

Abstract
Catalytic enantioselective mono-silylations of diols and polyols furnish valuable alcohol-containing molecules in high enantiomeric purity. These transformations, however, require high catalyst loadings (20-30 mol%) and long reaction times (2-5 days). Here, we report that a counterintuitive strategy – involving the use of an achiral co-catalyst that is structurally similar to the chiral catalyst – provides an effective solution to this problem. A combination of seemingly competitive Lewis-basic molecules can function in concert such that one serves as an achiral nucleophilic promoter while the other performs as a chiral Brønsted base. Upon addition of 7.5-20 mol % of commercially available N-heterocycle (5-ethylthiotetrazole), reactions typically proceed within one hour, delivering the desired products in high yields and enantiomeric ratios. In some instances, there is no reaction in the absence of the achiral base, yet presence of the achiral co-catalyst results in facile formation of products in high enantiomeric purity.
In designing a catalytic process, a chemist might use a promoter molecule that interacts with both reaction partners. Catalytic units are often tethered so that proper geometry can be achieved, easing the bond-forming event (bifunctional catalysis1); the attendant structural rigidity enhances stereoselectivity and discourages the promoter sites from interfering with one another. An alternative stratagem enlists independent and unattached co-catalysts2; repeated adjustment of connector chains is thus obviated, catalyst candidates are less structurally complex and screening studies can be expeditiously performed. Incorporation of more than one catalyst molecule in an enantioselective process when they are structurally and functionally similar but only one is chiral, raises an intriguing question: Can the two promoters be induced to perform strictly distinct tasks so that adventitious diminution of stereoselectivity is not caused by the achiral component?
Enantioselective reactions that proceed through the shared action of two catalysts have been reported3; many are promoted by a chiral Brønsted acid and an achiral Lewis acid (often a metal-based complex4); transformations catalyzed by two different metal complexes or an N-heterocyclic carbene and a metal salt have been outlined as well. In other developments, chiral iminium ions, generated in situ, undergo further reaction with a Brønsted acid- or metal-activated partner. In the above cases, the co-catalysts, while at times possessing generally related attributes, are structurally disparate and realize independent tasks; since there is minimal overlay in their modes of action, their simultaneous presence does not pose a complication. For instance, a chiral proton-donor that associates preferentially with a heteroatomic site operates differently from a Au-based complex with strong propensity towards interacting with Lewis basic π clouds5; it is much less likely that a Brønsted acid can firmly bind to and activate an alkyne unit while the “softer” transition metal complex interacts with a heteroatom in preference to a proton. Use of two Brønsted acids or two Au complexes, fully differentiating their roles as collaborating catalysts, would be a more challenging proposition.
In contemplating the design of a more efficient catalytic enantioselective diol silylation, we were led to consider the use of two heterocyclic Lewis base catalysts that are structurally similar but accomplish separate tasks. One chiral catalyst would have to serve as the Brønsted base by association with the diol substrate, enhancing reactivity of one of the enantiotopic alcohol sites, while an achiral Lewis base would be exclusively the nucleophilic promoter, generating an activated silyl species. Such a scenario is distinct from a recent approach where a chiral Brønsted acid and an achiral nucleophile transform an anhydride to an activated acylating complex that then reacts enantioselectively with meso diamines6 or racemic amines7. In the latter system, again, the co-catalysts are unable to replace one another; it is unlikely that structural alterations would transform the nucleophilic promoter into a competing achiral anion binder (vs. the chiral Brønsted acid), bringing diminution in enantioselectivity.
Results
Catalytic enantioselective alcohol silylation and its shortcomings
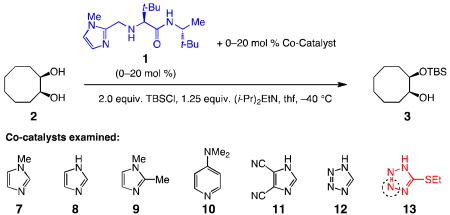
In 2006, we reported the first instances of catalytic enantioselective silylations of diols (Fig. 1)8; such processes are simple to operate and promoted by a readily accessible, robust and recyclable chiral catalyst (1). The utility of such processes9 has since been illustrated in several contexts, including reactions with triols,10 applications to natural product synthesis10,11 and kinetic resolutions12. Alternative strategies have subsequently been outlined; one involves an imidazole-based directing unit13 and in another a heterobicycle is used to effect kinetic resolution of cyclic aryl alcohols14. In spite of the high enantiomeric ratio (e.r.) values, as represented in Fig. 1a, a severe drawback of the transformations promoted by 1 is the low efficiency: up to 30 mol % loadings and reaction times that span several days are needed. Based on the initial model, depicted in Fig. 1b, extensive investigations were carried out concerning alterations of the catalyst structure to render the silylation reactions more efficient; all efforts proved unproductive, largely because solution heterogeneity led to variable and irreproducible data. Computational studies to locate the suggested transition structure (Fig. 1b), were likewise unsuccessful. Earlier investigations vis-à-vis the effect of structural changes on reactivity and, particularly, enantioselectivity8, suggested that 1 likely serves as a Brønsted base; for example, removal of the imidazole unit or its replacement with a less basic pyridine or thiazole moiety results in complete loss of activity. Furthermore, we judged the scenario that an achiral base [(i-Pr2)NEt] is operative and 1 acts as a chiral nucleophilic catalyst that induces enantioselectivity from a position distal to the reacting diol to be less tenable. We became doubtful, however, as to whether 1 can operate as a nucleophilic activator and a Brønsted base.
