

Abstract
Background & Aims
Vascular endothelial growth factor (VEGF)—induced angiogenesis is implicated in fibrogenesis and portal hypertension. However, the function of VEGF in fibrosis resolution has not been explored.
Methods
We developed a cholecystojejunostomy procedure to reconstruct biliary flow after bile duct ligation in C57BL/6 mice to generate a model of fibrosis resolution. These mice were then given injections of VEGF-neutralizing (mcr84) or control antibodies, and other mice received an adenovirus that expressed mouse VEGF or a control vector. The procedure was also performed on macro-phage fas-induced apoptosis mice, in which macrophages can be selectively depleted. Liver and blood samples were collected and analyzed in immunohistochemical, morphometric, vascular permeability, real-time polymerase chain reaction, and flow cytometry assays.
Results
VEGF-neutralizing antibodies prevented development of fibrosis but also disrupted hepatic tissue repair and fibrosis resolution. During fibrosis resolution, VEGF inhibition impaired liver sinusoidal permeability, which was associated with reduced monocyte migration, adhesion, and infiltration of fibrotic liver. Scar-associated macrophages contributed to this process by producing the chemokine (C-X-C motif) ligand 9 and matrix metalloproteinase 13. Resolution of fibrosis was impaired in macrophage fas-induced apoptosis mice but increased after overexpression of chemokine (C-X-C motif) ligand 9.
Conclusions
In a mouse model of liver fibrosis resolution, VEGF promoted fibrogenesis, but was also required for hepatic tissue repair and fibrosis resolution. We observed that VEGF regulates vascular permeability, monocyte infiltration, and scar-associated macrophages function.
Keywords: Sinusoidal Endothelial Cell, Hepatic Sinusoid, Extracellular Matrix, Liver Damage
Fibrogenesis is the common end point of chronic liver injury of diverse etiologies.1 Although hepatic stellate cell (HSC) activation into a myofibroblastic phenotype is viewed as the dominant mechanism of fibrogenesis, other nonparenchymal cells, including endothelial and Kupffer cells, are also central modulators of fibrosis progression.1,2 As a corollary, fibrosis resolution requires apoptosis or deactivation of myofibroblastic cells again with dominant roles for other nonparenchymal cells in the resolution process.3,4 Although recent studies have highlighted the importance of angiogenesis and proangiogenic molecules in fibrogenesis using both specific neutralizing antibodies as well as more general inhibitors,1,5–7 there is no detailed analysis of the effects of dominant angiogenic molecules during fibrosis resolution.
Vascular endothelial growth factor (VEGF), in addition to its angiogenic function,8,9 was first described as a vascular permeability factor based on its ability to generate tissue edema and regulate monocyte recruitment and infiltration.10–12 Earlier studies revealed a fibrogenic effect of VEGF through multiple mechanisms, including promotion of inflammation, release of fibrosis-enhancing molecules from VEGF-activated endothelial cells, and direct effects of VEGF on HSC.6,13 These studies suggest that VEGF inhibition might also have beneficial effects on fibrosis resolution. Therefore, VEGF-inhibition strategies that are already utilized to treat diverse human diseases are being considered for the treatment of fibrosis in liver and other organs.
In the present study, we establish and utilize a murine model of cholestatic fibrosis regression to test the hypothesis that VEGF neutralization in vivo can promote hepatic tissue repair and liver fibrosis resolution. Although confirming a role of VEGF in fibrogenesis, a combination of loss-and gain-of-function approaches unexpectedly revealed that VEGF promotes hepatic tissue repair and fibrosis resolution. This effect can be mediated through a VEGF-dependent sinusoidal permeability that facilitates monocyte recruitment and infiltration into scar tissue, which appears to be a requirement for fibrosis resolution. Mechanistically, we identify a chemokine (C-X-C motif) ligand 9 (CXCL9)—matrix metalloproteinase 13 (MMP13) axis that mediates the observed actions of VEGF. Collectively, the data uncover dual and opposing effects of VEGF on fibrogenesis and tissue repair/fibrosis resolution through effects of VEGF on sinusoidal permeability, monocyte infiltration, and scar-associated macrophage (SAM) function.
Materials and Methods
Animal Experiments
Bile duct ligation and cholecystojejunostomy
C57BL/6 mice (10–12 weeks) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were subjected to bile duct ligation (BDL) for 2 weeks to induce fibrosis as described previously.14 Cholecystojejunostomy (CJ) was performed to reconstruct biliary flow and stimulate fibrosis resolution based on a biliary reconstruction model previously described in the rat, except that the mouse gall bladder was used as an outflow conduit.15 After BDL and/or CJ, mice received a well-characterized VEGF-neutralizing antibody (anti-VEGF, mcr84)16 or control antibody (50 μg per injection [IP] ×2/week). In other experiments, mice received adenovirus-expressing mouse VEGF (AdVEGF) or LacZ (0.8 × 109 PFU/kg through tail vein injection × 1) after BDL and/or CJ. The dose regimen of AdVEGF was determined based on pilot experiments (Supplementary Figure 1). All animal work was performed under Mayo Institutional Animal Care and Use Committee oversight.
