

Abstract
Neovascularization is one of the hallmarks associated with tumor growth. In the recent years, a number of angiogenesis inhibitors have been approved for clinical use in cancer patients. However, the efficacy of antiangiogenic therapy is in most cases short-lasting, with likely drug resistance developing within a few months. It is becoming clear also that there are a subset of malignant tumors that are inherently resistant to angiogenesis inhibition. The knowledge regarding resistance mechanisms towards angiogenesis inhibitors is still evolving and here we propose some theories and in some cases provide experimental evidence.
Keywords: cancer, angiogenesis, antiangiogenic therapy, drug resistance mechanisms
Introduction
Since the seminal articles about the importance of tumor angiogenesis and the possibility of antiangiogenesis therapies in the early 1970's [1,2], the field of angiogenesis has expanded to become one of the major areas of cancer research today. Antiangiogenic drugs are currently approved by FDA for clinical use, in breast cancer, colorectal cancer, lung cancer and renal cell carcinoma, but only in patients with metastatic disease and without a curative potential [3-8]. Except for patients with renal cell carcinoma, the angiogenesis inhibitors have to be combined with chemotherapy in order to obtain any significant tumor response.
In the late 1990's, the curative potential of antiangiogenic therapy was warmly debated in the scientific community [9,10]. Theoretically, it was proposed that targeting the tumor endothelium would circumvent the problem of acquired drug resistance [11]. Experimentally, the initial studies indicated that drug resistance would not emerge with antiangiogenic therapy [12,13], but there are now a large number of studies which demonstrate that treatment failure is a common finding in animal models when angiogenesis inhibitors are given as a monotherapy [14-16]. Also the results of all clinical trials conducted with angiogenesis inhibitors to date show that drug resistance develops within a few months of the commencement of antiangiogenic therapy. Here we will discuss various experimental concepts that may explain resistance to angiogenesis inhibitors and in some cases provide scientific data that demonstrates such resistance mechanisms.
Mutations in Endothelial Cells
The neovasculature of a growing malignancy develops due to endothelial cells that are recruited from the surrounding normal tissues. This is demonstrated by genetic tracing of host endothelial cells by implanting a LacZ negative tumor in mice that are Tie2-LacZ positive [17] or by growing dsRed-labelled tumors in mice expressing enhanced green fluorescent protein (eGFP) in all normal tissues [18] (Fig. 1). While the unstable genome of the cancer cells commonly cause them to develop acquired drug resistance, the tumor endothelium presumably have a normal genome and lack the ability to circumvent drug inhibition [11,19,20]. This is supported by a number of experimental studies wherein angiogenesis inhibitors have been administered over long time periods without developing drug resistance [13,21,22].
Figure 1.
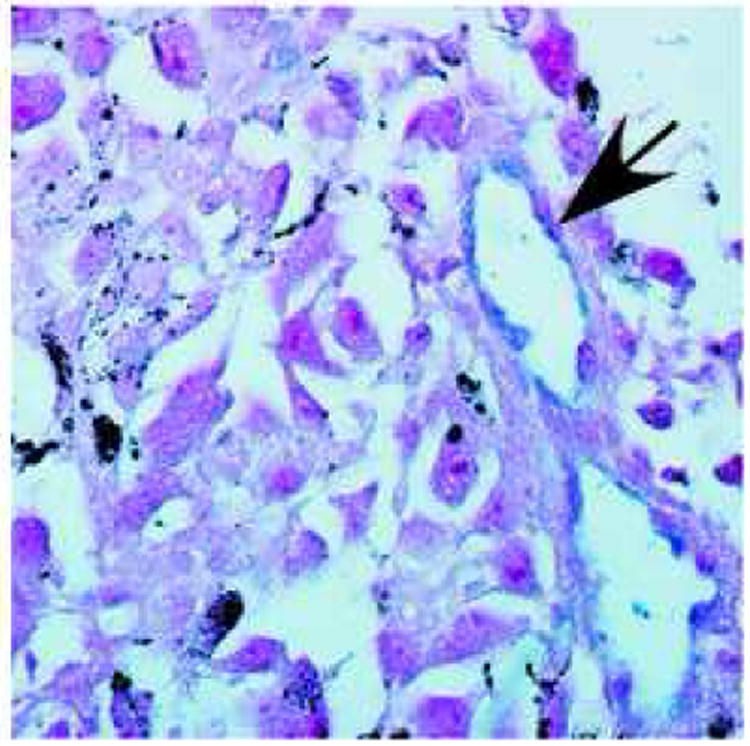
LacZ staining of a LacZ- B16F10 melanoma grown in a Tie2-LacZ+ mouse. All endothelial cells in the tumor vasculature are LacZ+ (blue), indicating that the neovasculature is recruited from the surrounding normal tissue. Arrow pointing to a blood vessel. Red: neutral red counterstain. LacZ staining was performed as described previously [17]. 600× magnification.
There are a few publications suggesting that the tumor endothelium may also harbor genetic aberrations in non-endothelial cancers [23-26]. In the article by Hida et al., endothelial cells isolated from human tumors grown in nude mice frequently have an abnormal karyotype with excessive number of centrosomes [23]. The probability of human tumor cell contamination in the analyzed mouse endothelial cultures is low as the human-specific difteria toxin was used to clear the cultures of human cells, and also, mouse-specific chromosomal probes were used for in situ hybridization analysis. In the same article, heterogenous chromosomal rearrangements in the endothelial cells indicate that the genetic alterations are non-clonal and probably a result of the permissive microenvironment within malignant tumors, or an in vitro phenomenon. Furthermore, the extent of aneuploidy increased upon passaging of tumor endothelial cells in vitro, compared to normal tissue endothelial cells, indicating an inherently unstable genome within the tumor endothelium [24]. In another article by the same group they show that in a spontaneous prostate cancer developing in mice, the tumor endothelium does not carry the SV40 T antigen that causes cancer development in the prostate epithelial cells [27].
In the article by Gunsilius et al., patients with chronic myelogenous leukemia reveal the BCR/ABL fusion gene within normal endothelial cells in the myocardium and in bone marrow-derived endothelial cells cultured in vitro [26]. The reason for mutations in the endothelium is likely due to a common bone marrow progenitor that harbors the fusion gene, and forwards it both to the leukemia cells and to circulating endothelial cells that are incorporated in normal tissue blood vessels [26].
In an analysis of tumor specimens from patients with B-cell lymphomas, lymphoma-specific chromosomal translocations were found in tumor endothelial cells [25]. The identification of endothelial cells was performed by immunostaining for CD31, von Willebrand factor or Ulex europaeus lectin. However, only 37% of the tumor endothelial cells were found to harbor chromosomal alterations. Given the inherent potential for non-specific immunoreactivity in antibody labelling experiments, there is a risk that some of the identified “endothelial” cells could be lymphoma cells instead.
However, if the findings in the above articles are indeed correct, the impact of antiangiogenic therapy could be far more limited then anticipated because drug resistance could develop due to evolving genomic instability of the tumor endothelium. Also, if tumor endothelial cells do indeed possess an unstable genome, it could explain why tumor endothelial cells in culture are more resistant to vincristine than normal endothelial cells [28]. All in all, the data supporting genetic alterations in the tumor endothelium is limited, and more research is needed in this area.
