

Abstract
Adoptive transfer of tumor-infiltrating lymphocytes (TILs) can mediate regression of metastatic melanoma; however, TILs are a heterogeneous population, and there are no effective markers to specifically identify and select the repertoire of tumor-reactive and mutation-specific CD8+ lymphocytes. The lack of biomarkers limits the ability to study these cells and develop strategies to enhance clinical efficacy and extend this therapy to other malignancies. Here, we evaluated unique phenotypic traits of CD8+ TILs and TCR β chain (TCRβ) clonotypic frequency in melanoma tumors to identify patient-specific repertoires of tumor-reactive CD8+ lymphocytes. In all 6 tumors studied, expression of the inhibitory receptors programmed cell death 1 (PD-1; also known as CD279), lymphocyte-activation gene 3 (LAG-3; also known as CD223), and T cell immunoglobulin and mucin domain 3 (TIM-3) on CD8+ TILs identified the autologous tumor-reactive repertoire, including mutated neoantigen-specific CD8+ lymphocytes, whereas only a fraction of the tumor-reactive population expressed the costimulatory receptor 4-1BB (also known as CD137). TCRβ deep sequencing revealed oligoclonal expansion of specific TCRβ clonotypes in CD8+PD-1+ compared with CD8+PD-1– TIL populations. Furthermore, the most highly expanded TCRβ clonotypes in the CD8+ and the CD8+PD-1+ populations recognized the autologous tumor and included clonotypes targeting mutated antigens. Thus, in addition to the well-documented negative regulatory role of PD-1 in T cells, our findings demonstrate that PD-1 expression on CD8+ TILs also accurately identifies the repertoire of clonally expanded tumor-reactive cells and reveal a dual importance of PD-1 expression in the tumor microenvironment.
Introduction
Cancer immunotherapy has experienced major progress in the last decade. Adoptive transfer of ex vivo–expanded tumor-infiltrating lymphocytes (TILs) can cause substantial regression of metastatic melanoma (1, 2). Blockade of the interaction of cytotoxic T lymphocyte antigen 4 (CTLA-4; also known as CD152) or programmed cell death 1 receptor (PD-1; also known as CD279) with their ligands using blocking antibodies alone or in combination have been shown to unleash an otherwise-ineffective immune response against melanoma (3–7), renal cell carcinoma (3), and non–small cell lung cancer (3). The antitumor responses observed in these clinical trials support the presence of naturally occurring tumor-reactive CD8+ T cells and their immunotherapeutic potential. In the particular case of TIL therapy, persistence of transferred tumor-specific T cell clones is associated with tumor regression (8). Moreover, retrospective clinical studies have shown an association of autologous tumor recognition by TILs and clinical response (9, 10), which suggests that enrichment of tumor-reactive cells could enhance clinical efficacy. However, the identification of the diverse repertoire of tumor-reactive cells limits the ability to study these cells, enhance clinical efficacy, and extend this therapy to other malignancies.
Melanoma TILs represent a heterogeneous population that can target a variety of antigens, including melanocyte differentiation antigens, cancer germline antigens, self-antigens overexpressed by the tumor, and mutated tumor neoantigens (11). The latter appear to be of critical importance for the antitumor responses observed after transfer of TILs, given the substantial regression of metastatic melanoma in up to 72% of patients in phase 2 clinical trials, in the absence of any autoimmune side effects in the great majority of patients (2). This contrasts with the modest antitumor activity but high prevalence of severe autoimmune manifestations observed after transfer of peripheral blood gene-engineered T cells expressing TCRs targeting shared melanocyte differentiation antigens MART1 and gp100 (12, 13). Furthermore, T cells targeting mutated neoepitopes are not subject to negative selection in the thymus and may constitute the predominant naturally occurring tumor-reactive population in cancer patients. In support of this notion, a recent study reported the frequent detection and dominance of T cell populations targeting mutated epitopes in melanoma-derived TILs (14). Conversely, T cells targeting shared melanocyte differentiation antigens and cancer germline antigens in bulk melanoma TILs were represented at a strikingly low frequency (15). These findings have shifted our interest from the more accessible and commonly studied T cells targeting melanocyte differentiation antigens to T cells targeting unique patient-specific mutations. However, the often rare availability of autologous tumor cell lines necessary to study these reactivities, and the hurdles associated with the identification of the unique mutations targeted, have thus far hindered immunobiological studies of these T cell populations in the tumor.
Naturally occurring tumor-reactive cells are exposed to their antigen at the tumor site. Thus, the immunobiological characterization of T cells infiltrating tumors represents a unique opportunity to study their function and to identify the patient-specific repertoire of tumor-reactive cells. TCR stimulation triggers simultaneous upregulation of both costimulatory and coinhibitory receptors, which can either promote or inhibit T cell activation and function. Expression of the inhibitory receptors PD-1, CTLA-4, lymphocyte-activation gene 3 (LAG-3; also known as CD223), and T cell immunoglobulin and mucin domain 3 (TIM-3) is regulated in response to activation and throughout differentiation (16, 17). Chronic antigen stimulation has been shown to induce coexpression of inhibitory receptors and is associated with T cell hyporesponsiveness, termed exhaustion (18). Exhaustion in response to persistent exposure to antigen was first delineated in a murine model of chronic lymphocytic choriomeningitis virus (19), but has been observed in multiple human chronic viral infections (20–22) as well as in tumor-reactive MART1-specific TILs (23, 24), and has provided the rationale for restoring immune function using immune checkpoint blockade. Conversely, 4-1BB (also known as CD137) is a costimulatory member of the TNF receptor family that has emerged as an important mediator of survival and proliferation, particularly in CD8+ T cells (25–27). 4-1BB is transiently expressed upon TCR stimulation, and its expression has been used to enrich for antigen-specific T cells in response to acute antigen stimulation (28). However, expression of this marker has not been extensively explored in CD8+ lymphocytes infiltrating human tumors. In addition to changes in the expression of cosignaling receptors on the surface of T cells, antigen-specific stimulation typically results in clonal expansion. TCR sequence immunoprofiling can be used to monitor T cell responses to a given immune challenge even without a priori knowledge of the specific epitope targeted, through determination of the abundance of specific clonotypes (29, 30). However, there is limited knowledge regarding the TCR repertoire and the frequency of tumor-reactive clonotypes infiltrating human tumors.
We hypothesized that the assessment of unique phenotypic traits expressed by CD8+ TILs and TCR β chain (TCRβ; encoded by TRB) clonotypic immunoprofiling of lymphocytes infiltrating the tumor could provide a powerful platform to study antitumor T cell responses and evaluated their usefulness in identifying the diverse repertoire of tumor-reactive cells. Despite the accepted negative regulatory role of PD-1 in T cells, our findings establish that expression of PD-1 on CD8+ melanoma TILs accurately identifies the repertoire of clonally expanded tumor-reactive, mutation-specific lymphocytes and suggest that cells derived from this population play a critical role in tumor regression after TIL administration.
Results
CD8+ lymphocytes in the tumor display distinct phenotypic traits with increased but variable expression of PD-1, LAG-3, TIM-3, and 4-1BB.
To investigate whether the state of differentiation of CD8+ TILs from freshly excised melanoma lesions or expression of markers characteristic of chronic or acute antigen-specific stimulation could guide the identification of tumor-reactive cells, we characterized the phenotype of CD8+ TILs from 24 melanomas. We measured cell surface expression of differentiation markers CD62L, CD45RO, CD27, and CD57 (31), inhibitory receptors PD-1, LAG-3, TIM-3, and BTLA, typically expressed on chronically stimulated CD8+ T cells (23, 24, 32, 33), and costimulatory receptor 4-1BB (28, 34) and IL-2 receptor alpha CD25 (35), which can be upregulated after TCR stimulation. A summary comparing the phenotypic traits of CD8+ TILs and PBLs is depicted in Figure 1A. Freshly isolated CD8+ TILs displayed a higher frequency of effector memory (EM) cells (CD62L–CD45RO+) compared with PBLs obtained from 21 of the 24 patients tested (Figure 1A). Despite this enhanced frequency of EM-like cells, CD8+ TILs did not appear to be terminally differentiated, as they maintained high expression of CD27, a marker that typically decreases throughout differentiation, and low expression of CD57, a receptor associated with terminal T cell differentiation (31). In addition, CD8+ TILs exhibited enhanced expression of PD-1, LAG-3, TIM-3, and 4-1BB, but not BTLA or CD25. PD-1 was the receptor most overexpressed in CD8+ TILs compared with PBLs, followed by TIM-3, 4-1BB, and LAG-3 (32.4% ± 5.0%, 13.0% ± 3.2%, 10.8% ± 2.4%, and 5.7% ± 2.4%, respectively). Notably, these 4 receptors displayed substantial coexpression on a subset of CD8+ TILs (Figure 1B). The cumulative frequency of T cells coexpressing 2, 3, or 4 markers accounted for 16.6% ± 3.6% of TILs, compared with 0.3% ± 0.1% of PBLs. Figure 1C shows the coexpression pattern of some of the markers studied on CD8+ PBLs and fresh tumor CD8+ TILs for 1 representative melanoma patient. This patient’s CD8+ TILs displayed coexpression of TIM-3, LAG-3, and 4-1BB with PD-1. TIM-3, LAG-3, and 4-1BB were almost exclusively expressed on PD-1+ cells: 82.8% ± 3.1% of CD8+TIM-3+ TILs, 79.3% ± 4.9% of CD8+LAG-3+ TILs, and 78.6% ± 3.8% of CD8+4-1BB+ TILs expressed PD-1 in the fresh tumor (Figure 1D and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI73639DS1). Additionally, PD-1 and CD27 were also consistently coexpressed on a fraction of CD8+ TILs (Figure 1C). However, CD27 was not exclusively expressed on PD-1+ TILs, as there was still a substantial percentage of CD27+ TILs that were PD-1–. On average, only 37.6% of CD8+CD27+ TILs expressed PD-1 in the fresh tumor (Figure 1D). Thus, the coexpression pattern of PD-1 with LAG-3, TIM-3, and 4-1BB appeared to be different than that observed with PD-1 and the differentiation marker CD27. Overall, the unique coexpression profile of PD-1 with LAG-3, TIM-3, and 4-1BB in CD8+ TILs prompted us to study these markers and their potential significance in greater detail.