Macrophage fas-induced apoptosis mice
Macrophage fas-induced apoptosis (MAFIA) mice on a C57BL/6 background were purchased from Jackson Laboratories. In this model, Csf1r promoter is used to drive expression of a mutant human FK506 binding protein 1A, 12 kDa (FKBP12), and green fluorescent protein (GFP). Inducible apoptosis of mononuclear cell linage ablation (monocyte, macrophage, and dendritic cells) is achieved by administering AP20187, which binds FKBP12 and stimulates macrophage Fas-induced apoptosis).17,18 Lyophilized AP20187 (Ariad Pharmaceuticals, Cambridge, MA) was dissolved in 100% ethanol to 13.75 mg/mL. One day after CJ, the stock solution was diluted to 0.55 mg/mL in 4% ethanol, with 10% PEG-400, and 1.7% Tween 80 in sterile water (vehicle) and injected IP; 120 μg on day 1, followed by 40 μg daily IP for 4 additional days before starting experiments.18 MAFIA mice receiving vehicle served as controls.
Carbon tetrachloride model
Mice received carbon tetrachloride (CCl4) treatment IP for 4 or 6 weeks as indicated in different experiments.14 One day after the final dose of CCl4, mice were administered anti-VEGF or control antibody (50 μg IP ×2/week). In other experiments, mice received murine AdVEGF (0.8 × 109 PFU/kg), adenovirus encoding murine CXCL9 (AdCXCL9; 4.0 × 1010 PFU/kg), or LacZ (control) through tail vein injection. Mice were sacrificed at 1, 2, or 4 weeks after final dose of CCl4, as indicated. For AdCXCL9 generation, mouse CXCL9 plasmid was subcloned into the AdEasy viral construction system using manufacturer instructions (Stratagene).
Cell Isolation and Culture
Kupffer cells and HSC were isolated from normal rats and mice as described previously19 and are detailed in the Supplementary Material.
Measurement of Hepatic Vascular Permeability in Vivo
Miles assay technique was modified to quantify hepatic vascular permeability as described.20 Detailed methods are in the Supplementary Material.
Real-Time Polymerase Chain Reaction
Messenger RNA (mRNA) levels were quantified by real-time reverse transcription polymerase chain reaction per the manufacturer's specifications. Detailed methods are in the Supplementary Material. Primer sequences are listed in Supplementary Table 1.
Immunohistochemistry and Morphometry
Liver tissues were embedded in OCT and sections (5 μm) were cut and fixed in 4% paraformaldehyde solution for 30 minutes. Samples were blocked with 10% goat serum in phosphate-buffered saline for 1 hour and then incubated with goat anti-human collagen I (Clontech; 1:50) and mouse anti-F4/80 (eBioscience, San Diego, CA; 1:50) overnight at 4°C, followed by incubation for 1 hour with Alexa Fluor 488—conjugated donkey anti-goat (1:250) and Alexa Fluor 546—conjugated goat anti-mouse (1:250) secondary antibodies, respectively. The slides were then counterstained with 4′,6-diamidino-2-phenylindole, and confocal microscopy was performed by LSM 5 Pascal (Zeiss), Macrophages associated with extracellular matrix were identified as F4/80-positive macrophage directly in contact with collagen I fibers and referred to as scar-associated macrophages. For morphometry, livers were fixed in 10% phosphate-buffered formalin for 48 hours at 4°C, washed twice with water, stored in 70% ethanol at 4°C, and embedded in paraffin. Five-micrometer sections were stained with picrosirius red (Sigma, St Louis, MO) and counterstained with fast green (Sigma). The proportion of tissue stained with picrosirius red content was quantified with MetaView software (Universal Imaging, Downingtown, PA) as described previously.19 Collagen was stained with Sirius Red and quantitated in randomly chosen sections (×20; 10 fields each from sample).
Hydroxyproline Assay
Hydroxyproline content in whole-liver specimens was quantified colorimetrically as described previously.19 Hydroxyproline concentration was calculated from a standard curve prepared with high-purity hydroxyproline (Sigma) and expressed as μg/mg liver tissue.
Monocyte-Endothelial Cell Adhesion
Monocytes were isolated from whole blood obtained from normal human volunteers as HLA-DR+CD14+/dimCD3−CD20−CD56− cells by fluorescence-activated cell sorting (Supplementary Material and Supplementary Figure 2). Human primary monocyte and endothelial (HUVEC) cell lines were used for cell-to-cell adhesion studies. Confluent HUVEC in 24-well plates were treated with control (bovine serum albumin) or VEGF (10 ng/mL) for 12 hours in 0.5% fetal bovine serum medium. HUVEC cells were washed with phosphate-buffered saline and then monocytes stained with calcein-AM were seeded as 1 × 105/well in 500 μL Dulbecco's modified Eagle medium with 10% fetal bovine serum on top of HUVEC. After 1 hour, HUVEC was washed thoroughly with phosphate-buffered saline ×3 and the adhered monocytes were measured in a spectrofluorometer (Spectrafluor; TECAN) with Delta Soft 3 software. Migration of human primary monocyte was evaluated by Boyden chamber assay in response to recombinant human VEGF. Detailed methods are in the Supplementary Material.
Statistical Analysis
Results are expressed as mean ± SE. Significance was established using the Student's t test and analysis of variance when appropriate. Differences were considered significant when P < .05.