Angiogenic Signaling Redundancy - When One Factor is Blocked, Another is Upregulated
In malignant cancer cells multiple signaling pathways are commonly dysregulated at the same time [29-31]. Moreover, distinct malignant cell clones may have different signaling pathways dysregulated, as demonstrated in acute myeloid leukemia [32]. This implies that redundancy is to be expected when targeting a single signaling pathway, as other pathways are likely to go unchecked and could compensate for the one being targeted (Fig. 2).
Figure 2.
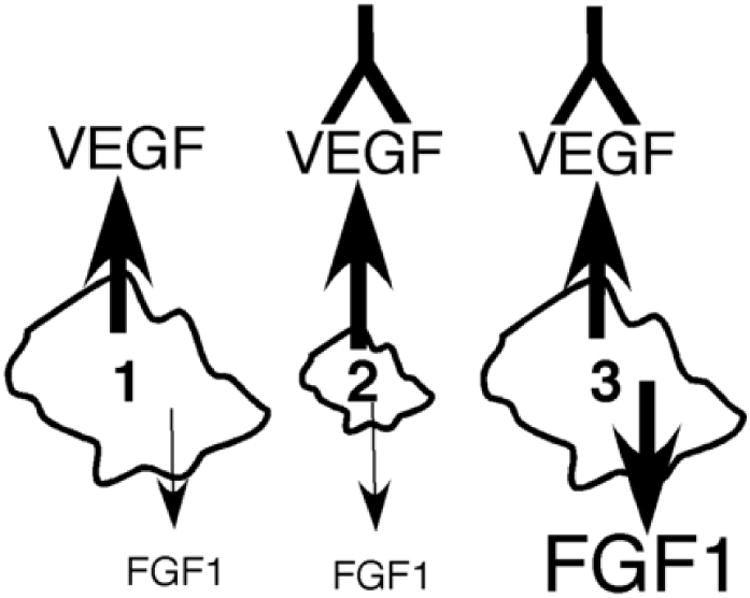
Signal redundancy in a tumor upon treatment with anti-VEGF antibody. 1. Before any treatment initiated, 2. Anti-VEGF treatment regresses tumor, 3. FGF1 production is upregulated in the tumor and the rescued angiogenic response causes the tumor to regrow.
If tumor growth is inhibited by overexpressing thrombospondin-1 (TSP-1), endostatin or tumstatin, these tumors finally escape and progress due to upregulation of angiogenic growth factors, in particular vascular endothelial growth factor (VEGF) [14]. Anti-VEGF receptor-2 (VEGFR-2) treatment in Rip1-Tag2 pancreatic tumors provides a transitory tumor response, followed by tumor regrowth with upregulation of fibroblast growth factors 1 and 2 (FGF1 and FGF2), ephrin A1 and A2, as well as angiopoietin-1 [33]. Such evasive resistance to anti-VEGFR-2 treatment could be suppressed by commencing therapy with FGF-trap to block FGF1 and FGF2 signaling [34]. Similarily, when a pan-VEGFR inhibitor was used in patients with glioblastoma multiforme, those that failed on therapy were found to have increased levels of FGF2, stromal cell-derived factor (SDF)-1 and circulating endothelial cells [35]. Experimentally, c-Met, platelet derived growth factor receptor (PDGFR)-α and epidermal growth factor receptor (EGFR) were all found to converge on and activate the phosphatidylinositol-3-kinase (PI3K) pathway in glioblastoma, lung and pancreatic tumor cell lines [36], indicating that such one-axis inhibition will be insufficient. In accordance with these preclinical data, signal transduction inhibitors targeting either EGFR or PDGFR were found to be ineffective in glioblastoma patients, and by using an antibody array the tumor samples were found to have multiple receptor tyrosine kinases (RTKs) upregulated simultaneously [36]. In another study signaling molecules such as extracellular signal-regulated kinase (ERK), Akt, mammalian target of rapamycin (mTOR) and signal transducer and activator of transcription protein 3 (STAT3) were all found to be activated in patients with pancreatic ductal adenocarcinoma [30].
It is difficult to interpret from the above studies whether the signaling redundancy is from within the malignant cell population or the supporting stroma or endothelium. However, heterogeneity is evident when endothelial cells from different normal organs and different tumor types are compared [37,38]. Moreover, the tumor endothelium differs from the surrounding normal tissue endothelial cells by having a multitude of genes upregulated [20,28], indicating that targeting a single cytokine or growth factor has a low probability of long-term therapeutic success. In a melanoma xenograft model, EGFR expression was found mainly on the tumor endothelial cells, and EGFR inhibition by gefitinib led to tumor growth inhibition, but subsequent upregulation of VEGFR-2 on the endothelium [39]. Thus, the addition of anti-VEGFR-2 to gefitinib treatment would be an appropriate strategy in this particular case.
In the VEGF/VEGFR family system, multiple ligands are encountered, with overlapping receptor specificities. For instance, when VEGF (VEGF-A) is blocked, VEGF-C and VEGF-D can take over its signaling function [40]. A feasible strategy to avoid such a resistance mechanism is the use of soluble VEGF receptors. Whereas antibodies directed at one specific VEGF receptor can be circumvented by ligand binding to other VEGF receptors, the administration of soluble VEGF receptor will effectively prevent such binding by absorbing the ligands [41]. Apart from the VEGF family, another example of crosstalk is between prostaglandin E2 and transforming growth factor β (TGFβ), both activating the activin-like kinase 5 (Alk5) receptor to stimulate angiogenesis [42].
Using cocktails of different angiogenesis inhibitors is a viable strategy to prevent signaling redundancy in tumor associated endothelial cells [34,41]. After transfecting tumor cells with angiostatin and endostatin, a synergistic antiangiogenic activity was found in both leukemia and melanomas in mice [43]. In human renal cell carcinoma xenografts, the combination of anti-VEGF and the endogenous tumstatin peptide gave a significant tumor growth delay, whereas each of the compounds individually yielded marginal tumor response [44].
The dependency of tumors on a particular angiogenic growth factor can change during antiangiogenic therapy. It has been shown that during prolonged anti-VEGF therapy, vascular remodeling occurs where tumor blood vessels get increased pericyte coverage due to upregulation of PDGF-B and ephrin B2 [45,46]. When these blood vessels acquire pericytes their dependency on VEGF decreases and they turn resistant to anti-VEGF therapy [46,47]. By using a combined inhibition of VEGF and PDGF receptors, one can circumvent this problem and enforce tumor vessel regression [48]. On the other hand absence of α-smooth muscle actin (αSMA) positive cells around the blood vessels correlates with hematogenous metastasis and poor prognosis in colorectal cancer patients [49], indicating that pericyte coverage on the tumor vasculature may protect against metastasis. There is in fact increasing evidence that a combined targeting of tumor endothelium and pericytes by combined VEGFR and PDGFR blockage facilitates the metastasis process [34].
Direct Versus Indirect Angiogenesis Inhibitors
Angiogenesis inhibitors can be divided into two groups, the direct and indirect angiogenesis inhibitors. Direct angiogenesis inhibitors target the endothelial cell behavior directly, whereas indirect inhibitors target cytokines or growth factors acting upon endothelial cells [50]. Theoretically, acquired drug resistance is expected to be a significant problem when indirect angiogenesis inhibitors, such as anti-VEGF antibodies are administered, as the tumor endothelial cell could still be stimulated by other growth factors that are not inhibited. Angiogenic growth factors are commonly produced by the tumor cells themselves, and the tumor cell population could easily evade such growth factor inhibition by shifting their signaling dependency. In human squamous cell carcinoma xenografts, anti-EGFR treatment initially downregulates VEGF production, but after two weeks of continous therapy, some tumor cell clones expand that continue to secrete VEGF despite anti-EGFR treatment [51].