Figure 1. CD8+ TILs exhibit unique phenotypic traits compared with PBLs.

(A) Phenotypic characterization of CD8+ PBLs and TILs in melanoma patients. Percentages of PBLs and TILs expressing individual or combination of markers are shown (mean ± SEM). Each dot represents 1 sample analyzed. **P < 0.01, ***P < 0.001, Mann-Whitney test. (B) Coexpression of PD-1, LAG-3, TIM-3, and 4-1BB in CD8+ PBLs and TILs. The frequency of cells expressing 0, 1, 2, 3, or 4 markers is shown. Bars represent maximum, minimum, and mean values. **P < 0.01, ***P < 0.001, Mann-Whitney test. (C) Coexpression pattern of PD-1 and LAG-3, TIM-3, 4-1BB, CD27, and CD57 on CD8+ PBLs and TILs. Dot plots display the phenotype of CD8+ lymphocytes from matched samples from 1 representative patient. The percentage of cells expressing each combination of receptors is shown. (D) Frequency of PD-1 expression on CD8+ TIL subpopulations. TIM-3+, LAG-3+, 4-1BB+, CD25+, BTLA+, CD57+, and CD27+ CD8+ TILs were gated, and the frequency of PD-1 expression within these populations is summarized. Markers are displayed in order and color coded according to frequency of PD-1 expression. Mean ± SEM are shown. *P < 0.005 vs. BTLA+, CD57+, and CD27+, Dunn test for multiple comparisons.
Expression of PD-1, LAG-3, and TIM-3 in the tumor identifies tumor-reactive and mutation-specific T cells.
In order to investigate whether CD8+ cells expressing PD-1, LAG-3, and TIM-3 in the tumor represented the tumor-reactive population, we isolated cells from 6 independent fresh tumors based on expression of PD-1, LAG-3, and TIM-3, expanded them in vitro for 15 days with IL-2, anti-CD3 stimulation, and irradiated feeders, and tested their ability to recognize autologous tumor cell lines. The availability of autologous tumor cell lines enabled us to assess the patient-specific repertoire of tumor-reactive cells, rather than the shared reactivities studied with HLA-I–matched tumor cell lines. We first studied fresh tumor 1913 (FrTu1913), a tumor from a patient who had objective clinical response after transfer of TIL1913. Notably, PD-1+, LAG-3+, and TIM-3+ FrTu1913-derived CD8+ TILs, but not the negative counterparts, contained all the tumor-reactive cells — as determined by IFN-γ secretion and 4-1BB upregulation — after coculture with autologous tumor cell line 1913 (TC1913) (Figure 2A). Recognition of autologous tumor appeared to be antigen specific and HLA-I restricted, as the HLA-I–blocking antibody W6/32 inhibited these responses, and TILs did not respond to HLA-I–matched or –mismatched tumor cell lines (Figure 2B). Tumor recognition by PD-1+, LAG-3+, and TIM-3+ derived TILs was not due to differences in the activation status of the T cells, since all the subsets were capable of responding to plate-bound anti-CD3 stimulation (Supplemental Table 1). Additionally, PD-1+, LAG-3+, and TIM-3+ derived cells were capable of lysing the autologous TC1913 (Figure 2C). With the exception of LAG-3+ cells isolated from FrTu2448, selection of PD-1+, LAG-3+, and TIM-3+ CD8+ TILs reproducibly separated the tumor-reactive cells from the non–tumor-reactive cells present in all the fresh tumors tested (FrTu1913, FrTu3289, FrTu3612, FrTu3713, FrTu3550, and FrTu2448; Figure 2D and Figure 3, A–H). Recognition of the autologous tumor cell line by these TIL subpopulations appeared to be HLA-I restricted and did not appear to be the result of differences in the activation status of the T cell populations (Supplemental Tables 2–5). Thus, expression of the inhibitory receptors PD-1, LAG-3, and TIM-3 in the fresh tumor could be used to prospectively identify and select the patient-specific repertoire of CD8+ tumor-reactive cells.
Figure 2. PD-1+, LAG-3+, and TIM-3+ derived CD8+ TILs, but not the negative counterparts, recognize and lyse their autologous tumor cell line.

Bulk CD3+CD8+ TILs were sorted to high purity from 6 tumors based on positive or negative expression of PD-1, LAG-3, and TIM-3 and expanded in vitro. (A) Response of FrTu1913-derived TILs to TC1913. TILs were cocultured with autologous TC1913, and tumor recognition was assessed by measuring IFN-γ release (duplicates, mean ± SD), and the frequency of CD8+4-1BB+ cells. (B) Reactivity of PD-1+ and PD-1– CD8+ TILs derived from FrTu1913 against a panel of targets: TC1913 with and without HLA-I blocking antibody (W6/32), TC2448 (matched HLA-A*0201), TC2301 (allogeneic), and plate-bound anti-CD3 (OKT3). Upregulation of 4-1BB (top) and IFN-γ release (bottom, duplicates, mean ± SD) are shown. (C) Lysis of TC1913 by FrTu1913-derived TILs. Cytotoxicity of TILs against TC1913 (mean ± SD). (D) Response of PD-1– and PD-1+ derived TILs to their autologous tumor targets from all the 6 fresh tumors studied. PD-1– and PD-1+ TILs were cocultured with their autologous tumor cell lines, and reactivity was assessed by measuring IFN-γ secretion (left) and 4-1BB upregulation (right). Each dot represents 1 patient’s sample. Mean ± SEM. *P ≤ 0.05, 2-tailed Wilcoxon signed-rank test.
Figure 3. Recognition and lysis of autologous tumor by CD8+ TILs sorted based on PD-1, LAG-3, and TIM-3 expression.

Bulk CD3+CD8+ TILs were sorted to high purity from FrTu3289, FrTu3612, FrTu3713, FrTu3550, and FrTu2448 based on positive or negative expression of PD-1, LAG-3 and/or TIM-3, and expanded in vitro for 15 days. (A–E) Response of fresh tumor–derived TILs to their respective autologous tumor cell lines, TC3289 (A), TC3612 (B), TC3713 (C), TC3550 (D) and TC2448 (E). Reactivity was assessed by measuring IFN-γ release (duplicates, mean ± SD) and frequency of 4-1BB upregulation. (F–H) Cytolytic activity of fresh tumor–derived TILs in response to their respective autologous tumor cell lines, TC3289 (F), TC3713 (G), and TC2448 (H). Percentage of specific lysis at different effector/target ratios is shown as mean ± SD.
Previous studies characterizing the autologous tumor cell line TIL1913, derived from FrTu1913, resulted in the isolation of clones targeting a HLA-A*1101–restricted epitope derived from a mutation in the tumor suppressor gene locus CDKN2A (referred to herein as CDKN2Amut) expressed by TC1913 (36). The FrTu1913-derived TILs offered a unique opportunity to study mutation-reactive cells. CDKN2Amut-specific CD8+ lymphocytes detected using a specific HLA-A*1101 tetramer complex accounted for 11.5% of the PD-1+ derived TILs (Figure 4A). These cells upregulated 4-1BB upon coculture with COS-7 cells stably transduced with HLA-A*1101 (referred to herein as COS-A11 cells) that were pulsed with the specific peptide, and were not detected in the PD-1– fraction. CDKN2Amut-specific T cells were detected using CDKN2Amut tetramer in the PD-1+, LAG-3+, and TIM-3+ FrTu1913-derived CD8+ TIL populations, but not in the negative counterparts (Figure 4B). CDKN2Amut-specific cells present in these populations secreted IFN-γ in response to CDKN2Amut peptide, but not in response to an irrelevant HLA-A*1101–restricted control peptide, CRKRSmut (Figure 4B). These findings indicate that T cells targeting mutated neoepitopes express PD-1, LAG-3, and TIM-3 in the tumor and that these receptors identify and select for these T cell populations.