Results
VEGF-Neutralizing Antibody Impairs Fibrosis Resolution in Vivo
We first established a murine model of fibrosis resolution by utilizing the gallbladder dilation that occurs after BDL in mice, to achieve an access to reconstruct bile flow by virtue of CJ. CJ or sham surgery was performed 2 weeks after BDL. Two weeks after CJ, the whole bile duct system was drained through the constructed anastomosis with almost complete hepatic tissue repair (Figure 1A–C). This model provides an effective surgical murine model for fibrosis resolution providing a technical advance to existing models.15,21 To evaluate the role of VEGF in fibrosis resolution, mice were treated with a neutralizing anti-mouse VEGF antibody (mcr84) or a control IgG after CJ. Contrary to our initial prediction that blockade of VEGF would enhance fibrosis resolution, we found that blockade of VEGF significantly delayed tissue repair (Figure 1D, E, and F). For gain-of-function, we administered an adenoviral vector-encoding murine VEGF into mice after BDL and CJ. Consistent with data obtained with the neutralizing antibody, forced expression of VEGF promoted tissue repair (Figure 2A and B) 1 week after virus administration. We also confirmed earlier studies6,13 that identified a fibrogenic effect of VEGF during fibrosis development by administering VEGF-neutralizing antibody for 2 weeks, commencing 1 day after BDL or sham surgery. Here anti-VEGF therapy significantly suppressed liver fibrosis as measured by Sirius Red (Figure 2C and D) and hydroxyproline content (Figure 2E). As dramatic changes were not observed in angiogenesis between the control IgG and anti-VEGF-treated groups after CJ in our fibrosis resolution analyses (Supplementary Figure 3), we turned our attention to potential effects of VEGF inhibition on permeability and inflammatory cell infiltration that occur during fibrosis resolution.
Figure 1.
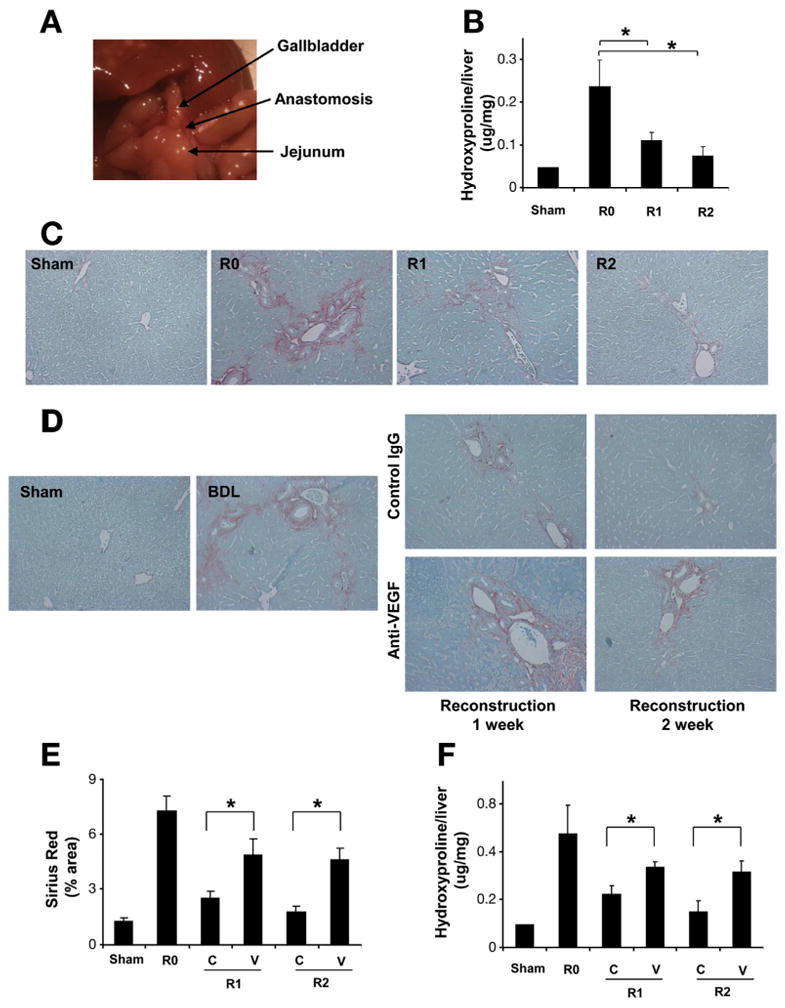
Anti-VEGF antibody disrupts fibrosis resolution. C57BL/6 mice were subjected to BDL for 2 weeks. CJ was performed to reconstruct biliary flow and induce fibrosis resolution. Reconstructed anatomy 2 weeks after CJ is shown (A). Hydroxyproline content (B) and Sirius Red staining (200×) (C) demonstrated almost complete fibrosis resolution two weeks after CJ. Mice received VEGF-neutralizing antibody or control antibody (IP ×2/week for 1 or 2 weeks), after 2 weeks of BDL followed by CJ. One week and 2 weeks after CJ, livers were harvested and subjected to analysis. Fibrosis resolution was attenuated by anti-VEGF assessed by Sirius Red staining (D), Sirius Red quantification (E), and hydroxyproline content (F). (R0: BDL 2 week without CJ; R1: BDL 2 weeks plus CJ for 1 more week; R2: BDL 2 weeks plus CJ for 2 more weeks; C: control IgG; V: anti-VEGF antibody; n = 10; *P < .05).
Figure 2.
