelet activation and thrombosis. Platelets also express functional levels of toll-like receptor 4 (TLR4) (24). We evaluated shear-induced platelet aggregation and thrombus formation in anticoagulated human whole blood in the presence of blocking antibodies against CD36, RAGE, or TLR4 (Figure 6). Anti-CD36 monoclonal antibody inhibited shear-induced thrombus formation (percentage of inhibition = 53.8 ± 16.9, P = 0.012) (Figure 6, A–C), and the extent of inhibition was comparable to that observed with anti–MRP-14 antibody (Figure 5E). In contrast, blocking antibodies against either RAGE or TLR4 had no effect on platelet aggregate formation under these experimental conditions (Figure 6, A–C).
Figure 6. CD36 is a putative receptor for MRP-14.

(A) Platelet thrombus formation under flow in anticoagulated human whole blood was evaluated in the presence of blocking monoclonal antibodies (10 μg/ml) against CD36, RAGE, TLR4, or control IgG. Original magnification, ×40; observation area, 360 × 270 mm. (B) Continuous, real-time thrombosis profiles of platelet thrombus formation under flow quantified (C) as a percentage of aggregation relative to control antibody (n = 3–5 per antibody).
Next, to determine whether CD36 is a candidate receptor for MRP-14, we examined the direct binding of MRP-14 to purified soluble CD36 in a plate binding assay. MRP-14 bound to soluble CD36-coated, but not BSA-coated, wells (Figure 7A).
Figure 7. CD36 is required for MRP-14 action.

(A) Binding of purified MRP-14 (0–2.5 μg/ml) to purified soluble CD36-coated or BSA-coated wells. (B) Thrombotic occlusion time after carotid artery photochemical injury in indicated mouse strains and occlusion time with intravenous infusion of saline or purified human MRP-14 (0.08 μg/g mouse) into Mrp14–/– or Mrp14–/– Cd36–/– recipient mice prior to photochemical injury. (C) Purified MRP-14 restores platelet thrombus formation under flow in Mrp14–/–, but not Mrp14–/–Cd36–/–, murine whole blood. Platelet thrombi on collagen-coated capillaries following perfusion (shear rate of 625 s–1) of rhodamine 6G–labeled Mrp14–/– blood from WT, Mrp14–/–, or Mrp14–/–Cd36–/– mice that was treated with purified human MRP-14 (5 μg/ml) or control buffer. Original magnification, ×40; observation area, 360 × 270 mm. (D) Continuous, real-time thrombosis profiles of the average fluorescence of three independent experiments. MRP-14 induced phosphorylation of VAV (E) and JNK (F) in platelets. Gel-filtered human platelets (2 × 108/ml) containing 2 mM CaCl2 and 1 mM MgCl2 were incubated with 50 μg/ml oxLDL, 1 μg/ml MRP-14, or a combination of these for 10 minutes, and platelet lysates were analyzed by immunoblotting with anti-phosphoprotein antibodies. The membranes were then stripped and reprobed with antibodies against the total relevant protein and actin. Results are representative of three independent experiments from different donors.
To establish whether CD36 is required for MRP-14 action, we crossed Mrp14–/– mice with CD36-deficient (Cd36–/–) mice to generate compound mutants (Mrp14–/–Cd36–/–) and then subjected them to carotid photochemical injury. Deficiency of CD36 alone had no effect on thrombotic occlusion time (CD36: 23.1 ± 8.3 minutes versus WT: 27.1 ± 5.9 minutes, P = 0.167) in this model (Figure 7B), a finding similar to that observed with the FeCl3 carotid injury model using 12.5% FeCl3 (25). Doubly deficient Mrp14–/–Cd36–/– mice have prolonged thrombotic occlusion time that is no different than that in singly deficient Mrp14–/– mice (44.7 ± 16.2 minutes versus 46.6 ± 22.9 minutes, respectively, P = 0.803; Figure 7B). In direct contrast to experiments performed with Mrp14–/– mice, in which infusion of purified MRP-14 (0.08 μg/g mouse) shortened the prolonged time to thrombotic occlusion (saline: 46.4 ± 20.5 minutes versus purified MRP-14: 30.9 ± 10.5 minutes, P = 0.026; Figure 7B), infusion of MRP-14 into doubly deficient Mrp14–/– Cd36–/– mice had no significant effect on the prolonged occlusion time (saline: 48.2 ± 15.3 minutes versus purified MRP-14: 47.2 ± 18.8 minutes, P = 0.884; Figure 7B). Infusion of a 5-fold increased amount of MRP-14 (0.4 μg/g mouse weight) into Mrp14–/– Cd36–/– mice also failed to shorten the prolonged occlusion time (43.0 ± 8.7 minutes, P = 0.40).