Figure 4. PD-1+, LAG-3+, and TIM-3+ derived CD8+ TILs prospectively identify tumor-reactive CD8+ TILs targeting a mutation in the CDKN2A gene locus.

CD3+CD8+ TILs were sorted to high purity from FrTu1913 based on positive or negative expression of PD-1, LAG-3, and TIM-3 and expanded in vitro. (A) Recognition of CDKN2Amut peptide pulsed COS-A11 cells by PD-1+ and PD-1– TILs. TILs were cocultured with COS-A11 cells pulsed with an irrelevant A*1101 restricted peptide, with CDKN2Amut peptide (1 μM), or with plate-bound anti-CD3. Dot plots display the frequency of expression of 4-1BB and CDKN2Amut peptide HLA-A*1101 tetramer complex binding of PD-1– (left) and PD-1+ (right) derived CD8+ TILs 24 hours after coculture. (B) IFN-γ secretion and frequency of CDKN2Amut tetramer+ cells in FrTu1913-derived TILs. TILs derived from FrTu1913 and control TIL3309, recognizing CRKRSmut HLA-A*1101 peptide, were cocultured with COS-A11 alone or pulsed with specific or irrelevant peptides; mean secretion of IFN-γ (duplicates) is represented. The frequency of CDKN2Amut-specific cells in the TIL populations was determined using a CDKN2Amut peptide HLA-A*1101 tetramer complex after gating on CD3+CD8+ cells (gray bars).
Expression of the inhibitory receptor PD-1, rather than 4-1BB, accurately defines the broad repertoire of tumor-reactive cells in the tumor.
In contrast to expression of the inhibitory receptors PD-1, LAG-3, and TIM-3, 4-1BB is a costimulatory receptor transiently expressed on the cell surface of CD8+ lymphocytes upon TCR stimulation. We found that 4-1BB expression was preferentially upregulated in a fraction of PD-1+ CD8+ TILs infiltrating freshly excised melanoma lesions (Figure 5A). PD-1+/4-1BB+ cells represented a small proportion of the CD8+ TILs in the 24 fresh tumors analyzed (9.8% ± 3.5%), whereas the PD-1+/4-1BB– population was substantially greater (22.1% ± 2.5%) (Figure 5B). To determine whether 4-1BB or PD-1 better identified the tumor-reactive TILs, we separated CD8+ TILs from 2 fresh tumors based on expression of PD-1 and 4-1BB into PD-1–/4-1BB–, PD-1+/4-1BB–, and PD-1+/4-1BB+, but not PD-1–/4-1BB+, due to its low frequency (Figure 5, A and B). We either established T cell clones for the 3 subpopulations sorted from FrTu1913 or bulk-expanded them from FrTu3713, and then tested their ability to recognize autologous tumor. Both the PD-1+/4-1BB+ and PD-1+/4-1BB– derived T cell clones from FrTu1913 and bulk-expanded cells from FrTu3713 contained tumor-reactive cells, as determined by 4-1BB upregulation after coculture with autologous tumor cell lines TC1913 and TC3713 (Figure 5, C and D). Tumor recognition was essentially undetectable in PD-1–/4-1BB– derived cells. PD-1+/4-1BB+ and PD-1+/4-1BB– expanded TILs showed comparable lysis of autologous tumor cell line TC3713 (Figure 5E). Given that a significant fraction of tumor-reactive T cells was found in the PD-1+/4-1BB– population, our data indicate that although the costimulatory receptor 4-1BB can be used to enrich for tumor-reactive cells, it in fact misses a large proportion of tumor-reactive T cells; thus, PD-1 expression in the fresh tumor more comprehensively defines the diverse repertoire of naturally occurring tumor-reactive CD8+ TILs.
Figure 5. Expression of PD-1, rather than 4-1BB, more comprehensively identifies the repertoire of tumor-reactive cells in human tumors.

(A) Representative dot plots showing PD-1 by 4-1BB expression of CD8+ TILs infiltrating FrTu1913 and FrTu3713. (B) Expression of PD-1 by 4-1BB on CD8+ TILs infiltrating melanoma tumors. The frequency of each combination of markers is shown as mean ± SEM. ***P < 0.001, Wilcoxon signed-rank test (n = 24). (C) Response of FrTu1913-derived clones to TC1913. CD8+ TILs were sorted from FrTu1913 into PD-1–/4-1BB–, PD-1+/4-1BB–, and PD-1+/4-1BB+, and clones were established. Clones derived from PD-1–/4-1BB– (n = 96), PD-1+/4-1BB– (n = 57), and PD-1+/4-1BB+ (n = 65) were cocultured with TC1913; 4-1BB upregulation upon coculture is plotted for each clone. The line represents the median. *P ≤ 0.05, ****P ≤ 0.0001, Dunn test for multiple comparisons. Pie charts depict the percentage of tumor-reactive and non–tumor-reactive clones in each population. Greater than 1% CD8+4-1BB+ and greater than twice the background percentage 4-1BB compared with no stimulation control was considered positive. (D) Response of bulk-expanded FrTu3713-derived TILs to TC3713. CD8+PD-1–/4-1BB–, PD-1+/4-1BB–, and PD-1+/4-1BB+ TILs were sorted from FrTu3713 and expanded. Cells were either left unstimulated, cocultured with TC3713, or stimulated with plate-bound anti-CD3. 4-1BB upregulation was measured to assess tumor recognition. Representative dot plots of CD3+CD8+-gated cells are shown. (E) Lysis of TC3713 by TILs derived from FrTu3713.
The skewed repertoire and binary distribution of CD8+ TIL TCRβ clonotypes based on PD-1 expression is indicative of local antigen-driven clonal expansion.
To further explore whether the predominant CD8+PD-1+ population infiltrating melanomas represented the tumor-reactive cells, we performed TCRβ deep sequencing of freshly isolated CD8+PD-1+ and CD8+PD-1– TILs. We reasoned that a less diverse TCRβ profile in the PD-1+ population would suggest antigen-driven clonal expansion. We studied FrTu1913 (Figure 6, A–C) and FrTu3713 (Figure 6, D–F), 2 tumors from patients who had objective clinical responses after transfer of TIL1913 or TIL3713. CD8+PD-1+ TILs infiltrating FrTu1913 (23%) and FrTu3713 (51.5%) were more oligoclonal compared with the PD-1– or bulk CD8+ TILs. The 30 most frequent clonotypes (unique TCRβ clonotypes; defined by hypervariable complementarity determining region 3 [CDR3β] nucleotide and amino acid sequences) in each population accounted for nearly 80% and 60% of the PD-1+ population — but only 28% and 37% of the PD-1– population — in FrTu1913 and FrTu3713, respectively (Figure 6, A and D). With rare exceptions, PD-1 expression separated the most frequent CD8+ clonotypes into 2 populations, given that they were either highly expanded in the PD-1+ TILs (PD-1+/PD-1– frequency ratio, >1) or dominant in the PD-1– population (PD-1+/PD-1– frequency ratio, <1) (Figure 6, B and E, and Tables 1 and 2). The 30 most highly expanded PD-1+ clonotypes were far less frequent in the PD-1– populations, and they only accounted for 1.2% and 3% of the PD-1– population in FrTu1913 and FrTu3713, respectively (Figure 6, C and F). The biased TCR repertoire displayed by CD8+PD-1+ TILs was also observed in FrTu3612, a tumor from a patient who did not respond following transfer of TIL3612 (Supplemental Figure 2). Notably, the binary distribution of TCRβ clonotypes observed based on PD-1 expression was less evident for CD8+ TILs separated based on 4-1BB expression, given that some of the most frequent clonotypes present in the 4-1BB+ population where still detected at a substantial frequency in the 4-1BB– population (Supplemental Figure 3, A and B). This is consistent with our previous results indicating that PD-1, rather than 4-1BB, more comprehensively identifies the diverse repertoire of tumor-reactive cells. Overall, the skewed TCRβ profile of the PD-1+ population and the binary distribution of TCRβ clonotypes based on PD-1 expression support the notion that this population underwent antigen-driven clonal expansion at the tumor site.
Figure 6. Skewed repertoire and binary distribution of CD8+ TIL TCRβ clonotypes based on expression of PD-1 in the tumor.