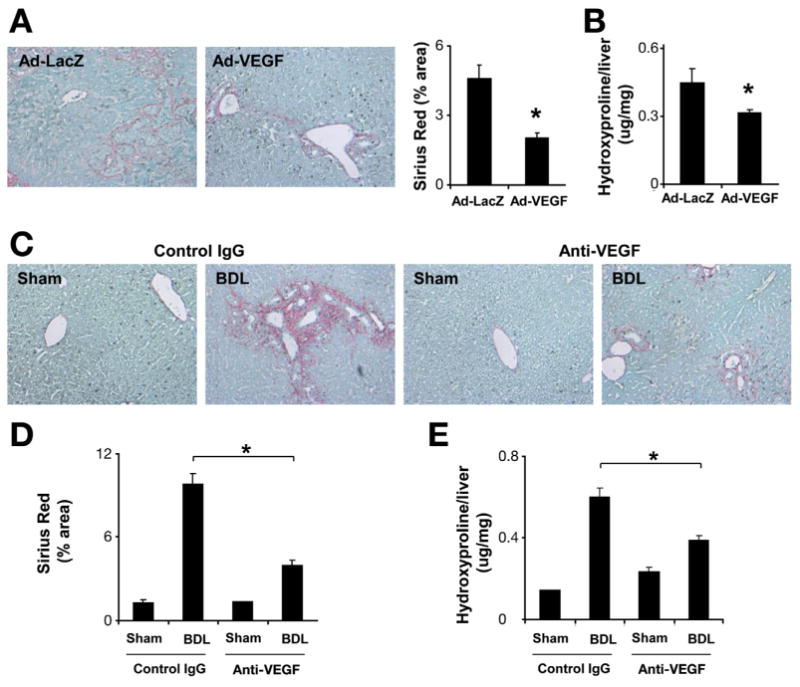
VEGF overexpression promotes fibrosis resolution. C57BL/6 mice were subjected to BDL for 2 weeks followed by CJ. One day after CJ, adenovirus-expressing mouse VEGF or LacZ (single dose 0.8 × 109 PFU/kg) was injected through tail vein injection. All animals were sacrificed 1 week after CJ. Fibrosis was assessed by Sirius Red staining (200×) (A) and hydroxyproline assay (B). VEGF overexpression was associated with enhanced fibrosis resolution (n = 7;*P < .05). C57BL/6 mice were subjected to BDL. One day after BDL, C57BL/6 mice received VEGF-neutralizing antibody or control antibody (IP ×2/week for 2 weeks). Two weeks after BDL, animals were sacrificed. Sirius Red staining (C) and quantification (D) showed decreased liver fibrosis after neutralizing anti-VEGF antibody treatment. Hydroxyproline quantification is shown as well (E) (n = 8; *P < .05).
VEGF-Neutralizing Antibody Impairs Monocyte Infiltration During Fibrosis Resolution
Fibrosis resolution is associated with inflammatory cell infiltration.12,22 We observed significant inflammatory cells within and adjacent to areas of fibrosis after BDL (Figure 3A). To further characterize effects of VEGF inhibition on inflammatory cell populations, we measured mRNA levels of macrophage and neutrophil cell surface markers; colony-stimulating factor 1 receptor (CSF1R) and the neutrophil cytosolic factor 1 (NCF1), respectively.23 Although no changes were observed in NCF1, CSF1r mRNA levels from tissue lysates were decreased after VEGF neutralization during fibrosis resolution (Figure 3B), indicating a decrease in SAM, a cell type implicated in scar fibrolysis. This finding was confirmed by double immunostaining for F4/80 and collagen to specifically identify SAM, which were also reduced in response to anti-VEGF antibody administration (Figure 3C). Similar results were observed with another macrophage marker CD68 as well (Supplementary Figure 4). Because SAM can be derived from blood monocytes,4,24 we hypothesized that VEGF-induced permeability and chemotaxis can promote monocyte adhesion to endothelium and infiltration into liver. This model was tested in vitro using the primary human monocyte and the endothelial cell line, HUVEC. VEGF stimulated monocyte migration (Figure 3D) in a Boyden chamber system by 2-fold, consistent with previous reports that VEGF promotes monocyte chemotaxis.25,26 Monocyte-endothelial cell adhesion is a key initiating event for monocyte extravasation from vasculature into tissues.27 Monocyte adhesion to VEGF-stimulated HUVEC was significantly elevated compared with vehicle (Figure 3E). These effects of VEGF on migration and adhesion of monocytes were also shown in assay with monocyte cell lines, THP-1 (Supplementary Figure 5). Finally, we modified the Mile's vascular permeability assay,20 to measure liver permeability during fibrosis resolution after anti-VEGF treatment. Three days after CJ, anti-VEGF therapy significantly reduced sinusoidal endothelial cell permeability (Figure 3F). In summary, VEGF promotes hepatic vascular permeability, monocyte-endothelial cell adhesion, and ensuing SAM accumulation in fibrotic liver during fibrosis resolution.
Figure 3.