Direct inhibitors, on the other hand, target signaling pathways within the endothelial cell, inducing apoptosis or inhibiting cell migration (Table 1). Endogenous angiogenesis inhibitors seem to exert direct endothelial cytotoxicity as a common characteristic. Apart from tumor cell cytotoxicity, several types of chemotherapeutic agents like cyclophosphamide and combretastatin A-4, also exhibit a direct cytotoxic effect on tumor endothelium [52-54]. Direct angiogenesis inhibitors in many cases actively induce endothelial cell apoptosis, therefore persistant signaling through other signaling pathways may not rescue the cell. Thus, the addition of direct inhibitors to indirect angiogenesis inhibitors as a therapy cocktail is likely to improve the therapeutic outcome [44,55].
Table 1.
Endogenous angiogenesis inhibitors with direct endothelial cytotoxicity.
| Angiogenesis inhibitor | Receptors and biological effect on endothelial cells | Reference |
|---|---|---|
| Angiostatin | Binds to angiomotin, αvβ3 integrin and other receptors and affects several signaling pathways to arrest the cell cycle and induce apoptosis. | [56-58] |
| Arresten | Binds to α1β1 integrin and inhibits migration and proliferation via effects on several signaling pathways. | [59,60] |
| Canstatin | Binds to αvβ3 and α3β1 integrin. Inhibits migration and induces apoptosis via FLIP downregulation. | [61,62] |
| Endorepellin | Binds to α2β1 integrin and inhibits cell migration via disassembly of actin cytoskeleton and focal adhesions. | [63,64] |
| Endostatin | Binds to α5β1 and other receptors and affects various signaling pathways to arrest the cell cycle and induce endothelial apoptosis. Disassembly of actin stress fibers. | [56,65-68] |
| Hexastatin | Not known. | [69] |
| Platelet factor-4 | Binds to heparin-like glycosaminoglycans. Inhibits cell cycle and matrix-associated proteases. | [56] |
| 16-kDa N-terminal fragment of prolactin | Unknown receptor. Affects several signaling pathways to arrest the cell cycle and induce apoptosis. | [56] |
| Thrombospondin | Binds to CD36 and αvβ3 integrin and inhibits several intracellular pathways. Inhibits matrix-associated proteases. | [56,70] |
| Tumstatin | Binds to αvβ3 and α6β1 integrin and inhibits protein synthesis. | [44,71] |
Tumors Can Grow Without Blood Vessels
It is generally thought that a tumor cannot grow beyond 2-3 mm without the recruitment of neovasculature [1]. Furthermore, some experiments have demonstrated that angiogenesis is initiated even in carcinomas in situ [72,73] and also in tumor nodules as small as 0.1 mm [74]. However, the notion that tumors require blood vessels in order to expand beyond 0.1 mm is not an absolute concept, according to some investigators. These studies propose that there are various methods by which cancer cells can grow without the recruitment of new blood vessels.
In glioblastoma multiforme, it has been known for a long time that cancer cells can migrate as diffuse colonies into the surrounding brain, without initiating angiogenesis [75]. Additionally, cancer cells can adapt to a hypoxic environment when tumor angiogenesis is inhibited, by selection of clones that are p53 negative and therefore become hypoxia-resistant [76]. Upon anti-VEGF or anti-VEGFR treatment, glioma cells in some cases are found to react by “vascular cooption” [77-79]. Vascular cooption is a term used when tumor cells grow around the pre-existing blood vessels in the normal tissue, receiving all the required nutrients and oxygen for further proliferation without the need for recruiting a new vasculature (Fig. 3). This phenomenon has been described both in gliomas and lung cancer [80-82].
Figure 3.
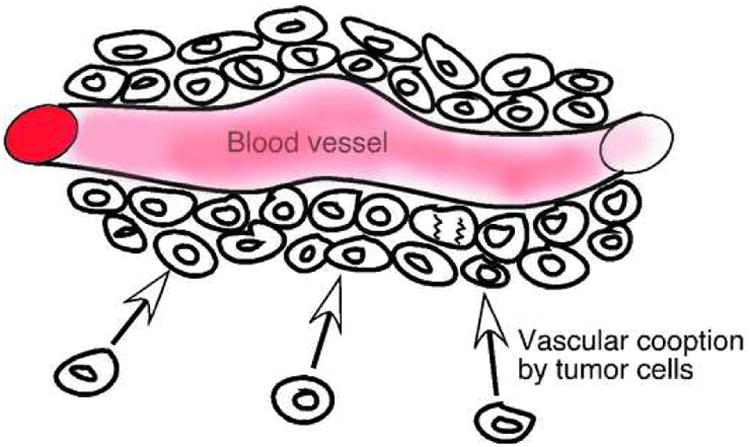
Vascular cooption, a phenomenon whereby tumor cells grows around pre-existing blood vessels in the normal tissue in stead of initiating angiogenesis.
Increased Vessel Density Without the Need for Endothelial Proliferation
Sprouting angiogenesis is a sequential process wherein endothelial cells proliferate, migrate and mature into new vessels. However, blood vessels can also split into new vessels without the need for endothelial proliferation, and this process is termed intussusception [83]. Intussusception is demonstrated in various types of malignant tumors, and has been suggested to occur in the absence of VEGF, as a fast and energy-conserving way of developing new tumor vessels [83]. Clinically, accumulations of tumor blood vessels generated by intussusceptive vessel growth correlate with a worse patient outcome for various types of cancers [84]. While anti-proliferative endothelial inhibitors (such as anti-VEGF antibodies) stop sprouting angiogenesis, intussusceptive vessel growth would probably not be affected by these inhibitors. Instead anti-migratory agents might be more effective in this setting.
Vasculogenic Mimicry
The target of antiangiogenic therapy is generally the endothelium within malignant tumors. Therefore, if tumor cells can develop blood conducting channels without the need for endothelial cells (i.e. vasculogenic mimicry), antiangiogenic treatment would potentially fail. Debates about the validity of vasculogenic mimicry within tumors still continue [85-87]. Some investigators express concerns regarding the interpretation of vasculogenic mimicry [87]. Tumor cells lining the vessel lumen could be due to apoptosis of the overlying endothelial cells, or simply a loss of endothelium ex vivo due to tissue preparation or cutting artefacts. Furthermore, they could be tumor cells in the process of invasion via the blood stream to metastasize. On the other hand, there is compelling evidence that vasculogenic mimicry is encoutered in some tumor types.
There are articles describing vasculogenic mimicry in malignant melanoma, sarcoma, glioma, breast cancer and many other cancer types [83,88-90]. It has been found that melanoma cells can dedifferentiate and display a vascular phenotype, induced by an ischemic microenvironment [86,91]. When analyzing erythrocyte-containing channels in intraocular melanoma tissue from cancer patients, endothelial cells in some tumors were not found by light microscopy, transmission electron microscopy or immunohistochemical staining [89]. In patients with choroidal melanomas these channels were found to be functional, i.e. perfused by blood [83]. In dsRed-expressing U87 glioma xenografts grown in eGFP mice, erythrocyte-containing channels were detected without an endothelial cell lining [18]. Also, in breast carcinoma xenografts, vascular channels composed of tumor cells were found to be perfused with blood when evaluated by MRI angiography, and tumor cells lining the channels expressed endothelial markers such as Flt-1 and Tie-2, but not CD31 [90]. However such data is not present for spontaneous mouse tumors. As a variant of vasculogenic mimicry, mosaic vessels, wherein a mixture of cancer cells and endothelial cells line the vessel wall, has been identified in various tumor types [92]. In this study, about 15% of the perfused tumor blood vessels show a mosaic pattern, including human tumors. How these type of vessels respond to anti-angiogenic therapy is still unknown.