To verify the importance of CD36 in modulating platelet function in response to extracellular MRP-14, we assessed the effect of purified MRP-14 on platelet thrombus formation under flow using Mrp14–/– Cd36–/– whole blood. While purified MRP-14 restored platelet thrombus formation of Mrp14–/– whole blood perfused through collagen-coated capillaries (Figure 7, C and D), purified MRP-14 had minimal enhancement of platelet thrombus formation of doubly deficient Mrp14–/– Cd36–/– whole blood (Figure 7, C and D). Further, we also examined whether MRP-14 is capable of activating platelets in a CD36-dependent manner. Oxidized LDL (oxLDL) initiates a CD36-mediated signaling cascade involving recruitment of Src family kinases (VAV, FYN, and LYN) and activation of JNK (26). Similarly to oxLDL, MRP-14 induces phosphorylation of VAV and JNK (Figure 7, E and F). Since hyperlipidemia increases both plasma oxLDL and MRP-14 concentrations (15), we examined the phosphorylation of VAV and JNK after stimulating platelets with both oxLDL and MRP-14. Interestingly, MRP-14 potentiated oxLDL-induced phosphorylation of both VAV and JNK. Taken together, these observations indicate that platelet CD36 is required for MRP-14 action (Figure 7, E and F).
Platelet MRP-8/14 in human coronary thrombus.
MI is most commonly caused by atherosclerotic plaque rupture and occlusive thrombus formation (1). To begin to examine the pathophysiological relevance of our findings of MRP-8/14 expression in platelets, we obtained coronary artery thrombi from patients (n = 4) presenting to the cardiac catheterization laboratory with acute STEMI. Angiography performed on one patient demonstrated thrombotic occlusion of the proximal right coronary artery (Figure 8A) that was treated with aspiration thrombectomy followed by balloon angioplasty and stent deployment (Figure 8B), resulting in a widely patent right coronary artery with no significant luminal narrowing (Figure 8C). The thrombectomy catheter retrieved multiple coronary artery thrombi (Figure 8D) that were then stained for platelets and MRP-8/14 using immunofluorescence microscopy. Platelet and MRP-8/14 staining were abundant and colocalized in this human coronary artery thrombus (Figure 8, E–H). These findings were confirmed in the examination of intracoronary thrombi from three additional STEMI patients (Supplemental Figure 5).
Figure 8. Platelet expression of MRP-8/14 in human coronary artery thrombus.
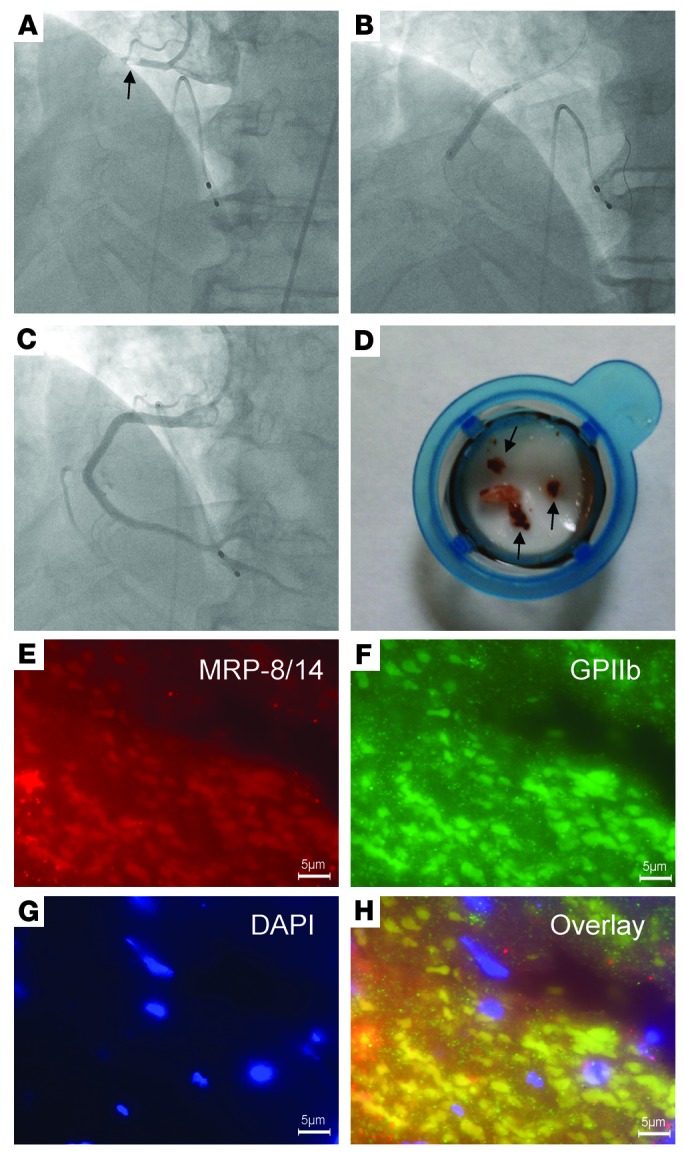
Coronary artery thrombus was obtained from a patient presenting to the cardiac catheterization laboratory with acute inferior STEMI. (A) Angiogram demonstrating thrombotic occlusion (arrow) of the proximal right coronary artery. (B) Deployment of stent at the site of the right coronary artery occlusion. (C) Final angiogram showing a widely patent right coronary artery with no significant luminal narrowing. (D) Thrombectomy catheter aspirate after passage through filter shows coronary artery thrombi (arrows). Immunofluorescence staining of coronary artery thrombus with anti–MRP-8/14 (E) and antiplatelet GPIIb (F) antibodies. Nuclei are stained with DAPI (G). Colocalization of MRP-8/14 and platelets are depicted in the overlay (H). Scale bars: 5 μm (E–H).