Figure 1. Previously reported enantioselective alcohol silylations, the initially proposed model, and the results of initial theoretical studies.

a, Generation of chiral mono-silyl derivatives of various cyclic and acyclic diols or triols proceeds with exceptional enantioselectivity but requires high catalyst loadings and long reaction times. b The initially proposed stereochemical model entails catalyst-substrate association through two H-bonds that involve the amide carbonyl and the amine linkage of the catalyst as well as activation of the silyl chloride by the resident N-methylimidazole (NMI) moiety. A wide range of catalyst modifications based on the aforementioned proposal did not lead to any increase in activity, casting doubt on the validity of the mechanistic model. c Theoretical studies suggest a dual role for the Lewis basic heterocycle: silyl activation and enhancement of substrate nucleophilicity. krel = relative rates of reaction by enantiomers.
Mechanistic analysis
We began with a computational study of a model transformation: silylation of MeOH with t-butyldimethylsilyl chloride (TBSCl) and imidazole; this combination is based on the original report15 regarding formation of silyl ethers with the same reagent and catalyst (used in stoichiometric amounts) in dimethylformamide (dmf) as the solvent. To understand better the role of the heterocyclic catalyst, we probed without dmf, which can serve as a weak nucleophilic catalyst, and since enantioselective silylation is performed in its absence. Theoretical investigations supported the critical notion that there are two imidazole molecules involved (Fig. 1c): one generates a highly electrophilic silyl complex and the other plays the role of Brønsted base, enhancing alcohol nucleophilicity (see the Supplementary Information for details) 16.
An implication of the model in Fig. 1c is the anti orientation of the promoter molecules; because of severe geometric constraints, therefore, 1 cannot serve as an effective bifunctional catalyst. Accordingly, we surmised that high enantioselectivities might originate from efficient enantiotopic group differentiation arising from 1 playing the role of a chiral Brønsted base (cf. Fig. 1b). We judged, however, that diol silylation through involvement of two molecules of N-methylimidazole-substituted 1 might be relatively unfavorable because nucleophilicity is sensitive to steric factors (vs. proton removal) and 1 might be too hindered to be a capable nucleophile. High enantioselectivities suggest that it is more likely that, although 1 is an effective chiral base for diols [vs. (i-Pr2)NEt], it is an inferior nucleophilic activator, leading to low silylation rates (Fig. 1a). If a second Lewis base were to perform as a co-catalyst that provides strong nucleophilic activation alongside 1 – but without interfering as a Brønsted base – then a more facile and equally enantioselective transformation could become feasible.
Examination of achiral co-catalysts
To establish feasibility, the experiments summarized in Table 1 were carried out. We set a limit of six hours; 20 mol % 1 was used at −40 °C with 1.25 equivalents of (i-Pr2)EtN to neutralize the generated HCl. There is 9% conversion to 3 with only 1 present (entry 1, Table 1), whereas the reaction is significantly more efficient (69% conv.) in forming rac-3 with N-methylimidazole (NMI; 7).
Table 1. The Effect ofHeterocyclic Co-Catalysts on Silylation of Diol 2.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Mol % 1 | Co-Catalyst; Mol % | Time (h) | Conv. (%)§ | e.r.† |
| 1 | 20 | none; NA | 6.0 | 9 | 93.5:6.5 |
| 2 | 0 | 7;20 | 6.0 | 69 | NA |
| 3 | 20 | 7;20 | 6.0 | 92 | 78:22 |
| 4 | 20 | 7;15 | 6.0 | 88 | 81.5:18.5 |
| 5 | 20 | 7;10 | 6.0 | 79 | 83:17 |
| 6 | 20 | 7;7.5 | 6.0 | 71 | 85.5:14.5 |
| 7 | 20 | 7;5.0 | 6.0 | 49 | 86.5:13.5 |
| 8 | 20 | 8;7.5 | 3.0 | 13 | 93:7 |
| 9 | 20 | 9;7.5 | 3.0 | 5 | 90.5:9.5 |
| 10 | 20 | 10; 7.5 | 3.0 | 8 | 86:14 |
| 11 | 20 | 11; 7.5 | 3.0 | 47 | 96:4 |
| 12 | 20 | 12; 7.5 | 3.0 | 45 | 98:2 |
| 13 | 20 | 13; 7.5 | 3.0 | >98 | 97:3 |
Reactions were carried out in a sealed vessel in air; see the Supplementary Information for details. NA = not applicable.