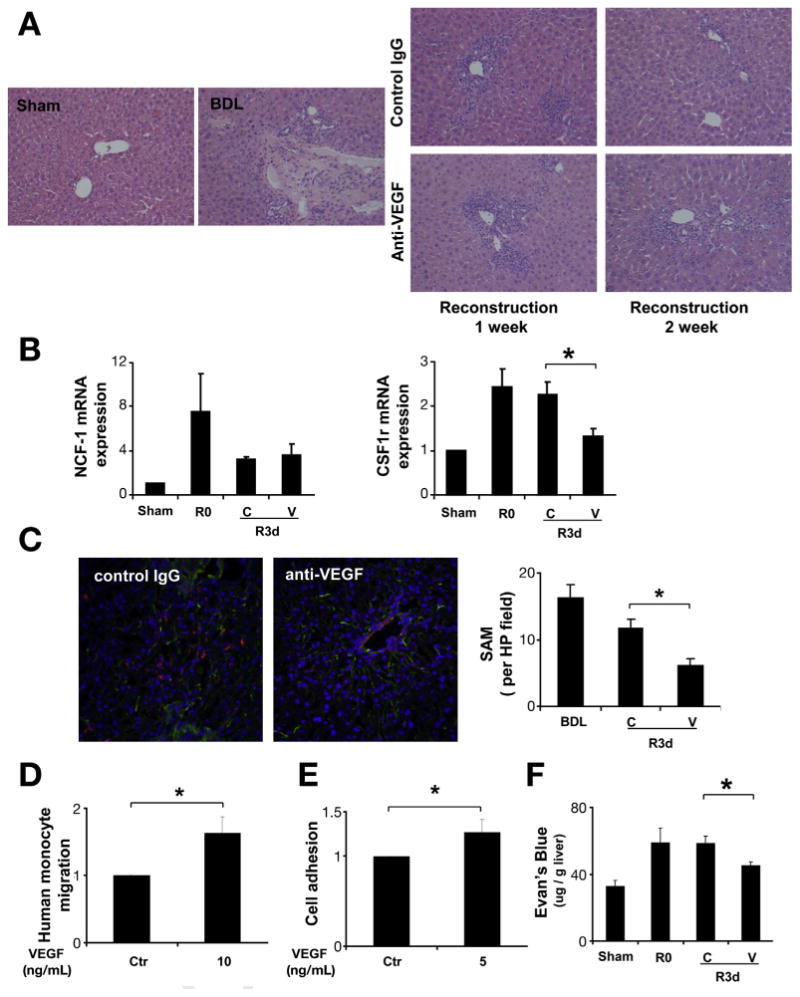
Anti-VEGF antibody decreased SAM during fibrosis resolution. C57BL/6 mice received VEGF-neutralizing antibody or control antibody (IP ×2/week for 1 or 2 weeks), after 2 weeks of BDL followed by CJ. One week and 2 weeks after CJ, livers were harvested and subjected to analysis. H&E staining (200×) showed significant inflammatory cell infiltration during scar resolution (A). Quantitative reverse transcription polymerase chain reaction from whole-liver mRNA (B) was performed to evaluate the expression level of the macrophage marker CSF1R, and the neutrophil marker NCF1. CSF1R mRNA levels were decreased after neutralizing VEGF antibody treatment during fibrosis resolution, and no change was seen in NCF1 mRNA levels. Immunofluorescence for collagen I (green) and macrophage marker F4–80 (red) were used to identify SAM in frozen section as describe in Materials and Methods (200×). VEGF neutralization significantly decreased SAM during liver fibrosis regression at day 3 (C). Cell migration was measured 3 hours later by Boyden chamber. VEGF (10 ng/mL) stimulated monocyte migration showed in (D). To evaluate the effect of VEGF on monocyte adhesion, HUVEC cell was treated with VEGF for 12 hours before co-culture with florescent dye labeled monocyte for an additional 1 hour. Cells were washed and fluorescence was measured to determine cell adhesion (E). Mice received one dose of VEGF-neutralizing antibody or control antibody after BDL + CJ. Three days later, mice received intravenous Evan's blue dye 30 minutes before sacrifice. Tissue Evans blue content in the liver (μg Evans blue/g liver tissue) was measured to assess vascular permeability (F). (R0: BDL 2 weeks without CJ; R3d: BDL 2 weeks plus CJ for 3 days; C: control IgG; V: anti-VEGF antibody; *P < .05).
SAM Are Required for Fibrosis Resolution
In earlier studies, selective depletion of macrophages showed distinct and opposing roles during liver injury and repair.4,28 To explore this in our models, MAFIA mice were subjected to sham, 2 weeks BDL and 2 weeks BDL plus CJ for 5 additional days. CSFR1-GFP–positive cell infiltration in scar tissue was measured during fibrogenesis and fibrosis resolution (Figure 4A). Monocyte and tissue macrophage depletion was confirmed by peripheral blood fluorescence-activated cell sorting analysis (Figure 4B) and in situ liver GFP and F4/80 (Figure 4C and Supplementary Figure 6). As predicted, macrophage depletion delayed liver fibrosis resolution, further supporting the concept that SAM are important for fibrosis regression after murine CJ (Figure 4D).
Figure 4.
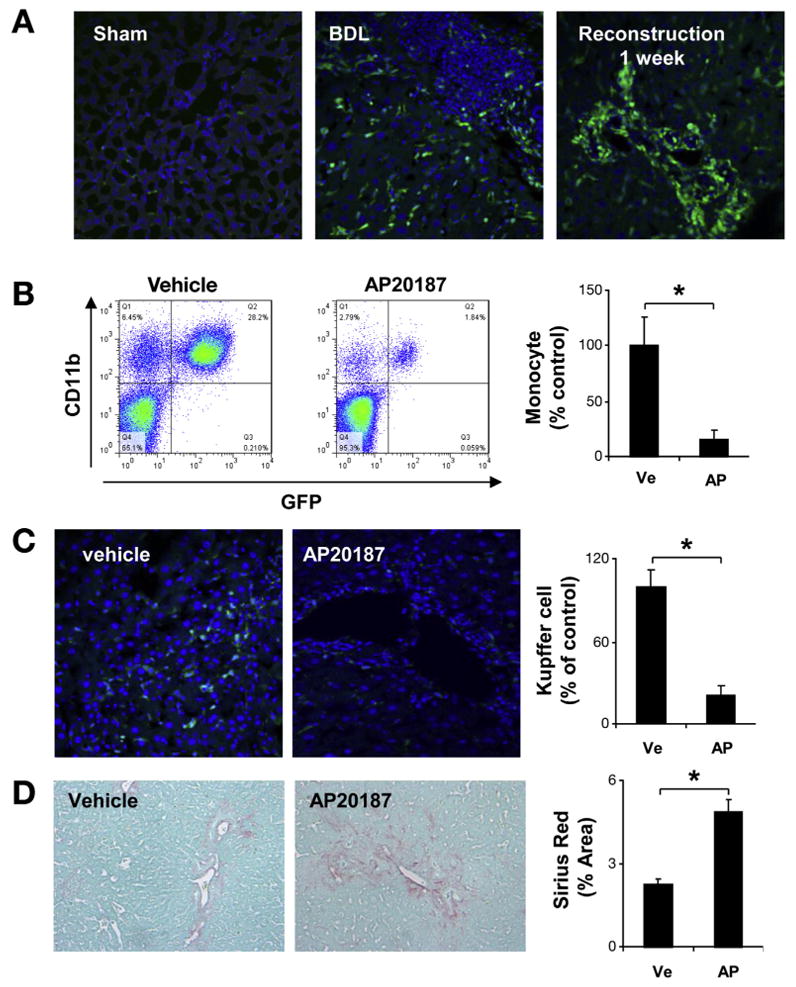
Macrophage depletion abrogates fibrosis resolution. MAFIA mice were subjected to sham, 2 weeks BDL, or 2 weeks BDL plus CJ for 5 days. In situ GFP (200×) showed CSFR1-positive cell infiltration in scar tissue during fibrosis resolution in MAFIA mice in absence of AP-20187 (A). After CJ + BDL, MAFIA mice were treated with vehicle or AP20187 daily for additional 5 days. Peripheral blood was collected and subject to fluorescence-activated cell sorting analysis. CD 11b and GFP double-positive cells were significantly decreased after AP20187 treatment compared with vehicle (B). GFP-positive cells were also significantly decreased in livers of MAFIA mice treated with AP20187 compared with vehicle (C). Sirius Red staining (D) showed attenuated fibrosis resolution (5 days after CJ) after macrophage depletion. (Ve, vehicle; AP, AP 20187; n = 7; P < .05).