Discussion
In this study, we identified a new pathway of thrombosis involving platelet MRP-14 and CD36 that does not affect bleeding time. This conclusion is supported by the following data: (a) the time to thrombotic occlusion was significantly prolonged in Mrp14–/– mice; (b) laser-induced platelet thrombi in the cremaster microvasculature were reduced and less stable in Mrp14–/– mice; (c) platelet thrombus formation under flow was reduced in whole blood from Mrp14–/– mice; (d) MRP-8 and MRP-14 were expressed in platelets, and agonist stimulation led to increased platelet surface expression and secretion of MRP-8/14; (e) platelet count, aPTT, thrombin generation, and bleeding time were similar in WT and Mrp14–/– mice; (f) transfusion of WT platelets or infusion of purified MRP-14 or MRP-8/14 into Mrp14–/–mice shortened the prolonged carotid artery occlusion time in Mrp14–/–mice; (g) compound deficiency of MRP-14 and CD36 resulted in a prolonged carotid artery occlusion time despite infusion of purified MRP-14; (h) MRP-14 was capable of activating platelets in a CD36-dependent manner (i.e., induces phosphorylation of VAV and JNK); and (i) robust expression of MRP-8/14 was evident in platelet-rich coronary artery thrombi, causing acute MI.
MRP-8/14 complexes are also present in mouse cells, and extensive biochemical characterization has confirmed that mouse MRP-14 is functionally equivalent to its human counterpart (27). Analyses of mice that lack MRP-8 and MRP-14 have provided important insights into the function of these proteins. Although homozygous deletion of MRP-8 results in embryonic lethality (28), deletion of the MRP14 gene does not affect viability and results in the additional loss of MRP-8 protein (5, 13). Failure to produce mature MRP-8 protein in the presence of normal MRP8 mRNA production likely results from an instability of MRP-8 in the absence of MRP-14. Thus, the Mrp14–/– mice used in this and previous studies lack both MRP-8 and MRP-14 protein and MRP-8/14 complexes.
MRP-8/14 is known to be expressed by various cell types, including neutrophils (29), monocytes (29), tissue macrophages under conditions of chronic inflammation (30), mucosal epithelium (31), and involved epidermis in psoriasis (32). Immunofluorescence, immunoblotting, and ELISA data from the present study now indicate that platelets also express and secrete MRP-8/14 protein. Immunofluorescence staining of purified platelets and coronary artery thrombi as well as flow cytometry of purified platelets indicate that MRP-8/14 protein is expressed in platelets. However, not all platelets appeared to express MRP-8/14. The basis for MRP-8/14 high versus low/negative platelet staining is unknown. Platelet age (young versus old) and activation status are known to influence platelet protein expression (33) and are the focus of ongoing studies.
The established roles of intracellular and extracellular MRP-8/14 in leukocyte function provide important clues to the putative action of MRP-8/14 in platelet function and thrombosis. Experiments with leukocytes isolated from Mrp14–/– mice have demonstrated aberrant calcium signaling and blunted calcium responses following chemokine stimulation (34). MRP-8/14 further appears to modulate calcium-coupled arachidonic acid signaling by binding to arachidonic acid in a calcium-dependent manner (35, 36) and by facilitating translocation of arachidonic acid to the cytoskeleton (37, 38). Intracellular MRP-8/14 also modulates cytoskeletal reorganization by promoting polymerization of microtubules (6).