Conversion (conv.) was determined by GC analysis; the variance of values is estimated to be <±2%.
Enantiomeric ratio (e.r.) values were determined by GC analysis; the variance of values is estimated to be <±2%. See the Supplementary Information for details.
We then chose to probe the reaction under an unusual set of conditions. We attempted silylation with 20 mol % 1 and NMI, an achiral and more active catalyst (cf. entries 1-2, Table 1); this is a combination that chemists in search of attaining maximum enantioselectivity would not likely consider. Avoiding “background reactions” by achiral catalyst competitors is a principle strictly adhered to in enantioselective catalysis. Nonetheless, with 20 mol % of the more strongly activating (cf. entries 1 vs. 2) achiral Lewis base 7 present alongside 20 mol % of less potent chiral Lewis basic 1, silyl ether 3 is remarkably obtained more rapidly and in 78:22 e.r (entry 3). Analysis of the findings in entries 1–3 of Table 1 reveal two crucial points: Firstly, the higher efficiency in entry 3 as opposed to lower conversions in entries 2–3 indicates that the combination of 1, almost certainly serving as an effective chiral base, and NMI, likely providing nucleophilic activation, leads to a more efficient process than when one is absent. Secondly, the chiral NMI-substituted 1 emerges as a superior Brønsted base compared to NMI and the more abundant (i-Pr)2EtN (20 mol % vs. 1.25 equiv.); otherwise, higher conversion and appreciable enantioselectivity would not be observed in entry 3 versus entry 2. We presumed that the lowering of e.r. is partly due to NMI (7) competitively serving as a Brønsted base (cf. Fig. 1c). Accordingly, silylations with reduced amounts of 7 were probed. As the concentration of 7 is decreased (entries 4–7, Table 1), silyl ether 3 is formed in higher enantioselectivity; with 20 mol % 1 and 5.0 mol % NMI (entry 7), there is nearly 50% conversion after six hours and the desired product is generated in 86.5:13.5 e.r. The latter findings show that (i-Pr)2EtN, present in excess amounts, performs as a weaker Brønsted base than NMI.
To establish whether the efficiency and enantioselectivity might be further improved, we investigated the effectiveness of several other N-heterocycles; we chose to carry out reactions with 7.5 mol % of a co-catalyst and measure progress within a span of three hours (vs. six hours with 7); representative findings are shown in entries 8–13 of Table 1. Neither imidazole (8, entry 8), nor 1,2-dimethylimidazole (9; entry 9) offer an improvement; the former probably generates an N-silylimidazole that is an inferior nucleophilic catalyst due to steric and electronic deactivation (the C–Si σ* orbital serving as electron acceptor17,18), whereas the latter is less effective because of its larger size (vs. NMI). The low activity exhibited with 2-methyl-substituted NMI 9 has noteworthy implications regarding the inefficiency of silylations with 1 as the catalyst (cf. Fig. 1a): if a methyl substituent hampers the ability of an NMI moiety as a nucleophilic activator, the sterically more demanding amino acid-based moiety of the chiral variant does so as well and likely to a larger extent. Inclusion of relatively basic 10 (entry 10, Table 1) does not lead to rate acceleration, likely as a result of the formation of the derived HCl salt [i.e., 10 competes effectively with (/-Pr)2EtN]. In contrast, with 4,5-dicyanoimidazole (11; entry 11) present, there is 45% conversion in three hours and 3 is generated with the same enantioselectivity as obtained without a co-catalyst (96:4 vs. 97.5:2.5 e.r.; cf. Fig. 1); reaction with 7.5 mol % tetrazole (12; entry 12) leads to similar rate acceleration and selectivity. The silylation proceeds to completion in three hours and 3 is obtained in 97:3 e.r. when 5-ethylthiotetrazole (13; entry 13, Table 1) is introduced. Similar roles (Brønsted base and nucleophilic activation) have been assigned to the imidazole moieties of histidine residues within bifunctional polypeptide catalysts used for enantioselective conversion of various alcohol families to carboxylic19,20, phosphoryl ester21 or sulfinate derivatives22. In such instances, Lewis base promoters perform their expected tasks as dictated by the geometrical constraints imposed by the structure of the bifunctional catalyst. Furthermore, examination of different promoter units in the aforementioned context requires the preparation of enantiomerically pure a-amino acid residues and subsequent synthesis of each polypeptide catalyst candidate.