Heterogenous Dependence of Tumors on Angiogenic Growth Factors
There are a large number of endogenous angiogenic and antiangiogenic factors identified as of today [83,93], and individual differences exist. Different mouse strains exhibit a distinct angiogenic response to FGF1 and VEGF [94]. Also, various mouse strains differed in their angiogenic response to FGF1 and VEGF when recruitment of circulating endothelial cells to the tumors are used as a read out [95].
Furthermore, different cancer types probably produce and depend on different angiogenic growth factors. This is suggested by preclinical data showing that anti-VEGF treatment was much less effective in neuroblastoma than Wilms tumor xenografts [96]. Therefore even if a drug is effective against one growth factor, the therapy can still fail if this factor is not important for the endothelium in that given tumor [37]. Also, the maturation of the vasculature within a particular tumor may determine the effectiveness of angiogenesis inhibition, for instance VEGF functions as a survival factor only in blood vessels that are not covered by pericytes [97].
Clinically, there are clear differences between different cancer types with respect to sensitivity towards antiangiogenic treatment [34]. While anti-VEGF monotherapy significantly inhibits the growth of renal cell carcinomas, the effect in many other tumor types is negligable when not combined with chemotherapy [98,99]. Furthermore, there are obvious differences between cancer patients with respect to their response to angiogenic stimulation. For example, about 20% of patients with metastatic renal cell carcinoma do not respond to anti-VEGF treatment [98,100]. On the other hand, there are a small subset of patients with metastatic renal cancer with impressive response to antiangiogenic therapy, with disease stabilization observed for 3-5 years [101]. Clearly, tumor tissue from these good responders will likely provide clues to the positive response.
Non-Endothelial Targets of Antiangiogenic Therapy
A malignant tumor consists of many different cell types, including tumor cells, endothelial cells, fibroblasts, macrophages and other leukocyte subpopulations. These cells are important contributors to the production of angiogenic growth factors as well as other protumorigenic cytokines [34,93]. If endothelial-specific inhibitors are used as antiangiogenic agents, the other cells in the tumors remain capable of producing angiogenic growth factors [34] (Fig. 4). One example of such a scenario is the use of VEGFR-2 signaling inhibitors. When VEGFR-2 is blocked on endothelial cells, fibroblasts and tumor cells can still secrete VEGF and stimulate the endothelium via VEGFR1 and VEGFR3 [102]. Moreover, tumors may evade antiangiogenic therapy by secreting SDF-1 from tumor-associated fibroblasts to attract circulating endothelial precursor cells and continue the process of neo-vascularization [103]. Macrophages seem to accumulate in hypoxic tumor areas and via production of hypoxia-inducible factor-1α (HIF1α) and VEGF contribute to tumor angiogenesis [104]. Furthermore, during pancreatic islet carcinogenesis, infiltrating neutrophils are important contributors when preneoplastic lesions switch to an angiogenic and invasive cancer phenotype [105].
Figure 4.
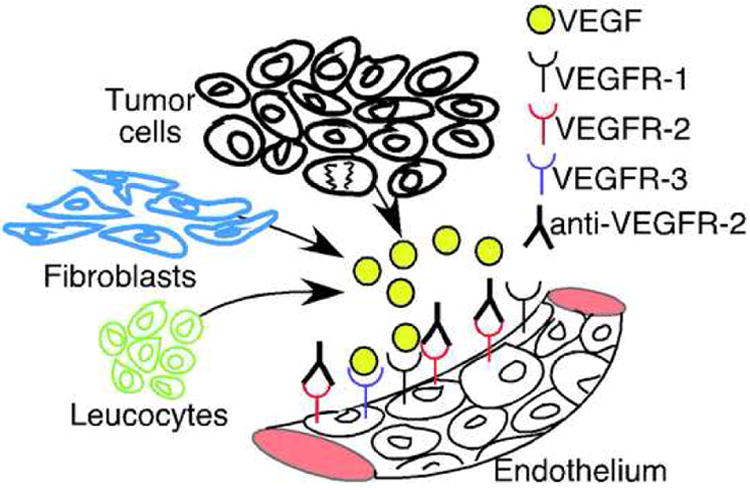
Fibroblasts, leucocytes and tumor cells secrete VEGF and can stimulate angiogenesis via VEGFR-1 and VEGFR-3, despite anti-VEGFR-2 therapy.
The Effective Dosage of Antiangiogenesis Agents Used in the Clinic
The traditional way of treating cancer patients has been to administer the maximal tolerated dose (MTD), with the bone marrow side effects often used as a limiting factor. After bone marrow recovery, repeated doses are given, commonly every three weeks, and the treatment is continued usually for 3-12 months. When angiogenesis inhibitors are used for cancer therapy, the MTD dosage schedule is not clearly defined.
Endostatin exposure of endothelial cells in vitro yieldes a U-shaped dose-response curve when proangiogenic molecules are measured [67] These experiments suggest that too little and too much of this drug leads to a loss of therapeutic effect. In a phase I clinical trial of endostatin, the same U-shaped dose-response curve was found when surrogate markers such as tumor blood flow and microvessel density were examined [106]. Additionally, other known endogenous angiogenesis inhibitors, such as TSP-1 and angiostatin, exhibit similar dose-dependent effects on endothelial cell and neutrophil migration in vitro [107-109]. However, in patients with advanced renal cell carcinoma, a bevacizumab (anti-VEGF) dose of 10 mg/kg gave significantly better results when compared to 3 mg/kg, despite prior preclinical studies showing that 3 mg/kg provides an optimal inhibition of VEGF signaling [101]. These results indicate that finding an optimal dose is not easy but yet critical for appropriate therapeutic response.
Another issue is the duration of antiangiogenic therapy, in patients that respond. In many cases, angiogenesis inhibitors will stop tumor growth, but the pre-existing tumor tissue does not disappear. Even if the endothelial cells are in fact eradicated by the treatment, the empty casts of the blood vessels still persist in the tumor matrix, and these casts can potentially function as tracks for regrowth of vessels once the drug treatment is halted [110]. Also, microscopic tumor nodules or micrometastases can persist for long periods of time, without the need for neovascularization, a process known as tumor dormancy [111]. Clinical data from long-term anti-VEGF therapy in patients with stage IV renal cell carcinoma indicate that treatment should be continued as long as the patients respond and tolerate it well [101].
An alternative strategy for angiogenesis inhibition is the use of vascular disrupting agents (VDA's). While antiangiogenic drugs arrest the growth of new vessels, VDA's destroy preformed blood vessels and therefore have a higher propensity to regress established solid neoplasms [112]. Cytotoxic drugs belonging to the class of tubulin disrupting agents, such as vinblastine and combretastatins, are potent vascular disrupting agents [112,113]. In lower doses these same drugs exhibit antiangiogenic activity and may reduce tumor blood flow by inducing endothelial edema within the tumors [15,53,54,114,115]. The combination of such agents with other treatment modalities, such as radiation or local heating of solitary tumors, can mediate increased vascular damage and tumor growth delay [54,112]. There is limited data to suggest that VDA's might augment the activity of angiogenesis inhibitors. In rhabdomyosarcoma xenografts, the effect of the angiogenesis inhibitor TNP-470 was not increased by combination with combretastatin A-4 [116]. However, in two other murine tumor models, increased tumor growth delay was observed when the angiogenesis inhibitor ZD6474 was combined with the VDA ZD6126 [117]. The potential of VDA therapy to promote improved survival in cancer patients is currently being assessed in various clinical trials [118].