Our observations that transfusion of WT platelets or infusion of purified MRP-14 or MRP-8/14 into Mrp14–/– mice shortened the prolonged carotid artery occlusion time of Mrp14–/– mice suggest that extracellular, rather than intracellular, MRP-14 is largely responsible for MRP-14 action in thrombosis. In response to cytokines or during contact with activated endothelium, myeloid cells secrete heterodimeric MRP-8/14, which is the dominant extracellular form (39–41), through a tubulin-dependent “alternative” secretion pathway (42). Extracellular MRP-8/14 is then able to bind to receptors on target cells, including CD36 (8), RAGE (10), TLR4 (9), special carboxylated N-glycans (43), and heparin-like glycoaminoglycans (44). Interestingly, both CD36 (22) and RAGE (23) signaling have been directly implicated in platelet activation and thrombosis. Platelets also express functional levels of TLR4 (24), which may contribute to thrombocytopenia through neutrophil-dependent pulmonary sequestration in response to LPS (45). Antibody-blocking experiments performed in the present study revealed CD36 as the most likely platelet receptor for MRP-14.
CD36 is an 88-kDa integral membrane protein expressed on platelets, monocytes/macrophages, microvascular endothelium, adipocytes, muscle cells, and specialized epithelium (e.g., retinal pigment epithelium) (46, 47). CD36 was first isolated and structurally characterized from platelets (48, 49), and a series of studies indicate that platelet CD36 binding to oxLDL (25) or endothelial cell–derived microparticles (22) contributes to platelet activation and thrombosis in mice. Despite having short intracytoplasmic domains, CD36 is capable of serving as a signaling receptor (50). oxLDL binding to CD36 induces platelet activation via a signaling cascade involving MAP kinase kinase 4 (MKK4), the MAP kinase JNK2, and Src kinases (e.g., VAV, FYN, LYN) (26). Interestingly, Cd36–/– mice have normal tail vein bleeding times and do not exhibit a bleeding diathesis (22). However, CD36 deficiency is associated with variable protection against thrombosis in the FeCl3 injury model that was dependent on the FeCl3 dose — namely, using a lower dose of FeCl3 (7.5% for 1 minute compared with 12.5% for 3 minutes, used by Podrez et al., ref. 25). Ghosh and colleagues concluded that CD36 ligands are generated during vascular injury and that the signals induced by these ligands contribute to thrombus formation by paracrine-like action (22). The results of our study indicate that MRP-14 is likely one such ligand. Our findings that transfusion of WT platelets into Mrp14–/– mice or infusion of purified MRP-14 into Mrp14–/–, but not Mrp14–/– Cd36–/– (Figure 7B), mice shortened the prolonged carotid artery occlusion time in Mrp14–/– mice strongly suggest that platelet MRP-14 influences thrombosis secondary to secretion and binding to platelet CD36. Furthermore, while purified MRP-14 restored platelet thrombus formation in Mrp14–/– whole blood perfused through collagen-coated capillaries (Figure 7, C and D), purified MRP-14 had little effect on platelet thrombus formation in doubly deficient Mrp14–/– Cd36–/– whole blood (Figure 7, C and D). We performed a plate binding assay, which demonstrated that purified MRP-14 is capable of binding directly to soluble CD36 (Figure 7A) and of activating platelets in a CD36-dependent manner, as indicated by the phosphorylation of VAV and JNK (Figure 7, E and F). Similarly to Podrez and coworkers, who used 12.5% FeCl3 (25), we did not observe a thrombosis defect in Cd36–/– mice using the photochemical injury model. In contrast, Ghosh and colleagues reported a thrombosis defect using 7.5% FeCl3 (22). The basis for the discrepant findings in Cd36–/– mice is uncertain, but they suggest that there may be model-dependent factors. The fact that the time to thrombotic occlusion after photochemical injury was prolonged in Mrp14–/–, but not Cd36–/–, mice raises the possibility that intracellular (e.g., cytoskeletal reorganization, calcium-coupled arachidonic acid signaling) as well as extracellular MRP-14 action modulates platelet function. Indeed, a role for intracellular MRP-14 is supported by Figure 7D, which shows that the addition of purified MRP-14 restored platelet thrombus formation in Mrp14–/– whole blood perfused through collagen-coated capillaries to a level below that of WT whole blood. Its role is also supported by the finding that arachidonic acid–induced platelet activation was attenuated in Mrp14–/– platelets (Supplemental Figure 3C). The possibility that MRP-14 binds to additional platelet receptors (e.g., TLR4, RAGE) to influence platelet and thrombotic functions cannot be definitively excluded in the present studies and will require the generation of doubly deficient MRP-14 and TLR4 or RAGE mice, respectively.