Enantioselective alcohol silylation with improved catalytic efficiency
The positive influence of 13 as a co-catalyst for enantioselective diol silylation is general (Table 2). Transformations depicted in entries 1–14 of Table 2 proceed with lower catalyst loadings (20 vs. 30 mol %; see below for further data), at the same or slightly lower temperature (entries 13 vs. 14) and in substantially shorter periods of time (1.0 h vs. 2–5 days) through the simple expediency of incorporating 7.5 mol % of 5-ethylthiotetrazole (13); mono-silylated diols are obtained in nearly identical yields and e.r. values as when only the chiral catalyst 1 is used. In one instance involving enantioselective synthesis of acyclic silyl ether 18 (entries 15–16, Table 2), there is minimal reaction in the absence of 13 (<10% yield after 24 h at −30 °C); when the co- catalyst is present, the desired product is obtained within 12 hours in 93% yield and 95:5 e.r.
Table 2. Catalytic Enantioselective Silylation of Various Diols With and Without 5-Ethylthiotetrazole (13) as the Co-Catalyst.
| Entry | Product | Mol % 1 | Mol % 13 | Time (h) | Temp. (°C) | Yield (%)§ | e.r.† |
|---|---|---|---|---|---|---|---|
| 1 |
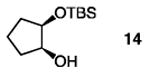
|
30 | none | 60 | −40 | 96 | 94:6 |
| 2 | 20 | 7.5 | 1.0 | −40 | 97 | 95.5:4.5 | |
| 3 |
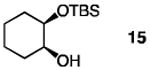
|
30 | none | 120 | −40 | 82 | 96:4 |
| 4 | 20 | 7.5 | 1.0 | −40 | 93 | 97:3 | |
| 5 |
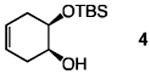
|
30 | none | 72 | −40 | 75 | 97:3 |
| 6 | 20 | 7.5 | 1.0 | −40 | 82 | 97.5:2.5 | |
| 7 |
|
30 | none | 120 | −40 | 93 | 96.5:3.5 |
| 8 | 20 | 7.5 | 1.0 | −40 | 93 | 96.5:3.5 | |
| 9 |
|
30 | none | 120 | −40 | 96 | 97.5:2.5 |
| 10 | 20 | 7.5 | 1.0 | −40 | 96 | 97.5:2.5 | |
| 11 |
|
30 | none | 48 | −78 | 82 | 98:2 |
| 12 | 20 | 7.5 | 1.0 | −78 | 78 | 94:6 | |
| 13 |
|
30 | none | 96 | −30 | 78 | 96.5:3.5 |
| 14 | 20 | 7.5 | 1.0 | −40 | 74 | 96:4 | |
| 15 |
|
30 | none | 24 | −30 | <10 | ND |
| 16 | 20 | 7.5 | 12 | −40 | 93 | 95:5 |
Reactions were carried out in a sealed vessel in air; see the Supplementary Information for details. ND = not determined.
Yield of isolated product after purification; the variance of values is estimated to be <±2%. All reactions proceeded to >98% conversion (except for entry 15).
Enantiomeric ratio (e.r.) values were determined by GC analysis; the variance of values is estimated to be <±2%. See the Supplementary Information for details.
The influence of 5-ethylthiotetrazole on catalytic kinetic resolutions23 is similarly notable (Table 3). Whereas transformations affording the silyl ethers in entries 1, 3 and 5 of Table 3 require 24–72 hours to proceed to 55–70% conversion, when 7.5 mol % 13 is introduced 47– 54% conversion is attained in one hour (entries 2,4 and 6), in spite of a lower loading of 1 and fewer equivalents of TBSCl in many cases. Faster rates are achieved on two occasions in spite of lower reaction temperature (entries 3 vs. 4 and 5 vs. 6). Moreover, the catalytic kinetic resolution that delivers silyl ether 6 is more enantioselective when both 1 and 13 are utilized (entries 5–6); such improvement is probably because in the presence of the co-catalyst, the reaction is sufficiently facile to be performed at −40 °C (vs. −15 °C). The presence of 20 mol % 13 facilitates silylation leading to mono-protected tertiary alcohol 21, a reaction that hardly occurs in its absence (entry 7 vs. 8), to proceed to 39% conversion in 12 hours with krel = >25.
Table 3. The Effect of Ethylthiotetrazole on Catalytic Kinetic Resolution through Enantioselective Silylation.
