

Abstract
Three years ago, two research groups independently identified a previously undescribed T cell cosignaling molecule; one referred to it as V-domain Ig suppressor of T cell activation (VISTA), and the other used the term programmed death-1 homolog (PD-1H). Recombinant and ectopically expressed PD-1H functions as a coinhibitory ligand for T cell responses. However, the function of endogenous PD-1H is not clear. In this issue of the JCI, Flies and colleagues demonstrate that endogenous PD-1H on both T cells and APCs serves as a coinhibitory molecule for T cell activation and provide further support for targeting PD-1H as a therapeutic strategy for transplantation and cancers.
Life is never dull, especially if you are interested in T cell cosignaling molecules. The identification of new members of this family can lead not only to new paradigms in immune recognition, but can also affect the treatment of cancer and autoimmune diseases (1–4). Among the newest additions to the T cell cosignaling family is an immunoglobulin superfamily member that bears two different names due to its independent discoveries three years ago: V-domain Ig suppressor of T cell activation (VISTA) and programmed death-1 homolog (PD-1H).
Wang et al. named this molecule VISTA (5), based on significant sequence homology with B7 homologue 1 (B7H1) and the ability of recombinant VISTA-Fc protein and ectopic expression of VISTA on APCs or tumor cells to inhibit T cell proliferation. In an independent study, Flies and colleagues identified the same molecule and named it PD-1H due to greater overall sequence and gene structure similarities with programmed death-1 (PD-1) (6).
Same target, different results
Remarkably, the antibodies produced against the molecule by Wang and colleagues and Flies et al. had starkly different effects. Whereas Wang et al. found that their anti-VISTA antibody exacerbated EAE, Flies et al. found that their anti–PD-1H antibody suppressed graft-versus-host diseases (GVDH) to such an extent that fully allogeneic bone marrow chimera mice (BALB/c to lethally irradiated C57BL/6 mice) could be obtained following a single dose of anti–PD-1H antibody at the onset of transplantation. The efficacy of anti–PD-1H in GVDH has not been achieved by other biologics.
Why is there such a difference in the effects of the anti–PD-1H and anti-VISTA antibodies, given that they target the same molecule? One of the first clues to reconcile the antibody-specific differences was the demonstration by Wang et al. that their anti-VISTA antibody shows almost undetectable levels of VISTA after T cell activation, while Flies et al. observed substantial elevation of PD-1H on both CD4+ and CD8+ T cells after activation. These fickle antibodies highlight what students of immunology have learned from mice: it is not a good idea to discern biology of a molecule by its antibodies (7–10). In this issue, Flies et al. (11) elucidate the biological function of PD-1H using mice with targeted mutation of the gene encoding PD-1H.
PD-1H on both T cells and APC inhibits T cell function
Flies et al. (11) obtained PD-1H–deficient mice from a regional resource center (Mutant Mouse Regional Resource Centers [MMRRC], UCD, Davis, California, USA). Despite broad expression of Pd1h in multiple lineages of hematopoietic cells, Flies and colleagues found no difference in the composition of any lineage of PD-1H–expressing cells, ruling out a major role for PD-1H in hematopoiesis and T cell development. Compared with their WT littermates, Pd1h–/– mice exhibited an increased frequency of activated T cells as they aged. This result is consistent with an overall T cell inhibitory function of PD-1H. Furthermore, Pd1h–/– mice had enhanced susceptibility to concanvalin A–induced hepatitis, but had greater resistance to ectopically transplanted glioma, especially after radiotherapy.
Using purified T cells from WT and Pd1h–/– mice, Flies and colleagues clearly demonstrated that T cells lacking PD-1H are more responsive to cognate antigen and surrogate T cell receptor ligands. In addition, the enhanced activation of T cells by Pd1h–/– APCs suggests that PD-1H on APCs inhibits T cells regardless of their Pd1h genotype. Therefore, PD-1H is a coinhibitory molecule regardless of its cellular distribution. Together, these results pave the way for immune modulation by anti–PD-1H antibodies for autoimmune diseases and transplantation as described in this issue (11) and previously by Flies et al. (6).
Remaining questions
The data reported to date on PD-1H have raised intriguing issues in relation to its fundamental mechanism of immune recognition, receptor, and function of unique intracellular domain.
First, in relation to the issue of immune recognition, Flies et al. mixed T cells and APCs from WT and Pd1h–/– mice and showed that PD-1H on both T cells and APCs can suppress T cell activation (11). Because T cells both express and respond negatively to PD-1H, it is easy to envision a suppressive T cell/T cell interaction directly through PD-1H (Figure 1A). This potentially direct T cell/T cell interaction is very different from the known mechanisms of T cell crosstalk, including those that mediate both immunological help and immune suppression, which are based solely on modification of APCs (Figure 1B). Th cells help induce costimulatory activity on APCs (12, 13). Conversely, Tregs suppress T cell activation by ripping off costimulatory molecules from APCs (14). A general mechanism for direct T cell/T cell suppression was previously invoked (15), but proof of such an interaction has eluded immunologists. With the identification of PD-1H as a coinhibitor that is abundantly expressed on activated T cells, it is not hard to envision that PD-1H on neighboring T cells enables them to tune each other’s response to cognate antigen (Figure 1A). Such a tuning mechanism can also be extended interactions between PD-1H–expressing T cells and APCs (Figure 1A).