CD8+ T cells as well as PD-1+ and PD-1– fractions were sorted to high purity from FrTu1913 (A–C) and FrTu3713 (D–F), mRNA was extracted, and TCRβ deep sequencing was performed. (A and D) Diversity of the TCRβ repertoire within the bulk CD8+, CD8+PD-1–, and CD8+PD-1+ TIL populations. The relative frequencies of the most frequent TCRβ clonotype (unique CDR3β amino acid sequences), the second most frequent, the 3rd to 30th most frequent, and the rest of the clonotypes are shown. (B and E) The 30 most frequent CD8+ TIL clonotypes in the fresh tumor are plotted based on their frequency in PD-1+ and PD-1– populations. (C and F) Frequency of the 30 most frequent TCRβ clonotypes in the CD8+PD-1+ TIL population. Their frequency in the PD-1+ and PD-1– subsets is represented. Each dot represents 1 unique TCRβ clonotype. The cumulative frequency (∑ freq.) of the clonotypes in each of the populations is shown below. ****P ≤ 0.0001, Wilcoxon signed-rank test.
Table 1.
TCRβ deep sequencing of CD8+ TILs infiltrating FrTu1913
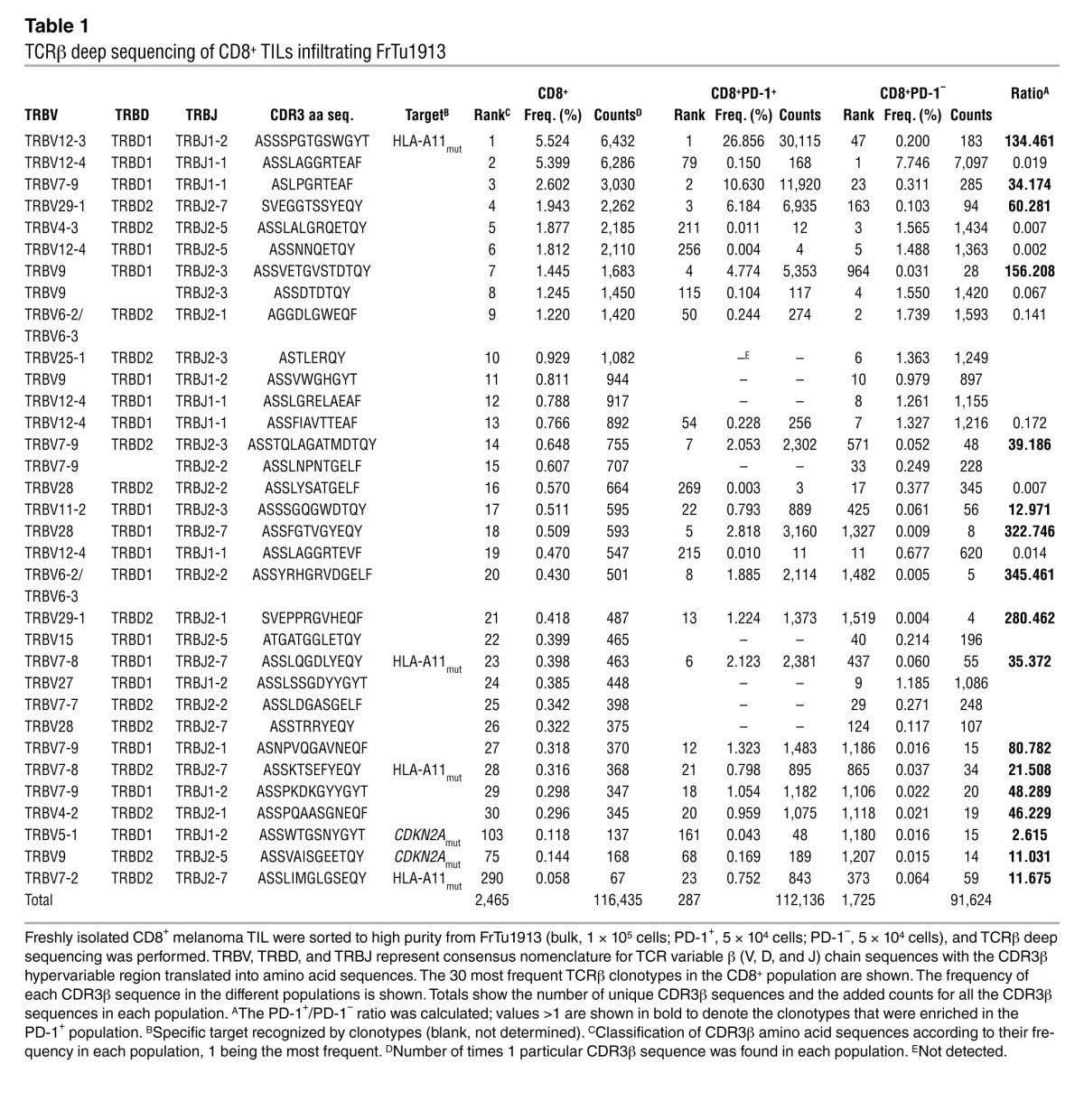
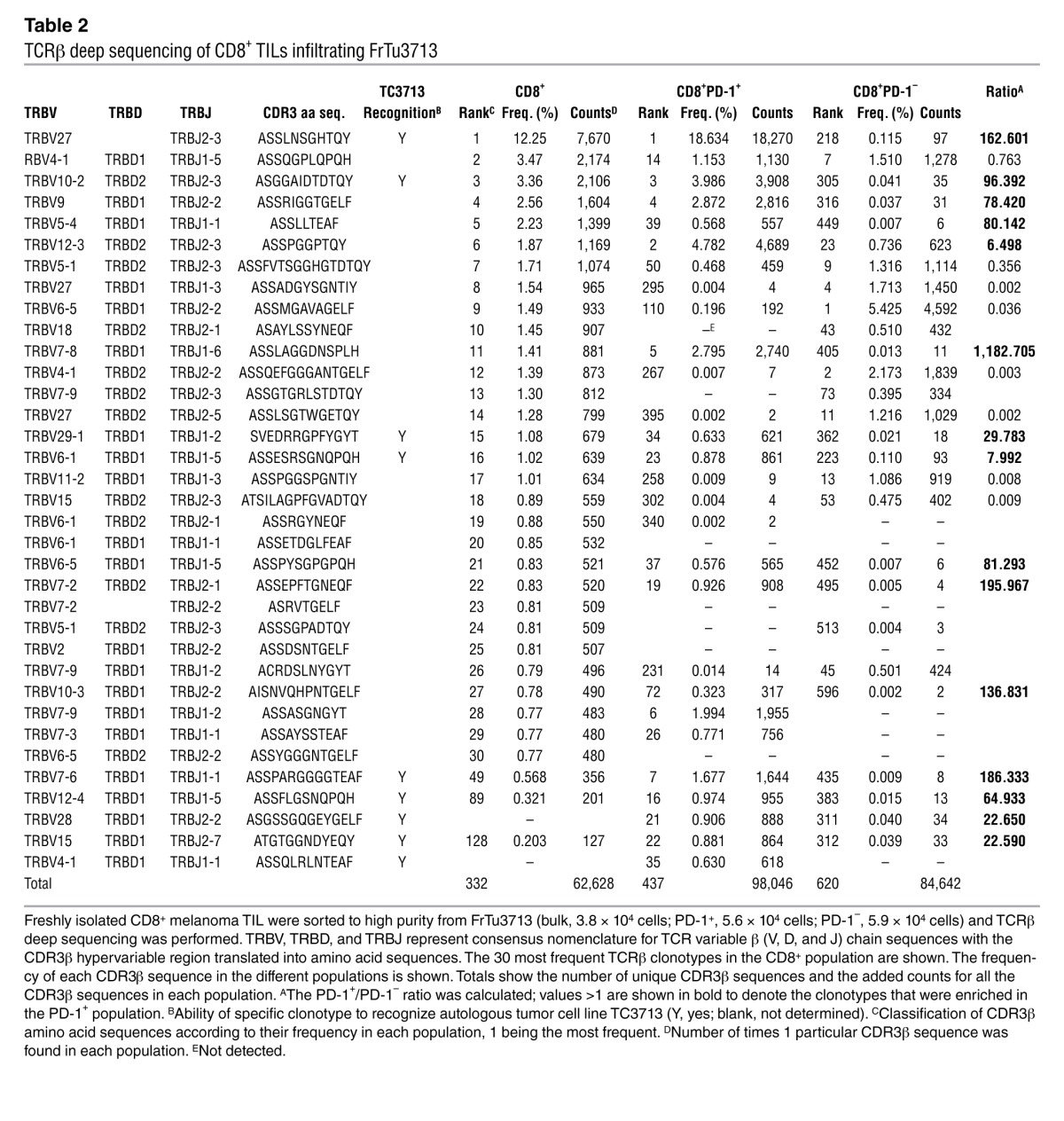
Table 2.
TCRβ deep sequencing of CD8+ TILs infiltrating FrTu3713
The frequency of specific clonotypes in the CD8+ and PD-1+ populations is associated with tumor recognition.
