

Abstract
Left ventricular hypertrophy is an initial compensatory mechanism in response to cardiac stress that can degenerate into heart failure and sudden cardiac death. Recent studies have shown that microRNAs (miRs) regulate several aspects of cardiovascular diseases. In this issue of the JCI, Bang and colleagues identified an exosome-mediated communication mechanism between cardiac fibroblasts and cardiomyocytes. Specifically, cardiac fibroblasts secrete miR-enriched exosomes, which are subsequently taken up by cardiomyocytes, in which they alter gene expression. In particular, a passenger strand miR, miR-21*, was identified as a potent paracrine factor that induces cardiomyocyte hypertrophy when shuttled through exosomes. These advanced comprehensive analyses represent a major step forward in our understanding of cardiovascular physiopathology, providing a promising adjunctive target for possible therapeutic approaches, namely the miR-mediated paracrine signaling network.
Left ventricular hypertrophy and microRNAs
Left ventricular hypertrophy (LVH) can be elicited by several clinical conditions, including systemic hypertension, cardiac valve disease, and myocardial infarction, that increase cardiac workload. This adaptation is one facet of cardiac remodeling and subsequent heart failure (HF), which is a major cause of death in Western countries (1). In 1970, the Framingham Heart Study revealed that the presence of LVH alone is associated with increased mortality (1). Moreover, recent appraisals of this cohort have also linked LVH to arrhythmic sudden cardiac death (2). Interestingly, inhibition of LVH after transverse aortic constriction prevents HF and increases survival in genetically modified animals (3, 4).
Because of the strong correlation between LVH and sudden cardiac death, the mechanisms underlying LVH etiology, and degeneration toward HF, have been studied extensively (4, 5). Several aspects of LVH-dependent HF still need to be clarified, and, in this regard, microRNAs (miRs) could provide additional insights into the mechanisms involved in LVH and HF. miRs are short (17–25 nucleotides) noncoding RNAs that function mainly to regulate gene expression by hindering the translation of specific mRNAs at the posttranscriptional level (6).
The biogenesis of miRs is a complex cascade. Briefly, from a transcript of the primary miR (pri-miR), approximately 60- to 70-nucleotide hairpin-structured precursor miR (pre-miR) is synthesized and exported to the cytoplasm, in which it is cleaved into the mature duplex miR (miR:miR*) by RNAse III. Subsequently, one of the two strands, the so-called passenger strand (also referred to as miR*), is released and degraded, whereas the other strand with the 5′ terminus that has less-stable base pairing is stripped away from the duplex and transferred to argonaute 2 (AGO) to become the guide strand or miR. While the guide strands target a miR-induced silencing complex (miRISC) to mRNAs with partially complementary sequences and silence them, the passenger strands are not generally considered to be of biological relevance (6).
With respect to adaptive growth regulation, miRs have been identified and associated with several cardiovascular diseases, including neointimal proliferation after vascular injury (7, 8), ischemic cardiomyopathy (9), and cardiac hypertrophy (10–14). The importance of miRs in cardiovascular biology has also been demonstrated in animals with conditional deletion of the essential miR-processing enzyme dicer1. Postnatal deletion of dicer1 in the myocardium results in spontaneous cardiac hypertrophy, interstitial fibrosis, and reactivation of fetal gene expression (15). In addition, specific miRs have been identified in LVH and HF. For example, loss of miR-208a, which is encoded by the α-myosin heavy chain (Myh6) transcript, prevents pathological hypertrophic remodeling and fibrosis (11). Cardiomyocyte-specific overexpression of the miR-212/132 family targets the antihypertrophic and proautophagic FOXO3 transcription factor and leads to pathological cardiac hypertrophy, HF, and death in mice, whereas animals lacking miR-212/132 are protected from pressure overload–induced HF (12). Members of the miR-34 family of miRs are upregulated in mouse models of left ventricular pressure overload, and miR-23a controls hypertrophic growth of cardiomyocytes through signaling that involves calcineurin and NFAT.
Other miRs will likely be uncovered as regulators of hypertrophy, and some information is already available about circulating miRs that affect both hypertrophy and fibrosis. Among these circulating miRs are miR-29a and miR-30, which have been proposed as possible markers for use in LVH diagnosis (16). Interestingly, miR-30 is downregulated upon angiotensin II administration, which promotes increased myocardial hypertrophy through excessive autophagy. In patients, miR-30 levels in the bloodstream are positively associated with left ventricular wall thickness. Intriguingly, miRs can be secreted from cells as vesicular bodies that arise from the plasma membrane or can be simply extruded by cells through membrane shedding (17). Once miRs are packed into specific multivesicular bodies, they can undergo unidirectional transfer as part of cell-cell communication strategies, establishing an intercellular trafficking network that exchanges genetic information and elicits a variety of phenotypic changes (18).