The binding of MRP-14 to platelet CD36 is analogous to other ligand-receptor interactions that act within the platelet-platelet contact zone after the initial aggregation event, including, among others, CD40L and its binding to platelet GPIIb/IIIa (19), GAS6 and its tyrosine kinase receptors MER, TYRO3, and AXL (51), and ephrins and their EPH kinase receptors (52). Indeed, it has been proposed that after initial aggregation, platelets form a synapse that facilitates signaling by membrane-tethered receptor-ligand pairs and localizes secreted and shed ligands that ultimately promote thrombus growth and stability (53). MRP-14 appears to function in this synapse.
Our studies show that MRP-14 deficiency did not interfere with tail bleeding time, platelet adhesion to and spreading on vWF or collagen, or plasma coagulation activity (i.e., aPTT and thrombin generation). The identification of a new platelet-dependent pathway for thrombosis that does not affect hemostatic parameters, such as bleeding time and platelet adhesion and spreading, has possible clinical implications. Thrombotic cardiovascular diseases, including MI and stroke, are the leading cause of death in developed countries (54). Total U.S. health care expenditures in 2009 for coronary heart disease and stroke were a staggering $165.4 billion and $68.9 billion, respectively (54), with the cost of pharmacologic therapies estimated to exceed $20 billion worldwide (54). Primary drug therapies include antiplatelet and anticoagulant agents. Antiplatelet agents and anticoagulants are used in the treatment of acute coronary syndromes and in primary and secondary prevention of coronary artery disease and stroke (55, 56). Current drugs are subject to significant bleeding risk, which is associated with increased mortality (57–59). While new antiplatelet (prasugrel, ticagrelor) and anticoagulant (dabigatran, rivaroxaban, apixaban) agents are being developed on the basis of superior efficacy, these therapeutic advances are associated with a 25% to 30% increase in the rate of bleeding (prasugrel, ticagrelor, apixaban) or transfusion (rivaroxaban, apixaban) in the treatment of acute coronary syndromes (60–62). There is emerging experimental evidence distinguishing the molecular and cellular mechanisms of hemostasis and thrombosis (63). Extracellular MRP-14 is now positioned as a novel and targetable mediator of thrombosis, but not hemostasis (i.e., reduced bleeding risk).
Finally, platelet MRP-8/14 has the potential to serve as a useful biomarker of coronary artery disease activity. The major causes of acute coronary syndromes, including MI and unstable angina, are plaque rupture and thrombosis (1). These ischemic events are typically precipitous and without warning symptoms. New approaches are required to identify patients who are at risk for the transition from “stable/chronic” to “unstable/acute” disease. Using platelet expression profiling, we reported previously that platelet MRP14 mRNA is increased in patients with STEMI compared with stable coronary artery disease patients (4). Ongoing studies are planned to determine whether platelet MRP-8/14 expression is predictive of coronary artery disease activity (i.e., stable angina versus acute coronary syndromes).
Methods
Materials.
Antibodies against human MRP-14, MRP-8, and MRP-8/14 were purchased from Abcam, BMA Biomedicals, and R&D Systems. Antibodies against mouse MRP-8 and MRP-14 were purchased from R&D Systems. Wug.E9 antibody against mouse P-selectin/CD62P conjugated with fluorescein isothiocyanate and JON/A (antibody against mouse-activated integrin GPIIb/IIIa) conjugated with R-phycoerythrin were purchased from emfret Analytics. Alexa Fluor secondary antibodies and Alexa Fluor 647–conjugated mouse anti-human CD42a were purchased from AbD Serotec. The following azide-free antibodies were used for the shear-induced platelet aggregation assays: control mouse IgG, anti-human CD36 (clone FA6-152), and TLR4 (clone HTA125) monoclonal antibodies from Abcam; anti-human MRP-14 (clone MRP1H9) from Biolegend; and anti-human RAGE (clone MAB11451) monoclonal antibodies from R&D Systems. Purified, recombinant human MRP-8 and MRP-14 were obtained from Novus Biologicals. Human α-thrombin was purchased from Haematologic Technologies and human fibrinogen from Enzyme Research Laboratories. Rose Bengal (4, 5, 6, 7-tetrachloro-3′, 6-dihydroxy-2, 4, 5, 7-tetraiodospiro (isobenzofuran-1(3H), 9 [9H] xanthan)-3-1 dipotassium salt) was purchased from Sigma-Aldrich.
Mice.
Mrp14–/– mice were generated in the laboratory of Nancy Hogg (5), and Cd36–/– mice were generated in the laboratory of Roy Silverstein (22). All mice had a congenic C57BL/6 background and were maintained in animal facilities at Case Western Reserve University School of Medicine. Eight- to 12-week-old male mice were used for all experiments.
Platelet preparation and coagulation assays.
For platelet isolation, aPTT, and thrombin generation, see Supplemental Methods.
MRP-8/14 expression in platelets.
For immunofluorescence micscroscopy, immunoblotting, and ELISA assays, see Supplemental Methods.