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Entry | Silylation Product | Mol % 1 Mol%13 | Equiv. TBSCI | Time(h); Temp. (°C) | Conv. (%)§ | Recovered Substrate Yield (%)§§; e.r.† | Product Yield (%)§§; e.r.† | Krel |
| 1 |
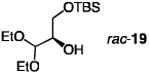
|
20;none | 1.0 | 24; −78 | 55 | 25; 92:8 | 55; 84:16 | 14 |
| 2 | 20; 7.5 | 1.0 | 1.0;−78 | 54 | 41; 91:9 | 49:85:15 | 14 | |
| 3 |
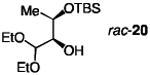
|
30;none | 1.0 | 24;−30 | 55 | 44; >98:2 | 52; 90:10 | >25 |
| 4 | 20; 7.5 | 0.6 | 1.0;−40 | 47 | 52; 89.5:10.5 | 35; 95:5 | >25 | |
| 5 |
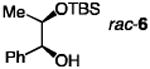
|
30; none | 1.0 | 72;−15 | 70 | 30; 98:2 | 68; 69.5:30.5 | 8 |
| 6 | 20; 7.5 | 0.6 | 1.0;−40 | 52 | 47; 96:4 | 52; 93:7 | >25 | |
| 7 |
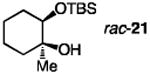
|
30;none | 1.0 | 24;−30 | <5 | >95%; ND | <5%; ND | NA |
| 8 | 20; 20 | 0.6 | 12; −40 | 39 | 62; 78.5:21.5 | 36; 95:5 | >25 | |
Reactions were carried out in thf in a sealed vessel in air; ND = not determined; NA = not applicable. krel = relative rates of reaction by enantiomers.
Conversion to the desired product as measured by GC analysis of unpurified mixtures; krel was measured based on a previously reported method23; the variance of values is estimated to be <±2%.
Yield of isolated product after purification; the variance of values is estimated to be <±2%.
Enantiomeric ratio (e.r.) values were determined by GC analysis; the variance of values is estimated to be <±2%. See the Supplementary Information for details.
An advantage of the catalytic system is that reactions reach completion within a few hours with lower catalyst loadings; two examples are presented in Fig. 2. With 5.0 mol % 1 and 5.0 mol % 13, one gram of diol 2 can be converted to silyl ether 3 in 96% yield and 96.5:3.5 e.r.; >98% conversion is observed in eight hours, constituting an improvement in turnover frequency of nearly two orders of magnitude compared to the transformation in Fig. 1a. The longer time required as a result of diminished catalyst loading supports the critical role of chiral 1 in what is likely the turnover-limiting step. In kinetic resolution of rac-22, 52% conversion is observed in two hours; silyl ether 20 and the recovered diol were isolated in 42% and 38% yield and 93:7 and 96:4 e.r., respectively.
Figure 2.

In the presence of commercially available 5-ethylthiotetrazole (13) as the co-catalyst, enantioselective silylation of alcohols, or the related kinetic resolution processes, can be performed with 5.0 mol % of chiral catalyst 1; the desired conversion levels (>98% and ∼50%, respectively) are achieved within eight hours. As exemplified through enantioselective preparation of silyl ether 3, the catalytic reactions are amenable to gram scale operations. See the Supplementary Information for experimental details; Krel = relative rates of reaction by enantiomers.
Mechanism and stereochemical models
The modes of reaction illustrated in Fig. 3, based on and supported by theoretical studies (see the Supplementary Information), put forth a plausible rationale for the enantioselectivity trends. Reaction via complex I leads to the major enantiomer; involvement of II entails unfavorable steric repulsion and less effective activation of the diol substrate by the chiral base (see below).
Figure 3. Mechanistic models to account for the observed sense and levels of enantioselectivity supported by DFT calculations (with deprotonated 13 serving as the nucleophilic co-catalyst).

Proper juxtaposition of the two basic units of the chiral co-catalyst, namely the imidazole and the secondary amine side chain, with the hydroxyl groups of thediol substrate, lead to efficient binding and activation of one of the enantiotopic hydroxyl groups for silylation. Computational studies reveal that the proposed transition structures are further stabilized byan ion-dipole interaction involving the carbonyl amide and the transferring proton. In one complex (II), there is a significant destabilizing steric interaction between the bound substrate and the co-catalyst backbone, resulting in a high degree of enantioselectivity. See the Supplementary Information for details of the computational studies.