The presence of clonotypes that were highly represented in the CD8+ TIL population and preferentially expanded in the CD8+PD-1+ population suggested that these may be tumor-reactive. To investigate this, we established CD8+ T cell clones by limiting dilution cloning from the PD-1+ and PD-1– populations from FrTu3713, and screened these clones for recognition of the autologous tumor cell line TC3713. Whereas 61% of the PD-1+ derived clones recognized TC3713, only 7% of the PD-1– clones were tumor-reactive (Figure 7A). We also generated clones from TIL3713, derived from FrTu3713, and sequenced 9 tumor-reactive clones without a priori knowledge of their PD-1 expression status in the tumor. Table 2 shows the frequency of the 30 most frequent CD8+ TCRβ clonotypes infiltrating FrTu3713, including the 9 tumor-reactive clones established, and the frequency of these clonotypes in the PD-1+ and PD-1– populations. Among the tumor-reactive clones sequenced, we detected the CDR3β sequence corresponding to the most highly expressed CD8+ TCRβ clonotype in FrTu3713, representing 12.3% (Table 2). Moreover, this clonotype was preferentially expanded in the PD-1+ population, but not in the PD-1– population (representing 18.6% and 0.1%, respectively). The cumulative frequency of all the tumor-reactive clones sequenced represented 29.2% of the PD-1+ fraction in FrTu3713 and only accounted for 0.4% of the PD-1– clonotypes. These data support that the increased clonotypic frequency in the CD8+ and PD-1+ populations in FrTu3713 can be used to predict autologous tumor recognition, regardless of the knowledge of the specific antigen targeted by these cells.
Figure 7. Tumor-reactive and mutation-specific clonotypes are highly expanded in the CD8+ and PD-1+ populations.

(A) Response of CD8+PD-1+ (n = 44) and CD8+PD-1– (n = 109) FrTu3713-derived T cell clones to TC3713. Each dot represents 1 clone; line represents median IFN-γ release for all clones tested. The percentage of tumor-reactive clones (>50 pg/ml IFN-γ) is shown above. ****P ≤ 0.0001, Mann-Whitney test. (B) Reactivity of FrTu1913-derived clones against autologous tumor TC1913 or allogeneic TC624 (matched HLA-A*0201). Clone 41 expressed the CDR3β amino acid sequence corresponding to the most frequent CD8+ clonotype in FrTu1913; clone 208 recognized CDKN2Amut HLA-A*1101 restricted epitope; clone 199 was a tumor-reactive clone of yet-unknown specificity; clone 88 did not recognize TC1913. Recognition of targets was measured by assessing 4-1BB upregulation. (C) Recognition of CDKN2Amut and HLA-A11mut by FrTu1913-derived T cell clones. COS-A2 cells were transiently transfected with plasmids encoding HLA-A11WT or HLA-A11mut, and recognition of CDKN2Amut peptide was assessed by measuring 4-1BB upregulation. (D) Frequency of the 30 most frequent TCRβ clonotypes in the CD8+PD-1+ population in FrTu1913. The cumulative frequency (Σ freq.) of the HLA-A11mut–specific clonotypes in each population is specified below.
CD8+ TCRβ clonotypes recognizing mutated tumor neoantigens are highly expanded in the CD8+ and PD-1+ populations infiltrating FrTu1913.
The previously reported tumor-reactive clonotypes specific for mutated HLA-A*1101 (referred to herein as HLA-A11mut) and CDKN2Amut, derived from FrTu1913 (36), allowed us to further investigate whether clonotypic frequency in the CD8+ and PD-1+ populations in the fresh tumor could predict tumor recognition. The 3 known CD8+ HLA-A11mut–specific clonotypes from FrTu1913 ranked among the 23 most frequent clonotypes in the PD-1+ population and were detected at lower frequencies in the PD-1– population (Table 1). The 2 known CDKN2Amut-specific clonotypes were also predominantly detected in the PD-1+ population (Table 1). However, none of these clonotypes accounted for the most frequent CD8+ TCRβ clonotype in FrTu1913, representing 26.9% and 0.2% of the PD-1+ and PD-1– populations, respectively.
We next sought to determine whether the most frequent clonotype in tumor FrTu1913 was capable of recognizing autologous tumor cell line TC1913 and either of the 2 known mutations expressed by this tumor. To identify this clonotype, we screened the clones established from FrTu1913 (Figure 5C) for the TRBV12-3 family region using a TCR Vβ 8–specific antibody and sequenced some of the clones. The CDR3β hypervariable region of clone 41 was identical to the most frequent clonotype expressed in FrTu1913. Assessment of autologous tumor recognition and recognition of CDKN2Amut and HLA-A11mut revealed that the most frequent clonotype in FrTu1913 was tumor reactive (Figure 7B) and specifically recognized the mutation in HLA-A*1101 uniquely expressed by TC1913 (Figure 7C). In FrTu1913, mutation-specific clonotypes were preferentially expanded in the PD-1+ population, and the most frequent clonotype in the CD8+ TIL and PD-1+ populations was tumor reactive and mutation specific (Figure 7D and Table 1).
Discussion
PD-1 was initially described to be expressed on a T cell hybridoma undergoing cell death (37). Its negative effect on T cell responses was first delineated in PD-1 knockout mice (38, 39). Since then, PD-1 expression and coexpression of other inhibitory receptors such as CTLA-4, TIM-3, BTLA, CD160, LAG-3, and 2B4 have become a hallmark of chronically stimulated T cells during chronic infection or in the tumor microenvironment. This altered phenotype, and the interaction of these receptors with their corresponding ligands on target cells, is associated with impaired proliferation and effector function frequently referred to as exhaustion (18, 24, 40). Expression of PD-1 in patients with chronic viral infections correlates with disease progression (22, 41). Additionally, CD8+ lymphocytes targeting melanoma differentiation antigens in the tumor express PD-1, CTLA-4, TIM-3, and LAG-3 and exhibit impaired IFN-γ and IL-2 secretion (23, 24), supporting a negative regulatory role of PD-1 and inhibitory receptors in naturally occurring T cell responses to cancer and providing a rationale for the treatment of cancer with immune checkpoint inhibitors.
In the present study, we found that expression of PD-1 on CD8+ melanoma TILs captured the diverse repertoire of clonally expanded tumor-reactive lymphocytes. TCRβ sequencing revealed that tumor-reactive and mutation-specific clonotypes were highly expanded in the CD8+ population and preferentially expanded in the PD-1+ population. This is consistent with the TCR stimulation-driven expression of this receptor on T cells (42). The inhibitory receptors TIM-3 and LAG-3 and the costimulatory receptor 4-1BB were also expressed on CD8+PD-1+ TILs and could also be used to enrich for tumor-reactive cells. PD-1 was consistently expressed at a higher frequency and was found to be more comprehensive at identifying the diverse repertoire of tumor-reactive cells infiltrating melanoma tumors, although the less frequent PD-1–/TIM-3+ and PD-1–/LAG-3+ subpopulations could also represent tumor-reactive cells (Supplemental Figure 4 and Supplemental Table 6). Additionally, previous studies from our laboratory showing coexpression of PD-1 and CTLA-4 (23), and our preliminary data supporting coexpression of PD-1 and ICOS (Supplemental Figure 5), suggest that other receptors may also be used to distinguish tumor-reactive cells. Our present results further support immunotherapeutic intervention using immune checkpoint blockade using PD-1, TIM-3, and LAG-3 blocking antibodies or 4-1BB agonistic antibody to restore the function of tumor-reactive lymphocytes, which is currently being actively pursued in the clinic (3, 4, 6, 7, 43). The potential cooperative mechanisms of inhibition of these receptors when engaged with their ligands (44, 45) suggests that the combined targeting of different inhibitory receptors can further enhance antitumor efficacy, as already shown with the combination of anti–PD-1 and anti–CTLA-4 (5). Our present results demonstrate that PD-1 identifies the clonally expanded CD8+ tumor-reactive population and suggest that expression of PD-1 on CD8+ TILs could function as a potential predictive biomarker of antitumor efficacy using immune checkpoint inhibitors.