VEGF Promotes CXCL9 and MMP13-Mediated Fibrosis Resolution
To investigate how VEGF neutralization affects SAM-mediated fibrosis resolution, a panel of macrophagerelated cytokines were measured by polymerase chain reaction analysis of liver tissues of mice undergoing fibrosis resolution (Supplementary Figure 7). Although a number of changes were observed, particularly notable was that VEGF neutralization abrogated CXCL9 mRNA levels during fibrosis resolution (Figure 5A). The changes in CXCL9 mRNA levels were confirmed at the protein level (Figure 5A) and therefore this target was selected for further study with the recognition that alternative targets might also be relevant. A large, although not statistically significant, change was also observed in serum (Supplementary Figure 7H). CXCL9 has been shown to ameliorate fibrogenesis.13,29 We extended these observations by evaluating mRNA levels of several MMPs, including 2, 9, and 13. From these, MMP13 mRNA levels were significantly elevated during fibrosis resolution, as reported previously,22 and VEGF neutralization significantly attenuated MMP13 elevation during fibrosis resolution (Figure 5A). After macrophage depletion in MAFIA mice, significant reductions in CXCL9 and MMP13 (Figure 5B) mRNA levels were also observed. We observed similar changes for CCL22 and interleukin-6 after VEGF neutralization or macrophage depletion (Supplementary Figures 7A and E and Figure 8A and B). In summary, VEGF neutralization and macrophage depletion attenuates CXCL9 and MMP13 production during hepatic tissue repair indicating that production of CXCL9 and MMP13 from SAM contributes to fibrosis resolution.
Figure 5.
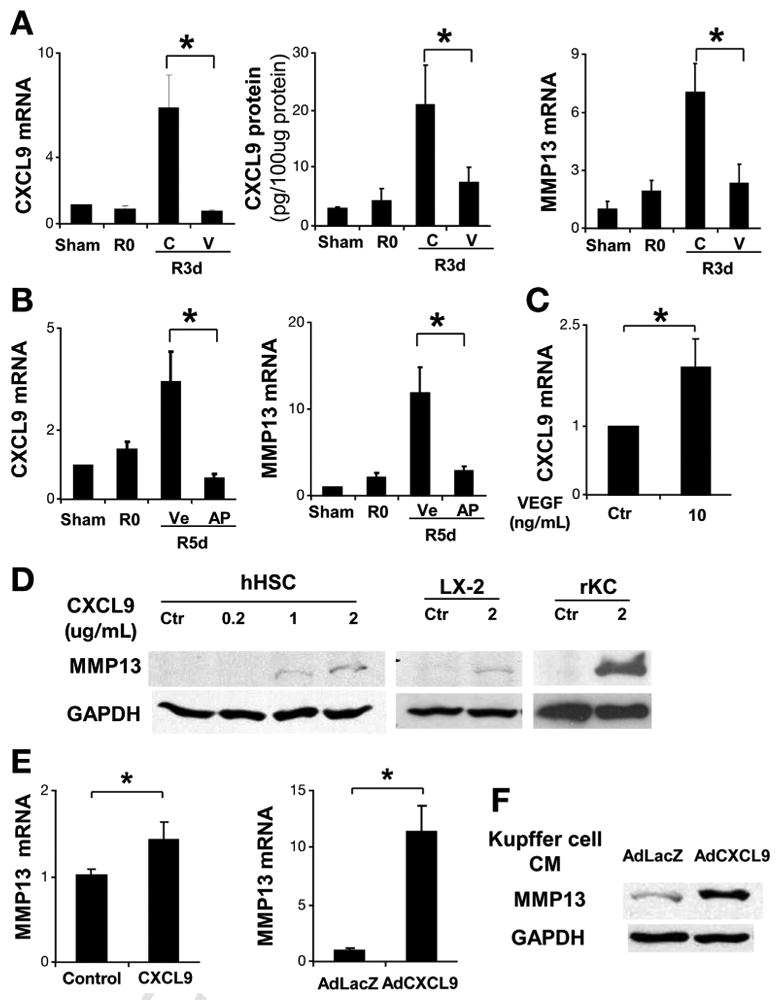
VEGF-neutralizing antibody abrogates CXCL9 and MMP13 production during fibrosis resolution. After BDL + CJ, C57BL/6 mice received 1 dose of VEGF-neutralizing antibody or control antibody. Three days later, mice were sacrificed and total liver mRNA and protein was isolated. CXCL9 mRNA and protein levels were significantly elevated during fibrosis resolution and this was abrogated by anti-VEGF antibody (A). MMP13 mRNA levels were elevated during fibrosis resolution and this was attenuated by anti-VEGF antibody (A). After CJ + BDL, MAFIA mice were treated with vehicle or AP20187 daily for additional 5 days. Total liver mRNA was subjected to real-time polymerase chain reaction for CXCL9 and MMP13. CXCL9 and MMP13 (B) elevation during fibrosis resolution was attenuated after Kupffer cell depletion with AP20187 in MAFIA mice. Fresh isolated Kupffer cells were treated with VEGF (10 ng/mL) or control for 3 hours. CXCL9 mRNA levels were elevated compared with control (C). Human HSC (hHSC) were treated with human recombinant CXCL9 for 48 hours. Total cell lysates were subjected to Western blot analysis. MMP13 increases in response to increasing concentrations of CXCL9 as shown (D). LX2 cell line was tested under same conditions with similar results (D). Freshly isolated rat Kupffer cells were treated with recombinant mouse CXCL9 for 48 hours. MMP13 production was evaluated by Western blot analysis (D). Human primary hepatic stellate cells (hHSC) were treated with recombinant CXCL9 for 48 hours after overnight serum