The role of exosomes in LVH
Exosomes are a specific subset of secreted membrane vesicles that are relatively homogeneous in size (30–100 nm) and differ from apoptotic bodies and microvesicles in their density and specific protein and nucleic acid composition. Multiple cells types secrete exosomes through fusion of multivesicular bodies with the plasma membrane. Recently, cell-to-cell exosome-dependent transfer of selected miRs from endothelial cells to vascular smooth muscle was reported within the vessel wall (19). Moreover, a similar mechanism was described to mediate the transfer of AGO/miR complexes from platelets to endothelial cells (20).
In this issue of the JCI, Bang and coworkers report a miR-containing exosome-mediated intercellular communication system that operates within the myocardium. The data provided by Bang and colleagues support previous observations of miR-containing exosomes mediating responses in other cell populations and confirm miR-mediated intercellular communication in cardiac tissue (21). In particular, Bang and colleagues revealed that miR-21*, a passenger strand microRNA that is usually degraded inside the cytoplasm, is specifically packaged into and shuttled by exosomes from cardiac fibroblasts to cardiomyocytes, in which it promotes a hypertrophic response (Figure 1 and ref. 21). Interestingly, Bang et al. demonstrated that fibroblast-derived exosomes are selectively enriched with miR-21* and exosome uptake by cardiomyocytes was dependent on temperature and actin. Once released within cardiomyocytes, exosome-derived miR-21* led to a substantial increase in cardiomyocyte cell size (Figure 1).
Figure 1. Dynamics of fibroblast-mediated exosome secretion and pivotal miRs involved in cardiac hypertrophy.
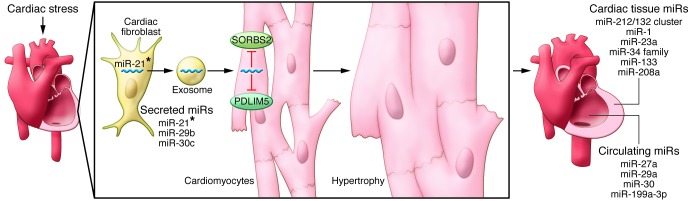
Following cardiac stress, fibroblasts secrete miR-enriched exosomes, which are enriched with miR-21*. Cardiomyocytes take up the fibroblast-secreted exosomes, which release miR-21*. Targeted knockdown of SORBS2 and PDLIM5 by miR-21* promotes hypertrophy. Many miRs that regulate hypertrophy have been identified and can be considered as tissue specific, circulating, and secreted. Among the latter, only miR-21* has been detected in pericardial fluid in the context of cardiac hypertrophy (21).
Using proteomic profiling, Bang and colleagues identified sarcoplasmic protein sorbin and SH3 domain-containing protein 2 (SORBS2) and PDZ and LIM domain 5 (PDLIM5) as targets of miR-21* in cardiomyocytes, since both the encoding genes were strongly silenced in miR-21*–transfected cardiomyocytes. Of note, in the same cardiomyocyte model, knock down of SORBS2 and PDLIM5 resulted in marked cellular hypertrophy. SORBS2 localizes to Z-disks, in which it influences the contractile and elastic properties of cardiac sarcomeres (22). Interestingly, release of SORBS2 from damaged cardiac tissue into the bloodstream upon fatal acute myocardial infarction has been described recently (23). PDLIM5 is highly conserved across species and, like SORBS2, colocalizes at Z-disks and is directly associated with dilated cardiomyopathy (24) and indirectly involved in human hypertrophic cardiomyopathy (25). Collectively, these findings raise the question of whether miR-21* plays a role in the development of HF in Z-disk disruption models.
The study by Bang and colleagues has also provided new insights for LVH. In fact, miR-21* was detected in pericardial fluid of mice with transverse aortic constriction–induced cardiac hypertrophy (21), thus confirming in vivo that miR-21* plays an important role in regulation of the cardiac fibroblast secretome and in determining a hypertrophic response (Figure 1). In addition, Bang and associates were able to demonstrate that systemic injection of 80 mg/kg of a miR-21* antagomir at day 0 and day 2 prevented LVH induction by angiotensin II administration.
Future implications
The most intriguing aspect of the study performed by Bang and coworkers is the identification of a unique mechanism in which cardiac fibroblasts influence the hypertrophic response of cardiomyocytes (21). If confirmed by further studies, these findings provide a potential therapeutic target to interfere with cardiovascular diseases at a physiopathological level. Although fibroblast-derived miR-21* promotes an undesirable pathologic hypertrophy of cardiomyocytes, the identification of the exosome-dependent miR secretion mechanism has been addressed extensively. These vesicles and their miR cargo will likely represent an exciting therapeutic venue in the years to come for cardiovascular biology.