Figure 1. Models of T cell/T cell interaction.
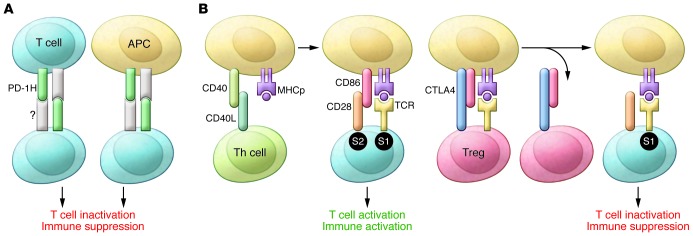
(A) A new PD-1H–centric model. PD-1H on the surface of T cells and APCs interacts with an unknown receptor(s) on T cells to directly inactivate T cells and confer immune suppression. (B) Classic models of immune activation and immune suppression through modulation of costimulatory activity of APCs. For immune activation, interaction of CD40L expressed on Th cells with CD40 on APCs induces expression of costimulatory molecules, including CD86 and others. The T cells that recognize the same APCs are activated as they are provided with both signal 1 (S1), the cognate antigens (MHC plus peptide [MHCp]), and signal 2 (S2), the costimulatory signal. During immune suppression, Tregs use CTLA-4 to rip CD28 ligands from the surface of APCs. The T cells that recognize the same APC are suppressed as they are provided with only S1, which inactivates T cells through death and anergy (17, 18).
The second unresolved issue is the nature of the PD-1H receptor. The elevated response of Pd1h–/– T cells to PD-1H as reported by Flies and colleagues, along with the data from Wang et al. demonstrating that recombinant VISTA-Fc robustly inhibits T cell activation, suggests that both T cells and APCs have a PD-1H/VISTA receptor or receptors. A direct binding partner of PD-1H has yet to be shown, and a putative receptor remains to be identified.
Third, notwithstanding the expression of PD-1H on T cells, most data to date can be accounted for by evoking PD-1H strictly as an inhibitory ligand, as previously suggested by Ceeraz et al. (16). This interpretation, while advantageous for its simplicity, fails to account for the existence of the large and very unique intracellular domain of the molecule. Wang et al. noted that VISTA is highly conserved in organisms ranging from fungi to humans (5); however, while the extracellular IgV domain of VISTA shows homology with B7H1, no relative of the large intracellular domain has been found, suggesting that this intracellular domain may have evolved independently of other cosignaling molecules. The unusual evolutionary conservation of the intracellular domain of PD-1H hints at a functional importance for this part of the molecule, but its uniqueness shields any clue as to what that function may be. With these outstanding issues and the strong effect of targeting PD-1H in transplantation, autoimmune diseases, and cancer, it is safe to assume that we have not heard the last from this protein.
Acknowledgments
I thank Pan Zheng and Dashuang Shi for helpful discussion. This work is supported by grants from the NIH (CA58033 and AI64350).
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(5):1891–1893. doi:10.1172/JCI75798.
See the related article beginning on page 1966.
References
- 1.Genovese MC, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med. 2005;353(11):1114–1123. doi: 10.1056/NEJMoa050524. [DOI] [PubMed] [Google Scholar]
- 2.Hwu P. Treating cancer by targeting the immune system. N Engl J Med. 2010;363(8):779–781. doi: 10.1056/NEJMe1006416. [DOI] [PubMed] [Google Scholar]
- 3.Kremer JM, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349(20):1907–1915. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- 4.Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366(26):2517–2519. doi: 10.1056/NEJMe1205943. [DOI] [PubMed] [Google Scholar]
- 5.Wang L, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208(3):577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187(4):1537–1541. doi: 10.4049/jimmunol.1100660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachmann MF, Gallimore A, Jones E, Ecabert B, Acha-Orbea H, Kopf M. Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur J Immunol. 2001;31(2):450–458. doi: 10.1002/1521-4141(200102)31:2<450::AID-IMMU450>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163(3):1128–1131. [PubMed] [Google Scholar]
- 9.Liu Y. Is CTLA-4 a negative regulator for T-cell activation? Immunol Today. 1997;18(12):569–572. doi: 10.1016/s0167-5699(97)01170-5. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. 2006;177(9):5990–5996. doi: 10.4049/jimmunol.177.9.5990. [DOI] [PubMed] [Google Scholar]
- 11.Flies DB, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell–mediated immunity. . J Clin Invest. 2014;124(5):1966–1975. doi: 10.1172/JCI74589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guerder S, Matzinger P. A fail-safe mechanism for maintaining self-tolerance. J Exp Med. 1992;176(2):553–564. doi: 10.1084/jem.176.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T- helper and a T-killer cell [see comments]. . Nature. 1998;393(6684):474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 14.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. . Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 15.Janeway CA. Autoimmune disease: immunotherapy by peptides? Nature. 1989;341(6242):482–483. doi: 10.1038/341482a0. [DOI] [PubMed] [Google Scholar]
- 16.Ceeraz S, Nowak EC, Noelle RJ. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013;34(11):556–563. doi: 10.1016/j.it.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Janeway CA, Janeway CA., Jr Interferon γ plays a critical role in induced cell death of effector T cell: a possible third mechanism of self-tolerance. J Exp Med. 1990;172(6):1735–1739. doi: 10.1084/jem.172.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–1068. doi: 10.1016/S0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]