Naturally occurring tumor-reactive cells play a pivotal role in mediating antitumor responses after TIL transfer. Currently, expansion of TILs for patient treatment involves nonspecific growth of TILs from tumor fragments in IL-2, and the diversity and frequency of antitumor T cells present in the final T cell product used for treatment remains largely uncharacterized. Prospective clinical studies have reported that in vitro recognition of autologous tumor by TILs is associated with a higher probability of clinical response (9, 10), which suggests that enrichment of tumor-reactive cells could enhance clinical efficacy. This is consistent with the idea that both tumor-reactive and non–tumor-reactive cells may compete for cytokines in vivo, especially in the absence of vaccination. However, the isolation of the patient-specific repertoire of tumor-reactive cells is not possible with current technologies (14, 28, 46–50). Our findings established that expression of PD-1, TIM-3, LAG-3, and 4-1BB in CD8+ TILs can be used to enrich for tumor-reactive cells, regardless of the specific antigen targeted. One potential concern with isolating T cells expressing inhibitory receptors for therapy is that these cells may be exhausted or functionally impaired (23, 24, 44, 51, 52). However, we found that PD-1+, TIM-3+, and LAG-3+ CD8+ cells expanded in IL-2 were capable of secreting IFN-γ and lyse tumor in vitro. This supports the notion that immune dysfunction associated with coexpression of inhibitory receptors on CD8+ TILs can be reversed (21, 41, 51, 53), and may enable the reproducible enrichment of tumor-reactive cells for patient treatment. Notably, in a preliminary experiment (n = 8 nonresponders; 14 responders), there was no association between the frequency of expression of any of the markers studied in the CD8+ TILs in the fresh tumor and the clinical response to TILs derived from these tumor samples. However, the fresh tumors included in this study belonged to patients treated in several TIL protocols over the course of 10 years, and TILs were generated from these tumors using different methods, which makes these data difficult to interpret. In addition, the frequency of cells initially expressing PD-1 in the tumor may not reflect the frequency of the PD-1 derived cells in the infusion bag. For example, a low frequency of PD-1+ cells may be highly enriched during the process of TIL culture as a result of the presence of tumor cells. Although in vivo antitumor activity of tumor-isolated TILs based on PD-1 expression requires testing in a clinical trial, the observation that the overwhelming majority of tumor-reactive cells were derived from cells expressing PD-1 suggests that cells expressing PD-1 and inhibitory receptors in the tumor play a critical role in tumor regression after TIL administration.
The functional implications of selecting PD-1–, LAG-3–, TIM-3–, or 4-1BB–expressing T cells to enrich for tumor-reactive cells for patient treatment remain unclear. Although previous studies have reported differential expression of PD-1, LAG-3, and TIM-3 throughout differentiation (17), or preferential expression of TIM-3 in IFN-γ–secreting cells (54), our preliminary results have failed to show consistent phenotypic or functional differences between PD-1+, LAG-3+, TIM-3+, and 4-1BB+ selected TILs, including cytokine secretion, proliferation, and susceptibility to apoptosis (data not shown). We found that PD-1 expression was almost completely lost in the PD-1+ derived populations upon in vitro culture in IL-2. Conversely, TIM-3 and LAG-3 expression increased in the TIM-3– and LAG-3– populations after expansion. Overall, there were no differences in the expression of PD-1, TIM-3, or LAG-3 between any the populations after expansion. Thus, in agreement with previous reports (55, 56), we conclude that expansion in IL-2 alters the expression of these markers and compromises the potential use of inhibitory receptors to select for tumor-reactive cells after in vitro expansion. Recent work in animal models suggests that chronic antigen stimulation (57–59) or a tolerizing microenvironment (60) may lead to permanent epigenetic changes in T cells, raising the possibility that the restoration of function observed in previously exhausted or tolerized cells in presence of cytokines may only be transient. These results have not yet been corroborated in human tumor-specific cells. However, given that the overwhelming majority of tumor-reactive cells appear to derive from cells expressing PD-1 in the tumor, studying permanent versus transient reversion of exhaustion may have important implications for adoptive cell transfer of TILs.
Tumor-reactive cells can also be found infiltrating other tumor malignancies, such as renal cell carcinoma (61) or ovarian (62), cervical (63), or gastrointestinal tract cancers (64), albeit at lower frequencies. Our findings provide alternatives to enrich and study tumor-reactive CD8+ TILs through selection of cells expressing the cell surface receptors PD-1, LAG-3, TIM-3, and 4-1BB, a hypothesis that we are actively investigating. Additionally, our present findings showed that the frequency of a specific clonotype in the CD8+ and PD-1+ populations can be used to predict its ability to recognize tumor and isolate tumor-specific TCRs, thus providing means to overcome potential irreversible functional impairments of TILs (52).
2 reports with opposing results have generated controversy regarding which may be the optimal marker for the identification of the tumor-reactive repertoire, PD-1 or 4-1BB. In one report studying PD-1 expression in the tumor, the authors showed promising although inconsistent ability to enrich for shared melanoma-reactive cells (55). In a more recent article studying the role of 4-1BB in fresh ovarian TILs, Ye et al. concluded that expression of 4-1BB, but not PD-1, on lymphocytes defines the population of tumor-reactive cells in the tumor (65). The results of Ye et al. appear to contradict our present findings, showing that expression of PD-1 rather than 4-1BB more comprehensively identifies the repertoire of tumor-reactive cells in the tumor. However, these inconsistencies can be explained by different experimental approaches undertaken to study the immunobiology of TILs. First, Ye et al. found that expression of 4-1BB in fresh ovarian TILs and tumor-associated lymphocytes was low, and thus exposed the tumor to IL-7 and IL-15 (65). In the 1 patient sample in which the authors enriched for tumor-reactive cells from fresh ovarian TILs or tumor-associated lymphocytes exposed to IL-7 and IL-15, expression of 4-1BB was dependent on in vitro activation, but no longer represented the natural expression of 4-1BB in the fresh tumor. Second, with the exception of the 1 experiment described above, the enrichment experiments reported were carried out with melanoma or ovarian TIL lines expanded in IL-2 and cocultured with tumor cell lines in vitro. It is well known that IL-2 can change the activation status and also the expression of inhibitory receptors on T cells (data not shown and ref. 56). Thus, the experiment comparing expression of PD-1 and 4-1BB performed by Ye et al. (65) addressed the significance of these receptors after in vitro coculture of a highly activated melanoma TIL line with a tumor cell line, rather than the role of PD-1 and 4-1BB expression in CD8+ lymphocytes in the fresh tumor. Finally, both Inozume et al. and Ye et al. used matched HLA-A2 cell lines to assess tumor reactivity (55, 65). However, the use of HLA-matched tumor cell lines does not enable the assessment of reactivities against unique mutations that are present only in the autologous tumor cell line. In our current study, we used fresh melanoma tumors for all our experiments, and these were rested in the absence of cytokines to preserve the phenotype of TILs. Moreover, we used autologous tumor cell lines to assess tumor recognition. We believe that our experimental approach overcomes the limitations described above, enabling us to conclude that tumor-reactive cells can be detected in both the PD-1+/4-1BB+ and PD-1+/4-1BB– CD8+ TIL populations.
In summary, expression of PD-1 in CD8+ TILs in the fresh tumor identified and selected for the diverse patient-specific repertoire of tumor-reactive cells, including mutation-specific cells. In addition, analysis of the CD8+ TIL TCRβ repertoire in 2 melanomas showed that the frequency of a specific TCRβ clonotype in the CD8+ and PD-1+ populations could be used to predict its ability to recognize the autologous tumor. The use of inhibitory receptors and the frequency of individual TCRs to prospectively identify and select the diverse repertoire of tumor-reactive cells holds promise for the personalized treatment of cancer with T cell therapies, but may also facilitate the dissection and understanding of the immune response in human cancer patients.
Methods
Patient characteristics and PBL and tumor samples.
Matched PBL (n = 21) and tumor specimens (n = 24) were obtained from patients with metastatic melanoma. Patients included in this study were not undergoing therapy when their samples were collected, and they all had metastatic melanoma. Patients had undergone a wide range of prior therapies, including surgery, chemotherapy, radiotherapy, and immunotherapy, or none of the above. See Supplemental Table 7 for patient characteristics. A fraction of the patients had received immunotherapy (including high-dose IL-2, anti–CTLA-4, or anti–PD-1) between 8 years to 2 months prior to tumor resection, but none had received adoptive cell therapy. See Supplemental Table 8 for details of the 6 tumors studied more in depth, specific immunotherapeutic regimens to which they were exposed, and times prior to phenotypic assessment. PBLs were obtained by either leukapheresis or venipuncture, prepared over Ficoll-Hypaque gradient (LSM; ICN Biomedicals Inc.), and cryopreserved until analysis. After surgical resection, tumor specimens were processed as previously described (66). Briefly, tumor specimens were minced under sterile conditions, followed by enzymatic digestion (RPMI-1640 with l-glutamine [Lonza], 1 mg/ml collagenase IV [Sigma-Aldrich], 30 U/ml DNAse [Genentech], and antibiotics) overnight at room temperature or for several hours at 37°C and intermittent mechanical tissue separation using gentleMACS (Miltenyi Biotech). Tumor single-cell suspensions were cryopreserved until further analysis.
Antibodies, flow cytometry, and cell sorting.