Many questions that pertain to the selective delivery of vesicle content to target cells, exosomes clearance in biologic fluids, and the possibility of a ligand-receptor interaction of surface membranes remain to be answered. The next steps in this field of research will be to further evaluate how miR-containing vesicles promote disease. Moreover, adjunctive efforts are needed to investigate miR-21* inhibition under pressure overload conditions on left ventricular function and survival. Finally, while the clinical use of antagomirs for human therapeutics looks to be part of future strategies for treating a variety of diseases (for example, miravirsen, a miR-122 antagomir, is currently being evaluated in a phase IIb trial for the treatment of virus C–related hepatitis; ref. 26), it remains unclear whether exosomes/microparticles can be exploited for selectively targeting and delivering biological therapies based on the modulation of miR effects.
Acknowledgments
We thank Salvatore De Rosa for precious insights and critical editing of the manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(5):1896–1898. doi:10.1172/JCI75801.
See the related article beginning on page 2136.
References
- 1.Kannel WB, Gordon T, Castelli WP, Margolis JR. Electrocardiographic left ventricular hypertrophy and risk of coronary heart disease. The Framingham study. Ann Intern Med. 1970;72(6):813–822. doi: 10.7326/0003-4819-72-6-813. [DOI] [PubMed] [Google Scholar]
- 2.Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32(5):1454–1459. doi: 10.1016/S0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- 3.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415(6868):206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 4.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Indolfi C, et al. Hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation. 2002;106(16):2118–2124. doi: 10.1161/01.CIR.0000034047.70205.97. [DOI] [PubMed] [Google Scholar]
- 6.De Rosa S, Curcio A, Indolfi C. Emerging role of microRNAs in cardiovascular diseases. Circ J. 2014;78(3):567–575. doi: 10.1253/circj.CJ-14-0086. [DOI] [PubMed] [Google Scholar]
- 7.Torella D, et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109(8):880–893. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 8.Iaconetti C, et al. Inhibition of miR-92a increases endothelial proliferation and migration in vitro as well as reduces neointimal proliferation in vivo after vascular injury. Basic Res Cardiol. 2012. 107 5296. [DOI] [PubMed] [Google Scholar]
- 9.De Rosa S, Fichtlscherer S, Lehmann R, Assmus B, Dimmeler S, Zeiher AM. Transcoronary concentration gradients of circulating microRNAs. Circulation. 2011;124(18):1936–1944. doi: 10.1161/CIRCULATIONAHA.111.037572. [DOI] [PubMed] [Google Scholar]
- 10.Carè A, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13(5):613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 11.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress dependent cardiac growth and gene expression by a microRNA. Science. 2007;316(5824):575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 12.Ucar A, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. doi: 10.1038/ncomms2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curcio A, et al. MicroRNA-1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PLoS One. 2013;8(7):e70158. doi: 10.1371/journal.pone.0070158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elia L, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120(23):2377–2385. doi: 10.1161/CIRCULATIONAHA.109.879429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.da Costa Martins PA, et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118(15):1567–1576. doi: 10.1161/CIRCULATIONAHA.108.769984. [DOI] [PubMed] [Google Scholar]
- 16.Pan W, et al. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One. 2013;8(1):e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abonnenc M, et al. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ Res. 2013;113(10):1138–1147. doi: 10.1161/CIRCRESAHA.113.302400. [DOI] [PubMed] [Google Scholar]
- 18.Huan J, et al. RNA trafficking by acute myelogenous leukemia exosomes. Cancer Res. 2013;73(2):918–929. doi: 10.1158/0008-5472.CAN-12-2184. [DOI] [PubMed] [Google Scholar]
- 19.Hergenreider E, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14(3):249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 20.Laffont B, et al. Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood. 2013;122(2):253–261. doi: 10.1182/blood-2013-03-492801. [DOI] [PubMed] [Google Scholar]
- 21.Bang C, et al. Cardiac fibroblast–derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124(5):2136–2146. doi: 10.1172/JCI70577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang B, Golemis EA, Kruh GD. ArgBP2, a multiple Src homology 3 domain-containing, Arg/Abl-interacting protein, is phosphorylated in v-Abl-transformed cells and localized in stress fibers and cardiocyte Z-disks. J Biol Chem. 1997;272(28):17542–17550. doi: 10.1074/jbc.272.28.17542. [DOI] [PubMed] [Google Scholar]
- 23.Kakimoto Y, et al. Sorbin and SH3 domain-containing protein 2 is released from infarcted heart in the very early phase: proteomic analysis of cardiac tissues from patients. J Am Heart Assoc. 2013;2(6):e000565. doi: 10.1161/JAHA.113.000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng H, et al. Loss of enigma homolog protein results in dilated cardiomyopathy. Circ Res. 2010;107(3):348–356. doi: 10.1161/CIRCRESAHA.110.218735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osio A, et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100(6):766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]