Deprotonated 13, generated through reaction with (i-Pr)2EtN, likely serves as the nucleophilic co-catalyst. The anionic species is turned exceptionally reactive towards a silyl chloride due to its high energy HOMO, caused by its several neighboring heteroatoms24, and the availability of a sterically accessible site for chloride displacement (cf. dotted circle in Table 1). Deprotonated 13 is the more effective nucleophilic promoter in spite of exhibiting the lowest degree of thermodynamic Brønsted basicity among those shown in Table 1 [pKa values for 11– 13 (in 50% EtOH) = 5.2, 4.9 and 4.2, respectively25,26]; the reason for such a trend rests in the distinction between basicity and nucleophilicity functions of a Lewis base. Supplanting a leaving group and forming an active electrophilic intermediate, unlike proton transfer, is not readily reversible; a nucleophilic catalyst is unable to serve as an effective Brønsted base, since although it might be rapidly converted to its conjugate acid, the resulting species would be short-lived (swift proton loss). Accordingly, there is minimal reaction when only 13 and 1.25 equivalents of (i-Pr)2EtN are present (<5% conv. after 6.0 h at −40 °C). The above considerations point to 1 as the enabling and enantiodifferentiating base.
Why is 1 such a capable Brønsted base for the diol substrates, and why does it outperform (i-Pr)2EtN, which is available in much larger amounts? The NMI-containing 1 is an effective Brønsted base catalyst because it can readily associate with diols by H-bonding. Proximity-induced reactivity enhancement is an acknowledged principle in catalysis27,28,29,30. Appropriate spatial fit involving the basic heteroatomic sites within the catalyst (N of imidazole and amine side chain) and substrate hydroxyl groups give rise to activation of one of the enantiotopic alcohol groups; NMI or derivatives of 1 that lack the heterocyclic moiety are thus ineffective in generating sufficient Brønsted basicity. Whereas achiral NMI readily catalyzes silylations of an alcohol (vs. a diol), 1 does not; in such cases, association of 1 with an alcohol, necessary for Brønsted base activation, is weaker (only one point of contact). Unlike imidazole-containing entities (e.g., 1 or NMI), (i-Pr)2EtN cannot delocalize the positive charge generated through protonation in a non-polar medium and is therefore an inferior base.
In the models in Fig. 3 one substrate hydroxyl group, rendered more basic by the NMI moiety of 1, can deprotonate and enhance the basicity of the chiral co-catalyst's secondary amine, empowering it, together with the Lewis basic amide carbonyl, to activate the adjacent carbinol for silylation. DFT calculations suggest that, alternatively, the secondary amine of the catalyst, with assistance from the Lewis basic amide terminus, might be the initial base, deprotonating the alcohol unit to be silylated; the resulting ammonium group is then either neutralized by an external molecule of general base (i-Pr)2NEt or internally by the neighboring imidazole unit. The considerably lower activity furnished by the carboxylic ester or thioamide derivatives of 18 is partly because the less Lewis basic carbonyl oxygens are ineffective relative to an amide, which can establish stabilizing electrostatic interaction with the nearby alcohol proton (ion-dipole stabilization; Fig. 3); such an association generates 2.4 kcal/mol of transition state stabilization compared to an ester group (see the Supplementary Information for details). Thus, overall, the significant improvement in efficiency of enantioselective catalytic silylation is the culmination of substituted imidazole 1, the deprotonated form of 5-ethylthiotetrazole 13 and (i-Pr)2NEt serving distinctly separate functions. The latter component, present in stoichiometric amounts, is a weak nucleophile that does not strongly bind to a diol or triol substrate, but can help re-form the chiral catalyst by quenching the HCl byproduct.
Conclusions
These investigations illustrate that it is feasible to design a catalytic enantioselective process by implementing the cooperative action of several Lewis basic entities, capable of possessing coinciding functions. By tuning various features of structurally related Lewis bases, their ability to serve as a Brønsted base or a nucleophilic promoter can be partitioned; as such, improvement in the efficiency of diol silylation can be attained without concomitant loss in enantioselectivity arising from the achiral co-catalyst serving as the Brønsted base. Efficient and selective substrate binding elevates basicity, giving rise to high enantioselectivity; features that promote substrate association render the Brønsted base catalyst more sizeable, curtailing its ability to be a nucleophilic activator. In contrast, an effective nucleophilic catalyst can be an inferior (thermodynamic) Brønsted base, since, unlike protonation, displacement of a leaving group is largely irreversible and kinetically controlled. The principles outlined herein serve as a conceptual framework for the development of new processes that demand separate and independently operational Lewis basic co-catalysts whose functions can easily overlap and that their simultaneous use might initially appear to be detrimental towards achieving high enantioselectivity. Such strategies will be valuable when two Lewis basic entities must operate in different capacities and distally via hypervalent electrophilic intermediates (e.g., with P- and Sn-based electrophiles).