Matrix Metalloproteinase Inhibitors and Their Lack of Efficacy in Clinical Trials
Endothelial cells and tumor cells use matrix metalloproteinases (MMPs) to degrade the extracellular matrix and basement membranes as the new blood vessels sprout inside a growing tumor [119,120]. Inhibitors of MMPs (MMPIs) to stop tumor angiogenesis and cancer progression, showed great promise in preclinical tumor experiments [121-123]. However, in large phase III clinical trials MMPIs failed to show prolongation of overall survival [121,123-125]. On the contrary, there was even a tendency towards increased metastasis risk and shorter survival for cancer patients receiving the drugs [123,126].
Looking back at the preclinical studies there were already indications that MMPIs were not effective. While halting solid tumor growth in a breast cancer model, the MMPI batimastat had no effect on ascites development or tumor cell invasion [127]. In a rat glioma model, batimastat also had insignificant effect on solid tumor growth [16].
The failure of MMPIs in clinical trials was suggested to be due to a lack of efficacy in large, advanced tumor masses, as compared to inhibition of angiogenesis in small tumors in an adjuvant setting [125,128]. Also, MMPs can in fact have both proangiogenic and antiangiogenic effects, via proteolytic activation of proangiogenic cytokines or liberation of endogenous angiogenesis inhibitors like tumstatin, arresten and canstatin [124,129]. Thus, the administration of MMPIs can inhibit angiogenesis by arresting proteolytic activation of proangiogenic cytokines, or promote angiogenesis by inihibiting the liberation of endogenous angiogenesis inhibitors from the extracellular matrix [37]. In this context, integrin α1 deficient mice exhibit diminished tumor angiogenesis and reduced vascularity in the skin due to increased activity of MMP7 and MMP9 which generates angiostatin from plasminogen. So an inhibitor that inactivates MMP7 and MMP9 would reduce the level of angiostatin and thus increase angiogenesis [130]. The potential response also depends on whether one uses a broad-spectrum or a narrow-spectrum MMPI [124]. Another possible reason for MMPIs failure to arrest tumor growth, might be connected to the upregulation of other proteases, such as serine and cysteine proteases, when MMPs are inactivated [131]. Finally, lack of patient compliance is a possible factor in the failure, as some patients stopped taking the MMPI drugs due to painful musculoskeletal side effects [125].
Future Aspects
It is evident that angiogenesis inhibitors currently approved for clinical use are not providing long term efficacy. Drug resistance commonly develops after a few months of therapy, and improvements are clearly needed. The use of combinations of drugs targeting different angiogenic growth factors (“antiangiogenic cocktails”), or drugs that target multiple angiogenesis pathways, such as sorafenib and sunitinib, might exhibit better efficacy.
Another strategy is to obtain tumor biopsies to analyze growth factors that are upregulated in a particular patient, in order to administer “tailor-made” antiangiogenesis treatment. For instance phosphatase and tensin homolog (PTEN) can be used as a biomarker. Loss of PTEN renders anti-EGFR therapy ineffective [132]. One can also assess the level of VEGFR-2+ circulating endothelial cells as a surrogate marker for the efficacy of antiangiogenic therapy [95,133]. The use of biomarkers for antiangiogenic response is a major focus of current research [134]. With the advancement of DCE-MRI and new ultrasound techniques, future clinical trials can potentially be designed so that these imaging modalities can guide the timing of chemotherapy administration related to induced changes in tumor blood perfusion.
Antiangiogenic therapy may be more potent if administered at an early stage as adjuvant therapy, after curative surgery, radiotherapy or high-dose chemotherapy. At this stage of disease, the antiangiogenic drugs may prolong patient survival substantially by keeping micrometastases in check. Currently, several clinical studies are being carried out to address these approaches [135,136]. In general, angiogenesis inhibitors are an important class of anti-cancer agents. But the current treatment response is transient, and more research is therefore required to address why drug resistance occurs.
Acknowledgments
This work was primarily supported by National Institutes of Health Grant DK62987 and partially by National Institutes of Health Grants DK55001, DK61688, AA13913, and CA12550 and funds from the Department of Medicine for the Division of Matrix Biology at Beth Israel Deaconness Medical Center. HPE was supported by grants from the University of Bergen, Helse Vest, the Eckbo Legacy and the Fulbright Association, Norway. We thank Dr. Elisabeth Zeisberg for providing us with the Tie2-LacZ picture.
Abbreviations
- eGFP
enhanced green fluorescent protein
- TSP-1
thrombospondin-1
- VEGF
vascular endothelial growth factor
- VEGFR-22
VEGF receptor-2
- FGF
fibroblast growth factor
- SDF-1
stromal cell-derived factor-1
- PDGFR-α
platelet derived growth factor receptor-α
- EGFR
epidermal growth factor receptor
- PI3K
phosphatidylinositol-3-kinase
- RTK
receptor tyrosine kinase
- ERK
extracellular signal-regulated kinase
- mTOR
mammalian target of rapamycin
- STAT3
signal transducer and activator of transcription protein 3
- TGFβ
transforming growth factor β
- Alk5
activin-like kinase 5
- αSMA
α-smooth muscle actin
- HIF-1α
hypoxia-inducible factor-1α
- MTD
maximal tolerated dose
- VDA
vascular disrupting agent
- MMP
matrix metalloproteinase
- MMPI
MMP inhibitor
- PTEN
phosphatase and tensin homolog
Footnotes
Conflict of Interest Statement: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175(3):409–16. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spalding BJ. Thumbs up for Avastin. Nat Biotechnol. 2008;26(4):365. doi: 10.1038/nbt0408-365b. [DOI] [PubMed] [Google Scholar]
- 4.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 5.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 6.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 7.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 8.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 9.Wadman M. Cancer ‘cure’ article stirs up hot debate. Nature. 1998;393(May 14):104–5. doi: 10.1038/30072. [DOI] [PubMed] [Google Scholar]
- 10.Marshall E. The power of the front page of The New York Times. Science. 1998;280(May 15):996–7. doi: 10.1126/science.280.5366.996. [DOI] [PubMed] [Google Scholar]
- 11.Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anticancer therapeutic agents. BioEssays. 1991;13(1):31–6. doi: 10.1002/bies.950130106. [DOI] [PubMed] [Google Scholar]
- 12.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(January 24):277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 13.O'Reilly MS, Holmgren L, Chen C, Folkman J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med. 1996;2(6):689–92. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- 14.Fernando NT, Koch M, Rothrock C, Gollogly LK, D'Amore PA, Ryeom S, et al. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin Cancer Res. 2008;14(5):1529–39. doi: 10.1158/1078-0432.CCR-07-4126. [DOI] [PubMed] [Google Scholar]