We purchased fluorescently conjugated antibodies from BD Biosciences (UCHT1, 1.6:100, CD3 PE-CF594; RPTA-T4, 3.4:100, CD4 V500; NK-1, 3:100, CD57 FITC, J168-540, 1.2:100, BTLA PE), eBioscience (MIH-4, 1.6:100, PD-1 APC), Biolegend (O323, 2:100, CD27 BV605; BD96, 1.4:100, CD25 BV650; EH12.2H7, 0.7:100, PD-1 BV421; c398.4A, 1:100, ICOS Pacific Blue), R&D (344823, 2.6:100, TIM-3 PE and APC), Enzo Life Sciences (17B4, 1:100, LAG-3 FITC), and Miltenyi (4B4-1, 2.6:100, 4-1BB PE). We carried out flow cytometry acquisition on a modified Fortessa instrument (BD Biosciences), equipped to detect 18 fluorescent parameters. We compensated and analyzed data with FlowJo software (Treestar). We sorted T cell subsets using a modified FACSAria instrument (BD Biosciences). Antibodies and tetramers were titrated for optimal staining.
For phenotypic characterization of CD8+ T cells and cell sorting using flow cytometry, PBMCs and tumor samples were thawed in the presence of 3 U/ml DNAse (Genentech Inc.), and rested 16–24 hours in a 1:1 mix of RPMI-1640 with l-glutamine (Lonza) and AIMV (Gibco) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 12.5 mM HEPES, and 5% human serum (noncommercial, prepared from healthy donors) without cytokines (T cell media) at a concentration of 2 × 106 live cells (determined by trypan blue exclusion) per well in a 24-well plate at 37°C in 5% CO2. This overnight resting period was carefully assessed and necessary for the recovery of expression cell surface receptors affected by cryopreservation or subject to cleavage during the enzymatic digest. Cells were harvested, counted, and blocked with Fc block for human cells (Miltenyi). 1 × 106 live cells were stained for 30 minutes in 50 μl volume of FACS buffer (PBS containing 0.5% BSA and 2 mM EDTA) with the antibodies specified, washed, and acquired. Propidium iodide (PI) was added to exclude dead cells. Phenotypic analysis and sorting of CD3+CD8+ subsets from the tumor single-cell suspensions were performed after gating on lymphocytes (FSC by SSC), excluding aggregates and dead cells (PI+). Gates were set according to isotype, and fluorescence minus one (FMO) controls. See Supplemental Figure 6 for 2 examples of the post-sort purity of CD3+CD8+ subsets sorted from FrTu1913 for TCRβ sequencing or for in vitro expansion.
RNA preparation and TCRβ deep sequencing.
Cells of interest were sorted into a 1:1 mix of PBS and FBS (Hyclone Defined, Logan), and cell pellet was resuspended in 200 μl RNAlater (Invitrogen). We extracted mRNA and amplified TRB gene products of sorted CD8+ T cell subsets using a template-switch anchored RT-PCR, as previously described (67), followed by Illumina sequencing. TCRβ annotation was performed by combining a custom Java program written in house and NCBI BLAST+. Briefly, BLAST+ was used to identify the V and J germline genes of a TCRβ read. The CDR3 was then determined by finding the conserved cysteine at the 5′ end of the CDR3 and the conserved phenylalanine at the 3′ end of the CDR3. Unique TCRβ species, in which species is defined as a unique TRBV-CDR3 (nucleotide)-TRBJ combination, were then collapsed to determine the count for each species.
Similar cell numbers were sorted from each subset from the fresh tumor to ensure comparable coverage (total number of TCRβ sequence counts from a population/initial cell input of population) to enable comparison of clonotypic diversity among different populations.
Expansion of CD8+ T cells and establishment of T cell clones.
FACS-sorted CD8+ T cells (5 × 103–1 × 105 cells) were expanded using a rapid expansion protocol (REP) in T25 flasks in T cell media containing 30 ng/ml soluble anti-CD3 (OKT3, Miltenyi), 3,000 IU/ml rhIL-2 (Chiron), and 3 × 107 irradiated PBMCs pooled from 3 allogeneic donors. For establishment of T cell clones, cells were plated at 2 cells/well in 96-well U-bottomed plates in T cell media containing OKT3 and IL-2 (as described above) and 7.5 × 104 irradiated allogeneic PBMCs/well. After day 5, media was replaced every other day with fresh media containing IL-2. Cells were split when confluent or when they reached a concentration exceeding 3 × 106 cells/ml. At day 14–15, bulk-expanded populations were counted and frozen down. Cell expansion was variable among different populations and patients (Supplemental Table 9). Cloning efficiency (percentage of clones plated at 2 cells/well that grew) ranged 9%–26%. T cell clones were maintained in 500 CU/ml IL-2 and tested for recognition of autologous tumor 3–6 weeks after cell sorting.
Assessment of target cell recognition: IFN-γ release, 4-1BB upregulation, and cytolysis assay.
Both IFN-γ release and 4-1BB upregulation were used to measure recognition of targets. After 15 days of in vitro expansion of clones or TILs, basal expression of 4-1BB was consistently negative. 4-1BB is upregulated transiently in response to TCR stimulation, regardless of the effector cytokines produced or the differentiation state of the cell (28), and its expression on stimulated CD8+ cells peaks at 24–36 hours. For assessment of target cell recognition by TILs after in vitro expansion, we thawed the effector cells 2 days prior to coculture and cultured them at 1 × 106 cells/ml in 3,000 IU/ml IL-2 at 37°C in 5% CO2. 2 days later, effector cells were washed and cultured, either alone or with target cells (1 × 105:1 × 105) or plate-bound anti-CD3 (OKT3, Miltenyi), in 96-well plates in T cell media without IL-2. In antibody-blocking experiments, we preincubated melanoma cells with 0.05 mg/ml HLA-I or HLA-II blocking antibodies (clones W6/32 and IVA12, respectively) for 3 hours, followed by coculture with T cells. 24 hours later, supernatants were harvested (duplicates) and analyzed as previously described (47), and cells were stained in 40 μl PBS containing 0.5% BSA and 2 mM EDTA with anti-CD3, anti-CD8, and anti–4-1BB antibodies and acquired on Canto II (BD Biosciences). Data were analyzed with FlowJo software (Treestar).
For cytolytic assays, targets cell lines were pulsed with 100 μCi (1 Ci = 37 GBq) 51Cr (PerkinElmer) and washed twice. We plated 1 × 104 labeled target cells per well with effector cells at the indicated effector/target ratios in a 96-well U-bottomed plate for 4 hours at 37°C. The amount of 51Cr released was determined by γ-counting, and specific lysis was calculated from triplicate samples as [(experimental cpm – spontaneous cpm)/(maximal cpm – spontaneous cpm)] and expressed as a percentage.
T cell lines, controls, cell lines, and reagents.
Infusion products TIL3309, TIL3289, and TIL1700 were expanded from fresh tumor digests as previously described (68). Briefly, single-cell suspensions of tumor digests were cultured in media containing 6,000 IU IL-2 (Chiron). TIL cultures that expanded were screened for recognition of autologous or HLA-matched tumor, and reactive TIL cultures were expanded further — using a REP with IL-2, anti-CD3 antibody, and irradiated feeder cells — to large numbers for patient infusion. A small portion of TILs underwent a second REP and were used as controls in coculture assays. PBMCs transduced with a MART126–35 A*0201 (F5 Td) or tyrosinase450–462 HLA-DR4 specific (CD4 T4) T cell receptors were generated as previously described (69). Evaluation of reactivity of FrTu1913-derived TILs or T cell clones was carried by pulsing peptides (1 μM for 1 hour) onto COS-A11 cells or COS-7 cells stably transduced with A*0201 (COS-A2 cells) and transiently transfected with HLA-A11WT or HLA-A11mut plasmids with Lipofectamine 2000 (Invitrogen). CDKN2Amut peptide and the control HLA-A*1101–restricted peptide CRKRSmut were purchased from Peptide 2.0.
Melanoma cell lines were established from tumor fragments or from mechanically or enzymatically separated tumor cells (as described above), which were cultured in RPMI 1640 plus 10% FBS (Hyclone Defined, Logan) at 37°C in 5% CO2. T2 cells and the human melanoma tumor cell lines 624, 624CIITA, TC526, TC1913, TC3289, TC3612, TC3713, TC3550, and TC2448 were cultured in RPMI 1640 plus 10% FBS (Sigma-Aldrich) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in 5% CO2. COS-7 cells and COS-7 cells stably transduced with HLA molecules were maintained in DMEM containing 10% FBS (Sigma-Aldrich) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in 5% CO2. Expression of HLA-II molecules on the surface of melanoma cell line 624 was upregulated by transduction with a retroviral construct encoding the HLA-II transactivator gene (CIITA) as previously described (70).
TC1913 and recognition by TIL1913 grown from FrTu1913 were reported previously (36). Briefly, a cDNA library was generated from autologous tumor cell line TC1913, and CD8+ T cell clones recognizing a frameshift mutation in CDKN2A locus that encodes for tumor suppressor proteins P14ARF and P16INK4a (CDKN2Amut) (3 different clonotypes), and a HLA-A11mut class I gene product (2 clonotypes) expressed by TC1913 were identified (36).