Platelet α-granule release and GPIIb/IIIa activation.
See Supplemental Methods.
Mouse bleeding times.
Tail bleeding times were measured by transecting the tails of anesthetized mice (50 mg/kg sodium pentobarbital) 5 mm from the tip, as previously described (64). Briefly, the transected tail tip was placed into a beaker containing saline at 37°C, and the time to complete cessation of bleeding for 30 seconds and 3 minutes was determined with a stopwatch.
Thrombus formation under laminar flow conditions.
The laminar flow chamber used in this assay has been described previously (20, 21). Factor Xa inhibitor (Portola Pharmaceutical Inc.) anticoagulated blood was incubated with 0.2 μg/ml rhodamine-6G (Sigma-Aldrich) and then perfused for 5 minutes through human type III collagen–coated rectangular capillaries at 625 s–1, resulting in the deposition of adherent platelets and platelet aggregates. Thrombus formation under flow was then analyzed in real time and quantified by measuring fluorescence, thrombus area, and thrombus volume over time using computer-assisted imaging analysis (original magnification, ×40 and observation area, 360 × 270 μm).
Photochemical carotid artery thrombosis.
See Supplemental Methods.
Laser-induced injury to the cremaster microcirculation using intravital microscopy.
See Supplemental Methods.
Plate binding assay.
See Supplemental Methods.
Adoptive transfer/platelet transfusion experiments.
Blood from the inferior vena cava of 2 mice was collected directly into 3.8% sodium citrate (9:1 blood/citrate ratio) and diluted with an equal amount of Tyrode’s buffer. For isolation of platelets, anticoagulated and diluted blood was centrifuged to obtain platelet-rich plasma and then applied to a column packed with Sepharose 2B to obtain gel-filtered platelets. WT or Mrp14–/– platelets (1 × 108 in 250 μl) were injected into recipient Mrp14–/– mice via the tail vein.
Histology and immunohistochemistry of tissue samples.
See Supplemental Methods.
Statistics.
Data are presented as the mean ± SD. Comparisons between groups were performed by an unpaired, 2-tailed Student’s t test. P values of less than 0.05 were considered statistically significant.
Study approval.
Animal care and procedures were reviewed and approved by the IACUC of Case Western Reserve University School of Medicine and were performed in accordance with the guidelines of the American Association for Accreditation of Laboratory Animal Care and the NIH.
Approval for use of human tissue for pathological examination was obtained by the University Hospitals Case Medical Center Institutional Review Board. Human platelets were prepared from the whole blood drawn from the antecubital vein of healthy volunteers after providing informed consent, in accordance with a University Hospitals Case Medical Center Institutional Review Board–approved protocol.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants to K. Croce (HL086672), A.H. Schmaier (HL052779), and D.I. Simon (HL085815 and HL057506 MERIT Award).
Footnotes
Conflict of interest: Yunmei Wang, Matthew L. Bilodeau, Kevin Croce, and Daniel I. Simon are co-inventors of technology related to MRP-14 and thrombosis that is assigned to Case Western Reserve University. Patrick Andre and Gillian Stephens were employees of Portola Pharmaceuticals and supplied collagen-coated capillaries for ex vivo thrombosis studies. Patrick Andre is presently employed by Merck Research Laboratories and Gillian Stephens by AstraZeneca.
Citation for this article:J Clin Invest. 2014;124(5):2160–2171. doi:10.1172/JCI70966.