- 15.Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest. 2000;105(8):R15–24. doi: 10.1172/JCI8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eikesdal HP, Bjorkhaug ST, Dahl O. Hyperthermia exhibits anti-vascular activity in the s.c. BT4An rat glioma -lack of interaction with the angiogenesis inhibitor batimastat. Int J Hyperthermia. 2002;18(2):141–52. doi: 10.1080/02656730110090712. [DOI] [PubMed] [Google Scholar]
- 17.Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8. doi: 10.1158/0008-5472.CAN-07-3127. [DOI] [PubMed] [Google Scholar]
- 18.Niclou SP, Danzeisen C, Eikesdal HP, Wiig H, Brons NH, Poli AM, et al. A novel eGFP-expressing immunodeficient mouse model to study tumor-host interactions. FASEB J. 2008;22(9):3120–8. doi: 10.1096/fj.08-109611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folkman J, Hahnfeldt P, Hlatky L. Cancer: looking outside the genome. Nat Rev Mol Cell Biol. 2000;1(1):76–9. doi: 10.1038/35036100. [DOI] [PubMed] [Google Scholar]
- 20.St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. 2000;289(5482):1197–202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- 21.Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390(6658):404–7. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 22.Brem H, Goto F, Budson A, Saunders L, Folkman J. Minimal drug resistance after prolonged antiangiogenic therapy with AGM-1470. Surg Forum. 1994;XLV:674–7. [Google Scholar]
- 23.Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64(22):8249–55. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- 24.Hida K, Klagsbrun M. A new perspective on tumor endothelial cells: unexpected chromosome and centrosome abnormalities. Cancer Res. 2005;65(7):2507–10. doi: 10.1158/0008-5472.CAN-05-0002. [DOI] [PubMed] [Google Scholar]
- 25.Streubel B, Chott A, Huber D, Exner M, Jager U, Wagner O, et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N Engl J Med. 2004;351(3):250–9. doi: 10.1056/NEJMoa033153. [DOI] [PubMed] [Google Scholar]
- 26.Gunsilius E, Duba HC, Petzer AL, Kahler CM, Grunewald K, Stockhammer G, et al. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet. 2000;355(9216):1688–91. doi: 10.1016/S0140-6736(00)02241-8. [DOI] [PubMed] [Google Scholar]
- 27.Dudley AC, Khan ZA, Shih SC, Kang SY, Zwaans BM, Bischoff J, et al. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14(3):201–11. doi: 10.1016/j.ccr.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003;17(9):1159–61. doi: 10.1096/fj.02-0557fje. [DOI] [PubMed] [Google Scholar]
- 29.Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin Oncol. 2006;33(4):407–20. doi: 10.1053/j.seminoncol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Pham NA, Schwock J, Iakovlev V, Pond G, Hedley DW, Tsao MS. Immunohistochemical analysis of changes in signaling pathway activation downstream of growth factor receptors in pancreatic duct cell carcinogenesis. BMC Cancer. 2008;8(Feb 6):43. doi: 10.1186/1471-2407-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997;57(5):963–9. [PubMed] [Google Scholar]
- 32.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118(2):217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 33.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a Pan-VEGF Receptor Tyrosine Kinase Inhibitor, Normalizes Tumor Vasculature and Alleviates Edema in Glioblastoma Patients. Cancer Cell. 2007;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 37.Miller KD, Sweeney CJ, Sledge GWJ. The Snark is a Boojum: the continuing problem of drug resistance in the antiangiogenic era. Ann Oncol. 2003;14(1):20–8. doi: 10.1093/annonc/mdg033. [DOI] [PubMed] [Google Scholar]
- 38.Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11(6):539–54. doi: 10.1016/j.ccr.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin DN, Bielenberg DR, Lifshits E, Heymach JV, Klagsbrun M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc Res. 2008;76(1):15–22. doi: 10.1016/j.mvr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438(7070):946–53. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 41.Zwaans BM, Bielenberg DR. Potential therapeutic strategies for lymphatic metastasis. Microvasc Res. 2007;74(2-3):145–58. doi: 10.1016/j.mvr.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfranca A, Lopez-Oliva JM, Genis L, Lopez-Maderuelo D, Mirones I, Salvado D, et al. PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFbeta/Alk5 signaling pathway. Blood. 2008;112(4):1120–8. doi: 10.1182/blood-2007-09-112268. [DOI] [PubMed] [Google Scholar]
- 43.Scappaticci FA, Smith R, Pathak A, Schloss D, Lum B, Cao Y, et al. Combination angiostatin and endostatin gene transfer induces synergistic antiangiogenic activity in vitro and antitumor efficacy in leukemia and solid tumors in mice. Mol Ther. 2001;3(2):186–96. doi: 10.1006/mthe.2000.0243. [DOI] [PubMed] [Google Scholar]
- 44.Eikesdal HP, Sugimoto H, Birrane G, Maeshima Y, Cooke VG, Kieran M, et al. Identification of amino acids essential for the antiangiogenic activity of tumstatin and its use in combination antitumor therapy. Proc Natl Acad Sci U S A. 2008;105(39):15040–5. doi: 10.1073/pnas.0807055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glade Bender J, Cooney EM, Kandel JJ, Yamashiro DJ. Vascular remodeling and clinical resistance to antiangiogenic cancer therapy. Drug Resist Updat. 2004;7(4-5):289–300. doi: 10.1016/j.drup.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, Soffer SZ, Kim ES, McCrudden KW, Huang J, New T, et al. Vascular remodeling marks tumors that recur during chronic suppression of angiogenesis. Mol Cancer Res. 2004;2(1):36–42. [PubMed] [Google Scholar]
- 47.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60(5):1388–93. [PubMed] [Google Scholar]
- 48.Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. Faseb J. 2004;18(2):338–40. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 49.Yonenaga Y, Mori A, Onodera H, Yasuda S, Oe H, Fujimoto A, et al. Absence of smooth muscle actin-positive pericyte coverage of tumor vessels correlates with hematogenous metastasis and prognosis of colorectal cancer patients. Oncology. 2005;69(2):159–66. doi: 10.1159/000087840. [DOI] [PubMed] [Google Scholar]
- 50.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2(10):727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 51.Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61(13):5090–101. [PubMed] [Google Scholar]
- 52.Browder T, Butterfield CE, Kraling BM, Shi B, Marshall B, O'Reilly MS, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60(7):1878–86. [PubMed] [Google Scholar]
- 53.Iyer S, Chaplin DJ, Rosenthal DS, Boulares AH, Li LY, Smulson ME. Induction of apoptosis in proliferating human endothelial cells by the tumor-specific antiangiogenesis agent combretastatin A-4. Cancer Res. 1998;58(20):4510–4. [PubMed] [Google Scholar]
- 54.Eikesdal HP, Bjerkvig R, Mella O, Dahl O. Combretastatin A-4 and hyperthermia; a potent combination for the treatment of solid tumors. Radiother Oncol. 2001;60(2):147–54. doi: 10.1016/s0167-8140(00)00318-2. [DOI] [PubMed] [Google Scholar]
- 55.Abdollahi A, Lipson KE, Sckell A, Zieher H, Klenke F, Poerschke D, et al. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 2003;63(24):8890–8. [PubMed] [Google Scholar]
- 56.Tabruyn SP, Griffioen AW. Molecular pathways of angiogenesis inhibition. Biochem Biophys Res Commun. 2007;355(1):1–5. doi: 10.1016/j.bbrc.2007.01.123. [DOI] [PubMed] [Google Scholar]