For some patients, CD4+CD25– cells were isolated from a pheresis pretreatment sample and used as normal autologous targets in cocultures, by doing CD25 depletion followed by CD4-positive selection using BD IMag cell separation system (BD Biosciences).
Statistics.
Values are reported as mean ± SEM, mean ± SD, or median, as indicated. We used Mann-Whitney test, Dunn test for multiple comparisons, and Wilcoxon signed-rank test to determine the statistical significance of the data. To determine the statistical difference in marker expression between CD8+ T cells in PBLs and TILs, we adjusted P values for the number of comparisons (Figure 1A; n = 11 comparisons). To determine whether there were differences in the frequency of PD-1 expression between CD8+ TIL populations, 21 multiple comparisons were performed, and only the ones that were statistically significantly different are shown (Figure 1D). P values were also adjusted for the number of comparisons. A P value of 0.05 or less was considered significant. We performed statistical calculations with Prism program 6.0 (GraphPad Software Inc.).
Study approval.
All patient samples were obtained in the course of a National Cancer Institute Institutional Review Board–approved clinical trial, and patients provided informed consent.
Supplementary Material
Acknowledgments
We thank Christian Hinrichs, Sanja Stevanovic, Anna Pasetto, Mojgan Ahmadzadeh, Pawel Muranski, and Mona El-Gamil for helpful discussion, revision of the manuscript, and technical advice. This work was supported in part by a generous contribution from the Milstein Family Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(5):2246–2259. doi:10.1172/JCI73639.
References
- 1.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamid O, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phan GQ, Weber JS, Sondak VK. CTLA-4 blockade with monoclonal antibodies in patients with metastatic cancer: surgical issues. Ann Surg Oncol. 2008;15(11):3014–3021. doi: 10.1245/s10434-008-0104-y. [DOI] [PubMed] [Google Scholar]
- 8.Robbins PF, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173(12):7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartzentruber DJ, et al. In vitro predictors of therapeutic response in melanoma patients receiving tumor-infiltrating lymphocytes and interleukin-2. J Clin Oncol. 1994;12(7):1475–1483. doi: 10.1200/JCO.1994.12.7.1475. [DOI] [PubMed] [Google Scholar]
- 10.Dudley ME, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. . J Clin Oncol. 2013;31(17):2152–2159. doi: 10.1200/JCO.2012.46.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. 2013;13:15. [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LA, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins PF, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kvistborg P, et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology. 2012;1(4):409–418. doi: 10.4161/onci.18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauce D, et al. PD-1 expression on human CD8 T cells depends on both state of differentiation and activation status. AIDS. 2007;21(15):2005–2013. doi: 10.1097/QAD.0b013e3282eee548. [DOI] [PubMed] [Google Scholar]
- 17.Baitsch L, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS One. 2012;7(2):e30852. doi: 10.1371/journal.pone.0030852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188(12):2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. . J Virol. 2007;81(17):9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. . Nat Med. 2006;12(10):1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 22.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443(7109):350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 23.Ahmadzadeh M, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baitsch L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev. 2009;229(1):192–215. doi: 10.1111/j.1600-065X.2009.00765.x. [DOI] [PubMed] [Google Scholar]
- 27.Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. . J Immunol. 2002;169(9):4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 28.Wolfl M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. . Blood. 2007;110(1):201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dziubianau M, et al. TCR repertoire analysis by next generation sequencing allows complex differential diagnosis of T cell-related pathology. Am J Transplant. 2013;13(11):2842–2854. doi: 10.1111/ajt.12431. [DOI] [PubMed] [Google Scholar]
- 30.Hohn H, et al. Longitudinal analysis of the T-cell receptor (TCR)-VA and -VB repertoire in CD8+ T cells from individuals immunized with recombinant hepatitis B surface antigen. . Clin Exp Immunol. 2002;129(2):309–317. doi: 10.1046/j.1365-2249.2002.01841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73(11):975–983. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 32.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 33.Fourcade J, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012;72(4):887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vinay DS, Kwon BS. Role of 4-1BB in immune responses. Semin Immunol. 1998;10(6):481–489. doi: 10.1006/smim.1998.0157. [DOI] [PubMed] [Google Scholar]
- 35.Waldmann TA. The multi-subunit interleukin-2 receptor. Annu Rev Biochem. 1989;58:875–911. doi: 10.1146/annurev.bi.58.070189.004303. [DOI] [PubMed] [Google Scholar]
- 36.Huang J, El-Gamil M, Dudley ME, Li YF, Rosenberg SA, Robbins PF. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol. 2004;172(10):6057–6064. doi: 10.4049/jimmunol.172.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 39.Nishimura H, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 40.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. . Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Kaufmann DE, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. . Nat Immunol. 2007;8(11):1246–1254. doi: 10.1038/ni1515. [DOI] [PubMed] [Google Scholar]
- 42.Agata Y, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 43.Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin Oncol. 2010;37(5):508–516. doi: 10.1053/j.seminoncol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox AL, et al. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science. 1994;264(5159):716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 47.Robbins PF, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183(3):1185–1192. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker C, et al. Adoptive tumor therapy with T lymphocytes enriched through an IFN-γ capture assay. Nat Med. 2001;7(10):1159–1162. doi: 10.1038/nm1001-1159. [DOI] [PubMed] [Google Scholar]
- 49.Knabel M, et al. Reversible MHC multimer staining for functional isolation of T-cell populations and effective adoptive transfer. Nat Med. 2002;8(6):631–637. doi: 10.1038/nm0602-631. [DOI] [PubMed] [Google Scholar]
- 50.Hombrink P, et al. High-throughput identification of potential minor histocompatibility antigens by MHC tetramer-based screening: feasibility and limitations. PLoS One. 2011;6(8):e22523. doi: 10.1371/journal.pone.0022523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang SF, et al. Early T cell signalling is reversibly altered in PD-1+ T lymphocytes infiltrating human tumors. . PLoS One. 2011;6(3):e17621. doi: 10.1371/journal.pone.0017621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289–295. doi: 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- 53.Zippelius A, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64(8):2865–2873. doi: 10.1158/0008-5472.CAN-03-3066. [DOI] [PubMed] [Google Scholar]
- 54.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inozume T, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. . J Immunother. 2010;33(9):956–964. doi: 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mujib S, et al. Antigen-independent induction of Tim-3 expression on human T cells by the common γ-chain cytokines IL-2, IL-7, IL-15, and IL-21 is associated with proliferation and is dependent on the phosphoinositide 3-kinase pathway. J Immunol. 2012;188(8):3745–3756. doi: 10.4049/jimmunol.1102609. [DOI] [PubMed] [Google Scholar]
- 57.Youngblood B, et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013;191(2):540–544. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Youngblood B, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011;35(3):400–412. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Utzschneider DT, et al. T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol. 2013;14(6):603–610. doi: 10.1038/ni.2606. [DOI] [PubMed] [Google Scholar]
- 60.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science. 2012;335(6069):723–727. doi: 10.1126/science.1214277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. 2004;427(6971):252–256. doi: 10.1038/nature02240. [DOI] [PubMed] [Google Scholar]
- 62.Ioannides CG, Freedman RS, Platsoucas CD, Rashed S, Kim YP. Cytotoxic T cell clones isolated from ovarian tumor-infiltrating lymphocytes recognize multiple antigenic epitopes on autologous tumor cells. J Immunol. 1991;146(5):1700–1707. [PubMed] [Google Scholar]
- 63.Piersma SJ, et al. Human papilloma virus specific T cells infiltrating cervical cancer and draining lymph nodes show remarkably frequent use of HLA-DQ and -DP as a restriction element. Int J Cancer. 2008;122(3):486–494. doi: 10.1002/ijc.23162. [DOI] [PubMed] [Google Scholar]
- 64.Turcotte S, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol. 2013;191(5):2217–2225. doi: 10.4049/jimmunol.1300538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ye Q, et al. CD137 accurately identifies and enriches for naturally-occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014;20(1):44–55. doi: 10.1158/1078-0432.CCR-13-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gros A, Turcotte S, Wunderlich JR, Ahmadzadeh M, Dudley ME, Rosenberg SA. Myeloid cells obtained from the blood but not from the tumor can suppress T-cell proliferation in patients with melanoma. Clin Cancer Res. 2012;18(19):5212–5223. doi: 10.1158/1078-0432.CCR-12-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Price DA, et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202(10):1349–1361. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dudley ME, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu Y, Boss JM, Hu SX, Xu HJ, Blanck G. Apoptosis-independent retinoblastoma protein rescue of HLA class II messenger RNA IFN-gamma inducibility in non-small cell lung carcinoma cells. Lack of surface class II expression associated with a specific defect in HLA-DRA induction. J Immunol. 1996;156(7):2495–2502. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.