References
- 1.Davies MJ, Thomas AC. Plaque fissuring-the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J. 1985;53(4):363–373. doi: 10.1136/hrt.53.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeWood MA, et al. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med. 1980;303(16):897–902. doi: 10.1056/NEJM198010163031601. [DOI] [PubMed] [Google Scholar]
- 3.Sherman CT, et al. Coronary angioscopy in patients with unstable angina pectoris. N Engl J Med. 1986;315(15):913–919. doi: 10.1056/NEJM198610093151501. [DOI] [PubMed] [Google Scholar]
- 4.Healy A, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113(19):2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 5.Hobbs JA, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23(7):2564–2576. doi: 10.1128/MCB.23.7.2564-2576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogl T, et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104(13):4260–4268. doi: 10.1182/blood-2004-02-0446. [DOI] [PubMed] [Google Scholar]
- 7.Lackmann M, et al. Identification of a chemotactic domain of the pro-inflammatory S100 protein CP-10. J Immunol. 1993;150(7):2981–2991. [PubMed] [Google Scholar]
- 8.Kerkhoff C, Sorg C, Tandon NN, Nacken W. Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry. 2001;40(1):241–248. doi: 10.1021/bi001791k. [DOI] [PubMed] [Google Scholar]
- 9.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 10.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102(10):1239–1246. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 11.Morrow DA, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravasatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) Trial. Am Heart J. 2008;155(1):49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altwegg LA, et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. 2007;28(8):941–948. doi: 10.1093/eurheartj/ehm078. [DOI] [PubMed] [Google Scholar]
- 13.Manitz MP, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23(3):1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnekenburger J, et al. The calcium binding protein S100A9 is essential for pancreatic leukocyte infiltration and induces disruption of cell-cell contacts. J Cell Physiol. 2008;216(2):558–567. doi: 10.1002/jcp.21433. [DOI] [PubMed] [Google Scholar]
- 15.Croce K, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120(5):427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagashima M, et al. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficiency is compatible with murine life. J Clin Invest. 2002;109(1):101–110. doi: 10.1172/JCI12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115(12):3355–3362. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 19.Andre P, et al. CD40L stabilizes arterial thrombi by a β3 integrin — dependent mechanism. Nat Med. 2002;8(3):247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 20.Andre P, et al. Anticoagulants (thrombin inhibitors) and aspirin synergize with P2Y12 receptor antagonism in thrombosis. Circulation. 2003;108(21):2697–2703. doi: 10.1161/01.CIR.0000093279.36628.12. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, et al. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112(19):2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh A, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J Clin Invest. 2008;118(5):1934–1943. doi: 10.1172/JCI34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gawlowski T, et al. Advanced glycation end products strongly activate platelets. Eur J Nutr. 2009;48(8):475–481. doi: 10.1007/s00394-009-0038-6. [DOI] [PubMed] [Google Scholar]
- 24.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83(2):196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 25.Podrez EA, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13(9):1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen K, Febbraio M, Li W, Silverstein RL. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008;102(12):1512–1519. doi: 10.1161/CIRCRESAHA.108.172064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nacken W, Sopalla C, Propper C, Sorg C, Kerkhoff C. Biochemical characterization of the murine S100A9 (MRP14) protein suggests that it is functionally equivalent to its human counterpart despite its low degree of sequence homology. Eur J Biochem. 2000;267(2):560–565. doi: 10.1046/j.1432-1327.2000.01040.x. [DOI] [PubMed] [Google Scholar]
- 28.Baker JR, et al. Distinct roles for S100a8 in early embryo development and in the maternal deciduum. Dev Dyn. 2011;240(9):2194–2203. doi: 10.1002/dvdy.22709. [DOI] [PubMed] [Google Scholar]
- 29.Hogg N, Allen C, Edgeworth J. Monoclonal antibody 5.5 reacts with p8,14, a myeloid molecule associated with some vascular endothelium. Eur J Immunol. 1989;19(6):1053–1061. doi: 10.1002/eji.1830190615. [DOI] [PubMed] [Google Scholar]
- 30.Hogg N, Palmer DG, Revell PA. Mononuclear phagocytes of normal and rheumatoid synovial membrane identified by monoclonal antibodies. Immunology. 1985;56(4):673–681. [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkinson MM, Busuttil A, Hayward C, Brock DJ, Dorin JR, Van Heyningen V. Expression pattern of two related cystic fibrosis-associated calcium-binding proteins in normal and abnormal tissues. J Cell Sci. 1988;91(pt 2):221–230. doi: 10.1242/jcs.91.2.221. [DOI] [PubMed] [Google Scholar]
- 32.Brandtzaeg P, Dale I, Fagerhol MK. Distribution of a formalin-resistant myelomonocytic antigen (L1) in human tissues. II. Normal and aberrant occurrence in various epithelia. Am J Clin Pathol. 1987;87(6):700–707. doi: 10.1093/ajcp/87.6.700. [DOI] [PubMed] [Google Scholar]