- 57.Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol. 2001;152(6):1247–54. doi: 10.1083/jcb.152.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wahl ML, Kenan DJ, Gonzalez-Gronow M, Pizzo SV. Angiostatin's molecular mechanism: aspects of specificity and regulation elucidated. J Cell Biochem. 2005;96(2):242–61. doi: 10.1002/jcb.20480. [DOI] [PubMed] [Google Scholar]
- 59.Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000;60(9):2520–6. [PubMed] [Google Scholar]
- 60.Sudhakar A, Nyberg P, Keshamouni VG, Mannam AP, Li J, Sugimoto H, et al. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J Clin Invest. 2005;115(10):2801–10. doi: 10.1172/JCI24813. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Magnon C, Galaup A, Mullan B, Rouffiac V, Bouquet C, Bidart JM, et al. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 2005;65(10):4353–61. doi: 10.1158/0008-5472.CAN-04-3536. [DOI] [PubMed] [Google Scholar]
- 62.Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, et al. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275(2):1209–15. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- 63.Mongiat M, Sweeney SM, San Antonio JD, Fu J, Iozzo RV. Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J Biol Chem. 2003;278(6):4238–49. doi: 10.1074/jbc.M210445200. [DOI] [PubMed] [Google Scholar]
- 64.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, et al. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004;166(1):97–109. doi: 10.1083/jcb.200401150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, et al. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274(17):11721–6. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- 66.Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002;62(19):5580–9. [PubMed] [Google Scholar]
- 67.Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W, et al. Endostatin's antiangiogenic signaling network. Mol Cell. 2004;13(5):649–63. doi: 10.1016/s1097-2765(04)00102-9. [DOI] [PubMed] [Google Scholar]
- 68.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003;100(8):4766–71. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 69.Mundel TM, Yliniemi AM, Maeshima Y, Sugimoto H, Kieran M, Kalluri R. Type IV collagen alpha6 chain-derived noncollagenous domain 1 (alpha6(IV)NC1) inhibits angiogenesis and tumor growth. Int J Cancer. 2008;122(8):1738–44. doi: 10.1002/ijc.23269. [DOI] [PubMed] [Google Scholar]
- 70.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6(1):1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295(5552):140–3. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- 72.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 73.Sledge GWJ, Miller KD. Angiogenesis and antiangiogenic therapy. Curr Probl Cancer. 2002;26(1):1–60. [PubMed] [Google Scholar]
- 74.Li CY, Shan S, Huang Q, Braun RD, Lanzen J, Hu K, et al. Initial stages of tumor cell-induced angiogenesis: evaluation via skin window chambers in rodent models. J Natl Cancer Inst. 2000;92(2):143–7. doi: 10.1093/jnci/92.2.143. [DOI] [PubMed] [Google Scholar]
- 75.Sakariassen PO, Prestegarden L, Wang J, Skaftnesmo KO, Mahesparan R, Molthoff C, et al. Angiogenesis-independent tumor growth mediated by stem-like cancer cells. Proc Natl Acad Sci U S A. 2006;103(44):16466–71. doi: 10.1073/pnas.0607668103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu JL, Rak JW, Coomber BL, Hicklin DJ, Kerbel RS. Effect of p53 status on tumor response to antiangiogenic therapy. Science. 2002;295(5559):1526–8. doi: 10.1126/science.1068327. [DOI] [PubMed] [Google Scholar]
- 77.Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2(4):306–14. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61(18):6624–8. [PubMed] [Google Scholar]
- 79.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284(5422):1994–8. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 81.Offersen BV, Pfeiffer P, Hamilton-Dutoit S, Overgaard J. Patterns of angiogenesis in nonsmall-cell lung carcinoma. Cancer. 2001;91(8):1500–9. [PubMed] [Google Scholar]
- 82.Passalidou E, Trivella M, Singh N, Ferguson M, Hu J, Cesario A, et al. Vascular phenotype in angiogenic and non-angiogenic lung non-small cell carcinomas. Br J Cancer. 2002;86(2):244–9. doi: 10.1038/sj.bjc.6600015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hillen F, Griffioen AW. Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007;26(3-4):489–502. doi: 10.1007/s10555-007-9094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Straume O, Chappuis PO, Salvesen HB, Halvorsen OJ, Haukaas SA, Goffin JR, et al. Prognostic importance of glomeruloid microvascular proliferation indicates an aggressive angiogenic phenotype in human cancers. Cancer Res. 2002;62(23):6808–11. [PubMed] [Google Scholar]
- 85.McDonald DM, Munn L, Jain RK. Vasculogenic mimicry: how convincing, how novel, and how significant? Am J Pathol. 2000;156(2):383–8. doi: 10.1016/S0002-9440(10)64740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–21. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 87.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 88.Konerding MA, Steinberg F, Budach V. The vascular system of xenotransplanted tumors--scanning electron and light microscopic studies. Scanning Microsc. 1989;3(1):327–35. discussion 335-6. [PubMed] [Google Scholar]
- 89.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–52. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shirakawa K, Kobayashi H, Heike Y, Kawamoto S, Brechbiel MW, Kasumi F, et al. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002;62(2):560–6. [PubMed] [Google Scholar]
- 91.Hendrix MJ, Seftor RE, Seftor EA, Gruman LM, Lee LM, Nickoloff BJ, et al. Transendothelial function of human metastatic melanoma cells: role of the microenvironment in cell-fate determination. Cancer Res. 2002;62(3):665–8. [PubMed] [Google Scholar]
- 92.Chang YS, di Tomaso E, McDonald DM, Jones R, Jain RK, Munn LL. Mosaic blood vessels in tumors: frequency of cancer cells in contact with flowing blood. Proc Natl Acad Sci U S A. 2000;97(26):14608–13. doi: 10.1073/pnas.97.26.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nyberg P, Salo T, Kalluri R. Tumor microenvironment and angiogenesis. Front Biosci. 2008;13:6537–53. doi: 10.2741/3173. [DOI] [PubMed] [Google Scholar]
- 94.Rohan RM, Fernandez A, Udagawa T, Yuan J, D'Amato RJ. Genetic heterogeneity of angiogenesis in mice. Faseb J. 2000;14(7):871–6. doi: 10.1096/fasebj.14.7.871. [DOI] [PubMed] [Google Scholar]
- 95.Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, et al. Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis; Implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell. 2005;7(1):101–11. doi: 10.1016/j.ccr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 96.Kim E, Moore J, Huang J, Soffer S, Manley CA, O'Toole K, et al. All angiogenesis is not the same: Distinct patterns of response to antiangiogenic therapy in experimental neuroblastoma and Wilms tumor. J Pediatr Surg. 2001;36(2):287–90. doi: 10.1053/jpsu.2001.20691. [DOI] [PubMed] [Google Scholar]
- 97.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103(2):159–65. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349(5):427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glade-Bender J, Kandel JJ, Yamashiro DJ. VEGF blocking therapy in the treatment of cancer. Expert Opin Biol Ther. 2003;3(2):263–76. doi: 10.1517/14712598.3.2.263. [DOI] [PubMed] [Google Scholar]