starvation in 0.5% fetal bovine serum Dulbecco's modified Eagle medium. MMP13 mRNA levels were elevated in response to CXCL9 (E). hHSC were transfected with control Ad-LacZ or Ad-CXCL9 (multiplicity of infection = 40). After 48 hours, MMP13 mRNA expression was increased in response to Ad-CXCL9 (E). Culture-activated rat HSC was treated with Kupffer cell condition medium infected with Ad-LacZ and Ad-CXCL9. Western blot for CXCL9 was shown in (F). R0: BDL 2 weeks without CJ; R3d: BDL 2 weeks plus CJ for 3 days; R5d: BDL 2 weeks plus CJ for 5 days; C, control IgG; V, anti-VEGF antibody; Ve, vehicle; AP, AP 20187; n = ; *P <.05).
We next delineated the relationship between the VEGF, CXCL9, and MMP13 in vitro. As previously reported, Kupffer cells as resident macrophage in liver are a major source of CXCL9 during infection and inflammation.30 As VEGF-neutralizing antibody blocked CXCL9 production during fibrosis resolution in vivo, we hypothesized that VEGF can directly stimulate CXCL9 production in Kupffer cells. Recombinant VEGF increased CXCL9 mRNA levels in freshly isolated Kupffer cells (Figure 5C). Macrophages and HSC have been implicated as producers of MMP13 during fibrosis regression.3,22,27 Recombinant CXCL9 significantly up-regulated MMP13 mRNA and protein levels from Kupffer cells and HSC (Figure 5D and E). In addition, conditioned media from Kupffer cells infected with AdCXCL9 promoted MMP13 production in rat HSC (Figure 5F), suggesting an autocrine and paracrine effect of CXCL9 on MMP13 production involving Kupffer cells and HSC. These data indicate that VEGF stimulates a CXCL9/MMP13 pathway that results in macrophage-driven fibrosis regression.
VEGF Promotes CCl4-Induced Fibrosis Resolution
To further evaluate if a similar paradigm involving CXCL9 and MMP13 can be generalized to other fibrosis resolution models, we treated mice with CCl4 for 6 weeks to induce advanced liver fibrosis and allowed them to recover in the absence of CCl4 for 4 weeks. Although collagen degradation as assessed by hydroxyproline assay could not be detected after 4 weeks of recovery, fibrotic septal remodeling was detected as decreased Sirius Red staining, characterized by dissipation, widening, and splitting of septa into multiple strands of thin fibrils, in accord with earlier data20,21 (Supplementary Figure 9A and B). We evaluated CXCL9 in this alternative model and again, we found an increase in CXCL9 expression (Supplementary Figure 9C) during fibrosis resolution, although MMP13 levels were not increased in this model during the resolution phase unlike the BDL/CJ resolution model (Supplementary Figure 9D). Next, we evaluated the VEGF effect on tissue repair in a 4-week CCl4 model. Mice were treated with a neutralizing anti-mouse VEGF antibody (mcr84) or a control IgG after cessation of CCl4. Blockade of VEGF significantly delayed CCl4-induced tissue repair as assessed by Sirius Red staining (Figure 6A). Consistent with data obtained in the BLD/CJ model, forced expression of VEGF promoted fibrosis regression 2 weeks after virus administration in CCl4-induced fibrosis (Figure 6B). VEGF overexpression also promoted CSF1R, CXCL9, and MMP13 production in this model as well as NCF1 (Figure 6C).
Figure 6.