- 33.Weyrich AS, et al. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci U S A. 1998;95(10):5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNeill E, Conway SJ, Roderick HL, Bootman MD, Hogg N. Defective chemoattractant-induced calcium signalling in S100A9 null neutrophils. Cell Calcium. 2007;41(2):107–121. doi: 10.1016/j.ceca.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Klempt M, Melkonyan H, Nacken W, Wiesmann D, Holtkemper U, Sorg C. The heterodimer of the Ca2+-binding proteins MRP8 and MRP14 binds to arachidonic acid. . FEBS Lett. 1997;408(1):81–84. doi: 10.1016/S0014-5793(97)00394-3. [DOI] [PubMed] [Google Scholar]
- 36.Siegenthaler G, et al. A heterocomplex formed by the calcium-binding proteins MRP8 (S100A8) and MRP14 (S100A9) binds unsaturated fatty acids with high affinity. J Biol Chem. 1997;272(14):9371–9377. doi: 10.1074/jbc.272.14.9371. [DOI] [PubMed] [Google Scholar]
- 37.Kerkhoff C, Eue I, Sorg C. The regulatory role of MRP8 (S100A8) and MRP14 (S100A9) in the transendothelial migration of human leukocytes. Pathobiology. 1999;67(5–6):230–232. doi: 10.1159/000028098. [DOI] [PubMed] [Google Scholar]
- 38.Roulin K, Hagens G, Hotz R, Saurat JH, Veerkamp JH, Siegenthaler G. The fatty acid-binding heterocomplex FA-p34 formed by S100A8 and S100A9 is the major fatty acid carrier in neutrophils and translocates from the cytosol to the membrane upon stimulation. Exp Cell Res. 1999;247(2):410–421. doi: 10.1006/excr.1998.4382. [DOI] [PubMed] [Google Scholar]
- 39.Hetland G, Talgo GJ, Fagerhol MK. Chemotaxins C5a and fMLP induce release of calprotectin (leucocyte L1 protein) from polymorphonuclear cells in vitro. Mol Pathol. 1998;51(3):143–148. doi: 10.1136/mp.51.3.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol. 2001;70(1):130–134. [PubMed] [Google Scholar]
- 41.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266(12):7706–7713. [PubMed] [Google Scholar]
- 42.Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272(14):9496–9502. doi: 10.1074/jbc.272.14.9496. [DOI] [PubMed] [Google Scholar]
- 43.Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol. 2001;166(7):4678–4688. doi: 10.4049/jimmunol.166.7.4678. [DOI] [PubMed] [Google Scholar]
- 44.Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2002;277(5):3658–3665. doi: 10.1074/jbc.M102950200. [DOI] [PubMed] [Google Scholar]
- 45.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005;106(7):2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 46.Silverstein RL, Febbraio M. CD36 and atherosclerosis. Curr Opin Lipidol. 2000;11(5):483–491. doi: 10.1097/00041433-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 47.Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108(6):785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clemetson KJ, Pfueller SL, Luscher EF, Jenkins CS. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. Biochim Biophys Acta. 1977;464(3):493–508. doi: 10.1016/0005-2736(77)90025-6. [DOI] [PubMed] [Google Scholar]
- 49.Rhinehart-Jones T, Greenwalt DE. A detergent-sensitive 113-kDa conformer/complex of CD36 exists on the platelet surface. Arch Biochem Biophys. 1996;326(1):115–118. doi: 10.1006/abbi.1996.0054. [DOI] [PubMed] [Google Scholar]
- 50.Huang MM, Bolen JB, Barnwell JW, Shattil SJ, Brugge JS. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci U S A. 1991;88(17):7844–7848. doi: 10.1073/pnas.88.17.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angelillo-Scherrer A, et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J Clin Invest. 2005;115(2):237–246. doi: 10.1172/JCI22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prevost N, et al. Signaling by ephrinB1 and Eph kinases in platelets promotes Rap1 activation, platelet adhesion, and aggregation via effector pathways that do not require phosphorylation of ephrinB1. Blood. 2004;103(4):1348–1355. doi: 10.1182/blood-2003-06-1781. [DOI] [PubMed] [Google Scholar]
- 53.Brass LF, Zhu L, Stalker TJ. Minding the gaps to promote thrombus growth and stability. J Clin Invest. 2005;115(12):3385–3392. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lloyd-Jones D, et al. Heart disease and stroke statistics — 2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):e21–e181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 55.Anderson JL, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50(7):e1–e157. doi: 10.1016/j.jacc.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 56.Kushner FG, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2009;54(23):2205–2241. doi: 10.1016/j.jacc.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 57.Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation. 2006;114(8):774–782. doi: 10.1161/CIRCULATIONAHA.106.612812. [DOI] [PubMed] [Google Scholar]
- 58.Stone GW, et al. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008;358(21):2218–2230. doi: 10.1056/NEJMoa0708191. [DOI] [PubMed] [Google Scholar]
- 59.Mehran R, et al. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet. 2009;374(9696):1149–1159. doi: 10.1016/S0140-6736(09)61484-7. [DOI] [PubMed] [Google Scholar]
- 60.Wiviott SD, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 61.Wallentin L, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 62.Connolly SJ, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151. doi: 10.1056/NEJMoa0905561. [DOI] [PubMed] [Google Scholar]
- 63.Sachs UJ, Nieswandt B. In vivo thrombus formation in murine models. Circ Res. 2007;100(7):979–991. doi: 10.1161/01.RES.0000261936.85776.5f. [DOI] [PubMed] [Google Scholar]
- 64.Nieman MT, et al. The preparation and characterization of novel peptide antagonists to thrombin and factor VIIa and activation of protease-activated receptor 1. J Pharmacol Exp Ther. 2004;311(2):492–501. doi: 10.1124/jpet.104.069229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.