Methods
Procedure for enantioselective alcohol silylation with 5-ethylthiotetrazole as co-catalyst
A mixture of cyclooctane diol 2 (1.00 g, 6.93 mmol), chiral catalyst 1 (107 mg, 0.347 mmol) and co-catalyst 5-ethylthiotetrazole 13 (45.2 mg, 0.347 mmol) was placed in a 25 ml round-bottom flask, to which diisopropylethylamine (DIPEA; 1.5 ml, 8.67 mmol) was added. The contents were dissolved in 5.5 ml of tetrahydrofuran, the flask was capped with a rubber septum, and the solution was allowed to cool to −40 °C (cryocool apparatus). In a separate vessel, tert-butyldimethylsilyl chloride (TBSCl; 2.09 g, 13.9 mmol) was dissolved in 4.8 ml THF (total volume ∼10.3 ml) and the solution was allowed to cool to −40 °C, after which it was added to the first mixture, which was allowed to stir for 8 h (at −40 °C). The reaction was quenched by addition of DIPEA (1.21 ml, 6.93 mmol) followed by methanol (578 μl). The resulting mixture was allowed to warm to 22 °C, diluted with CH2Cl2 (15 ml) and washed with 20 ml of a 10 wt.% solution of aqueous citric acid; the aqueous layer was washed with CH2Cl2 (2 × 15 ml) and the combined organic layers were dried over anhydrous MgSO4. The solids were removed by filtration and the solution was concentrated in vacuo to afford a yellow oil, which was purified by silica gel chromatography to afford 1.72 g of silyl ether 3 (6.66 mmol, 96% yield); GC analysis (Supelco Beta or Gamma Dex 120 column) indicated a 96.5:3.5 enantiomeric ratio (e.r.). The chiral co-catalyst (1) was recovered (>98%) in the following manner: The aqueous layer was treated with an aqueous solution of 3.0 N NaOH until pH = 12, and was then washed with 3 x 15 ml portions of CH2Cl2. The combined organic layers were dried over MgSO4, solids were removed by filtration and the solution was concentrated in vacuo to afford 1 as white solid.
Supplementary Material
Acknowledgments
This research was supported by the United States National Institutes of Health, Institute of General Medical Sciences (Grant GM-57212). We thank D. Silverio, V. Rendina and B. Potter for many helpful discussions and experimental assistance and Boston College for providing access to computational facilities.
Footnotes
Author contributions N. M. and H. A. were involved in the development of the catalytic protocol. F. H. designed and performed the theoretical studies. M. L. S. and A. H. H. directed the investigations. A. H. H. wrote the manuscript with revisions provided by the other authors.
Author information Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
The authors declare no competing financial interests.
References
- 1.Shibasaki M, Kanai M. In: New Frontiers in Asymmetric Catalysis. Mikami K, Lautens M, editors. Wiley; 2007. pp. 383–405. [Google Scholar]
- 2.Allen AE, MacMillan DWC. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem Sci. 2012;3:633–658. doi: 10.1039/C2SC00907B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patil NT, Shinde VS, Gajula B. A one-pot synthesis: the strategic classification with some recent examples. Org Biomol Chem. 2012;10:211–224. doi: 10.1039/c1ob06432k. [DOI] [PubMed] [Google Scholar]
- 4.Shao Z, Zhang H. Combining transition metal catalysis and organocatalysis: a broad new concept for catalysis. Chem Soc Rev. 2009;38:2745–2755. doi: 10.1039/b901258n. [DOI] [PubMed] [Google Scholar]
- 5.Patil NT, Mutyala AK, Konala A, Tella RB. Tuning the reactivity of Au-complexes in an Au(I)/chiral Brønsted acid cooperative catalytic system: an approach to optically active fused 1,2-dihydroisoquinolines. Chem Commun. 2012;48:3094–3096. doi: 10.1039/c2cc17450b. [DOI] [PubMed] [Google Scholar]
- 6.De CK, Seidel D. Catalytic enantioselective desymmetrization of meso-diamines: A dual small-molecule catalysis approach. J Am Chem Soc. 2011;133:14538–14541. doi: 10.1021/ja2060462. [DOI] [PubMed] [Google Scholar]
- 7.Mittal N, Sun DX, Seidel D. Kinetic resolution of amines via dual catalysis: Remarkable dependence of selectivity on the achiral co-catalyst. Org Lett. 2012;14:3084–3087. doi: 10.1021/ol301155b. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Enantioselective silyl protection of alcohols catalysed by an amino-acid-based small molecule. Nature. 2006;443:67–70. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- 9.Díaz-de-Villegas MD, Gálvez JA, Badorrey R, López-Ram-de-Víu MP. Organocatalyzed enantioselective desymmetrization of diols in the preparation of chiral building blocks. Chem Eur J. 2012;18:13920–13935. doi: 10.1002/chem.201202264. [DOI] [PubMed] [Google Scholar]