- 100.Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370(9605):2103–11. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 101.Yang JC. Bevacizumab for patients with metastatic renal cancer: an update. Clin Cancer Res. 2004;10(18 Pt 2):6367S–70S. doi: 10.1158/1078-0432.CCR-050006. [DOI] [PubMed] [Google Scholar]
- 102.Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94(6):715–25. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 103.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 104.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 105.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493–8. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Davis DW, Shen Y, Mullani NA, Wen S, Herbst RS, O'Reilly M, et al. Quantitative analysis of biomarkers defines an optimal biological dose for recombinant human endostatin in primary human tumors. Clin Cancer Res. 2004;10(1 Pt 1):33–42. doi: 10.1158/1078-0432.ccr-0736-3. [DOI] [PubMed] [Google Scholar]
- 107.Motegi K, Harada K, Pazouki S, Baillie R, Schor AM. Evidence of a bi-phasic effect of thrombospondin-1 on angiogenesis. Histochem J. 2002;34(8-9):411–21. doi: 10.1023/a:1023687505139. [DOI] [PubMed] [Google Scholar]
- 108.Benelli R, Morini M, Brigati C, Noonan DM, Albini A. Angiostatin inhibits extracellular HIV-Tat-induced inflammatory angiogenesis. Int J Oncol. 2003;22(1):87–91. [PubMed] [Google Scholar]
- 109.Celik I, Surucu O, Dietz C, Heymach JV, Force J, Hoschele I, et al. Therapeutic efficacy of endostatin exhibits a biphasic dose-response curve. Cancer Res. 2005;65(23):11044–50. doi: 10.1158/0008-5472.CAN-05-2617. [DOI] [PubMed] [Google Scholar]
- 110.Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165(1):35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Folkman J, Kalluri R. Cancer without disease. Nature. 2004;427(6977):787. doi: 10.1038/427787a. [DOI] [PubMed] [Google Scholar]
- 112.Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006;66(24):11520–39. doi: 10.1158/0008-5472.CAN-06-2848. [DOI] [PubMed] [Google Scholar]
- 113.Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10(2):415–27. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- 114.Eikesdal HP, Bjerkvig R, Dahl O. Vinblastine and hyperthermia target the neovasculature in BT4An rat gliomas: therapeutic implications of the vascular phenotype. Int J Radiat Oncol Biol Phys. 2001;51(2):535–44. doi: 10.1016/s0360-3016(01)01693-5. [DOI] [PubMed] [Google Scholar]
- 115.Galbraith SM, Maxwell RJ, Lodge MA, Tozer GM, Wilson J, Taylor NJ, et al. Combretastatin A4 phosphate has tumor antivascular activity in rat and man as demonstrated by dynamic magnetic resonance imaging. J Clin Oncol. 2003;21(15):2831–42. doi: 10.1200/JCO.2003.05.187. [DOI] [PubMed] [Google Scholar]
- 116.Landuyt W, Ahmed B, Nuyts S, Theys J, Op de Beeck M, Rijnders A, et al. In vivo antitumor effect of vascular targeting combined with either ionizing radiation or anti-angiogenesis treatment. Int J Radiat Oncol Biol Phys. 2001;49(2):443–50. doi: 10.1016/s0360-3016(00)01470-x. [DOI] [PubMed] [Google Scholar]
- 117.Siemann DW, Shi W. Efficacy of combined antiangiogenic and vascular disrupting agents in treatment of solid tumors. Int J Radiat Oncol Biol Phys. 2004;60(4):1233–40. doi: 10.1016/j.ijrobp.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 118.Hinnen P, Eskens FA. Vascular disrupting agents in clinical development. Br J Cancer. 2007;96(8):1159–65. doi: 10.1038/sj.bjc.6603694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taraboletti G, Garofalo A, Belotti D, Drudis T, Borsotti P, Scanziani E, et al. Inhibition of angiogenesis and murine hemangioma growth by batimastat, a synthetic inhibitor of matrix metalloproteinases. J Natl Cancer Inst. 1995;87(4):293–8. doi: 10.1093/jnci/87.4.293. [DOI] [PubMed] [Google Scholar]
- 120.Chung AS, Yoon SO, Park SJ, Yun CH. Roles of matrix metalloproteinases in tumor metastasis and angiogenesis. J Biochem Mol Biol. 2003;36(1):128–37. doi: 10.5483/bmbrep.2003.36.1.128. [DOI] [PubMed] [Google Scholar]
- 121.Yip D, Ahmad A, Karapetis CS, Hawkins CA, Harper PG. Matrix metalloproteinase inhibitors: applications in oncology. Invest New Drugs. 1999;17(4):387–99. doi: 10.1023/a:1006386406584. [DOI] [PubMed] [Google Scholar]
- 122.Talbot DC, Brown PD. Experimental and clinical studies on the use of matrix metalloproteinase inhibitors for the treatment of cancer. Eur J Cancer. 1996;32A(14):2528–33. doi: 10.1016/s0959-8049(96)00398-x. [DOI] [PubMed] [Google Scholar]
- 123.Pavlaki M, Zucker S. Matrix metalloproteinase inhibitors (MMPIs): the beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003;22(2-3):177–203. doi: 10.1023/a:1023047431869. [DOI] [PubMed] [Google Scholar]
- 124.Raza SL, Cornelius LA. Matrix metalloproteinases: pro- and anti-angiogenic activities. J Investig Dermatol Symp Proc. 2000;5(1):47–54. doi: 10.1046/j.1087-0024.2000.00004.x. [DOI] [PubMed] [Google Scholar]
- 125.Cristofanilli M, Charnsangavej C, Hortobagyi GN. Angiogenesis modulation in cancer research: novel clinical approaches. Nat Rev Drug Discov. 2002;1(6):415–26. doi: 10.1038/nrd819. [DOI] [PubMed] [Google Scholar]
- 126.Deplanque G, Harris AL. Anti-angiogenic agents. Clinical trial design and therapies in development. Eur J Cancer. 2000;36(13 Spec No):1713–24. doi: 10.1016/s0959-8049(00)00149-0. [DOI] [PubMed] [Google Scholar]
- 127.Low JA, Johnson MD, Bone EA, Dickson RB. The matrix metalloproteinase inhibitor batimastat (BB-94) retards human breast cancer solid tumor growth but not ascites formation in nude mice. Clin Cancer Res. 1996;2(7):1207–14. [PubMed] [Google Scholar]
- 128.Brown PD. Clinical studies with matrix metalloproteinase inhibitors. Apmis. 1999;107(1):174–80. doi: 10.1111/j.1699-0463.1999.tb01541.x. [DOI] [PubMed] [Google Scholar]
- 129.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3(6):589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97(5):2202–7. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holst-Hansen C, Low JA, Stephens RW, Johnson MD, Carmeliet P, Frandsen TL, et al. Increased stromal expression of murine urokinase plasminogen activator in a human breast cancer xenograft model following treatment with the matrix metalloprotease inhibitor, batimastat. Breast Cancer Res Treat. 2001;68(3):225–37. doi: 10.1023/a:1012217820507. [DOI] [PubMed] [Google Scholar]
- 132.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 133.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci U S A. 2007;104(43):17069–74. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jubb AM, Oates AJ, Holden S, Koeppen H. Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer. 2006;6(8):626–35. doi: 10.1038/nrc1946. [DOI] [PubMed] [Google Scholar]
- 135.Benson AB., 3rd New approaches to assessing and treating early-stage colon and rectal cancers: cooperative group strategies for assessing optimal approaches in early-stage disease. Clin Cancer Res. 2007;13(22 Pt 2):6913s–20s. doi: 10.1158/1078-0432.CCR-07-1188. [DOI] [PubMed] [Google Scholar]
- 136.Hayes DF, Miller K, Sledge G. Angiogenesis as targeted breast cancer therapy. Breast. 2007;16(Suppl 2):S17–9. doi: 10.1016/j.breast.2007.07.003. [DOI] [PubMed] [Google Scholar]