- 10.Yu Z, Hoveyda AH, Snapper ML. Catalytic enantioselective silylation of acyclic and cyclic triols: Application to total syntheses of cleroindicins D, F and C. Angew Chem Int Edn. 2009;48:547–550. doi: 10.1002/anie.200805338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodrigo J, Zhao Y, Hoveyda AH, Snapper ML. Regiodivergent reactions through catalytic enantioselective silylation of chiral diols. Synthesis of sapinfuranone A. Org Lett. 2011;13:3778–3781. doi: 10.1021/ol2010819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Mitra AW, Hoveyda AH, Snapper ML. Kinetic resolution of 1,2-diols through highly site- and enantioselective catalytic silylation. Angew Chem Int Edn. 2007;46:8471–8474. doi: 10.1002/anie.200703650. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Worthy AD, Tan KL. Scaffolding catalysts: highly enantioselective desymmetrization reactions. Angew Chem Int Edn. 2011;50:8167–8171. doi: 10.1002/anie.201103470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheppard CI, Taylor JL, Wiskur SL. Silylation-based kinetic resolution of monofunctional secondary alcohols. Org Lett. 2011;13:3794–3797. doi: 10.1021/ol2012617. [DOI] [PubMed] [Google Scholar]
- 15.Corey EJ, Venkateswarlu A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J Am Chem Soc. 1972;94:6190–6191. [Google Scholar]
- 16.Dietze PE, Xu Y. Mechanism for the general base catalyzed solvolysis of silyl ethers. J Org Chem. 1994;59:5010–5016. [Google Scholar]
- 17.Denmark SE, Beutner GL. Lewis base catalysis in organic synthesis. Angew Chem Int Edn. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 18.Tandura SN, Vronkov NG, Alekseev NV. Molecular and electronic structure of penta- and hexacoordinate silicon compounds. Top Curr Chem. 1986;131:99–189. [Google Scholar]
- 19.Copeland GT, Miller SJ. Selection of enantioselective acyl transfer catalysts from a pooled peptide library through a fluorescence-based activity assay: An approach to kinetic resolution of secondary alcohols of broad substrate scope. J Am Chem Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- 20.Fierman MB, O'Leary DJ, Steinmetz WE, Miller SJ. Structure-selectivity relationships and structure for a peptide-based enantioselective acylation catalyst. J Am Chem Soc. 2004;126:6967–6971. doi: 10.1021/ja049661c. [DOI] [PubMed] [Google Scholar]
- 21.Sculimbrene BR, Morgan AJ, Miller SJ. Enantiodivergence in small-molecule catalysis of asymmetric phosphorylation: Concise total syntheses of the enantiomeric D-myo-inositol-1-phosphate and D-myo-inositol-3-phosphate. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- 22.Evans JW, Fierman MB, Miller SJ, Ellman JA. Catalytic enantioselective synthesis of sulfinate esters through the dynamic resolution of tert-butanesulfinyl chloride. J Am Chem Soc. 2004;126:8134–8135. doi: 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- 23.Vedejs E, Jure M. Efficiency in nonenzymatic kinetic resolution. Angew Chem Int Edn. 2005;44:3974–4001. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 24.Trifonov RE, Ostrovskii VA. Protolytic equilibria in tetrazoles. Russ J Org Chem. 2006;42:1585–1605. [Google Scholar]
- 25.Lieber E, Enkoji T. Synthesis and properties of 5-(substituted) mercaptotetrazoles. J Org Chem. 1961;26:4472–4479. [Google Scholar]
- 26.Welz R, Müller S. 5-(Benzylmercapto)-1H-tetrazole as activator for 2′-O-TBDMS phosphoramidite building blocks in RNA synthesis. Tetrahedron Lett. 2002;43:795–797. [Google Scholar]
- 27.Page MI, Jencks WP. Entropic contributions to rate accelerations in enzymatic and intramolecular reactions and the chelate effect. Proc Nat Acad Sci. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menger FM. Enzyme reactivity from an organic perspective. Acc Chem Res. 1993;26:206–212. [Google Scholar]
- 29.Bruice TC, Lightstone FC. Ground state and transition state contributions to the rates of intramolecular and enzymatic reactions. Acc Chem Res. 1999;32:127–136. [Google Scholar]
- 30.Kennan AJ, Whitlock HW. Host-catalyzed isoxazole ring opening: A rationally designed artificial enzyme. J Am Chem Soc. 1996;118:3027–3028. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.