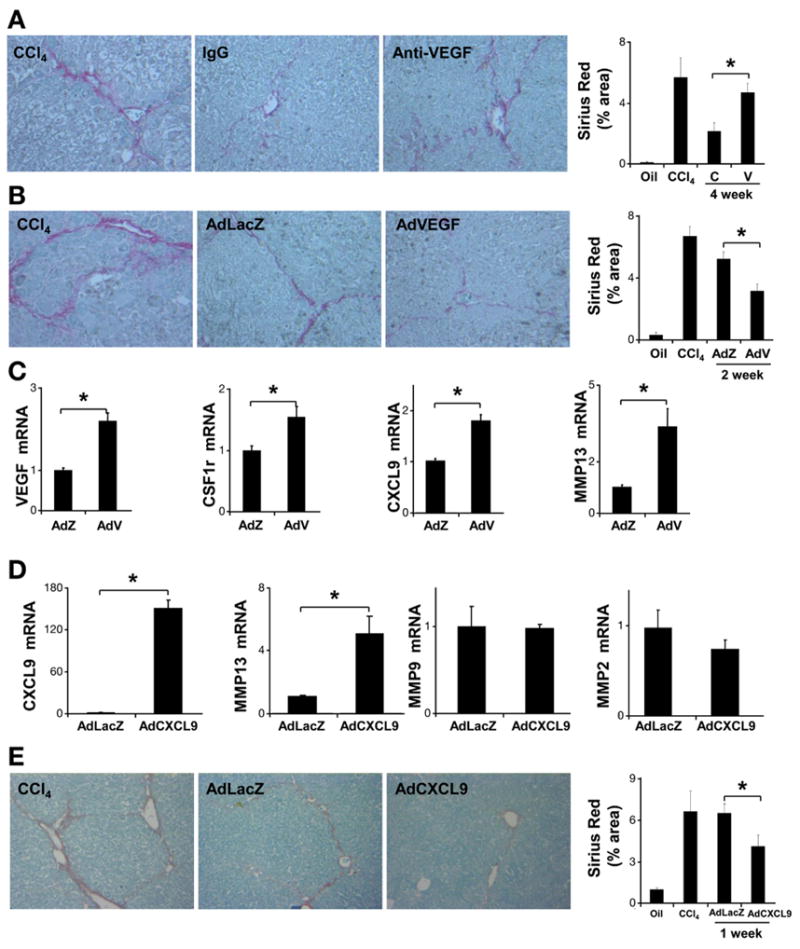
VEGF and CXCL9 modulation in resolution after CCl4-induced injury. C57BL/6 mice were treated with CCl4 for 4 weeks. One day after the final dose of CCl4, mice were administered anti-VEGF or control antibody (50 μg per injection, IP ×2/week). Mice were sacrificed 4 weeks after discontinuation of CCl4. Fibrosis resolution was attenuated by anti-VEGF assessed by Sirius Red staining (A) (n = 10, *P < .05). Mice received adenovirus-expressing mouse VEGF (AdVEGF) (0.8 × 109 PFU/kg) or LacZ through tail vein injection. Mice were sacrificed at 2 weeks after final dose of CCl4. Fibrosis was assessed using Sirius Red stain (B). VEGF overexpression was associated with enhanced fibrosis resolution (n = 10; *P < .05). Total liver mRNA was subjected to real-time polymerase chain reaction analysis to evaluate VEGF, CSF1r, NCF1, CXCL9, and MMP13 mRNA levels (C). C57BL/6 mice were treated with CCl4 for 6 weeks. One day after the final CCl4 dose, mice were injected with AdCXCL9 or AdLacZ (single dose 4 × 1010 PFU/kg through tail vein). Mice were sacrificed 1 week after discontinuation of CCl4. Total liver mRNA was subjected to real-time polymerase chain reaction analysis to evaluate CXCL9, MMP13, MMP9, and MMP2 mRNA levels (D). Liver sections were stained with Sirius Red (200×) for morphometric quantification, which showed that AdCXCL9 promotes tissue repair (E) (n = 7; *P < .05). (C, control IgG; V, anti-VEGF antibody; AdZ, AdLacZ; AdV, AdVEGF; *P < .05).
CXCL9 Overexpression Promotes CCl4-Induced Fibrosis Resolution
We next examined the mechanistic role of CXCL9 in this alternative model. AdCXCL9 or control AdLacZ virus were injected after the last dose of CCl4 and animals were sacrificed after 1 week. The dose of AdCXCL9 was predetermined in pilot studies (Supplementary Figure 10A and B). Significantly increased CXCL9 and MMP13 mRNA levels were detected in animals receiving AdCXCL9, while no changes were observed in other MMPs, such as MMP2 or MMP9 (Figure 6D). Additional evaluation of matrix dynamics using Sirius Red staining (Figure 6E) showed enhanced fibrotic tissue repair in animals receiving AdCXCL9. Similar experiments were performed using lower viral titers (1 × 1010 PFU/kg) and extended duration of recovery (2 weeks) with similar results (Supplementary Figure 11A and B). Thus, CXCL9 promotes fibrotic tissue repair after CCl4 withdrawal.
Discussion
Despite experimental progress in identifying mechanisms of fibrogenesis, progress in fibrosis regression has been more challenging. In this regard, we developed a novel murine mole of reversion of biliary fibrosis using a cholecystic biliary conduit in which fibrosis is almost entirely repaired. This technique is an important addition to currently available fibrosis resolution models,3,15,21 many of which require extended timelines for recovery, do not fully resolve fibrosis, or are not feasible in mice. We used this model to make the fundamental observation that VEGF promotes hepatic tissue repair. In addition, we identify a number of mechanisms by which this is achieved including salutary effects of VEGF on hepatic vascular permeability, monocyte infiltration into liver, and actions of fibrolytic SAM. SAM-derived CXCL9 in turn leads to extracellular matrix degradation through MMP13 production. Together, our findings indicate a dual action of VEGF on fibrosis involving the stimulation of both fibrogenesis and fibrosis resolution.
VEGF is a sentinel regulator of angiogenesis and vascular remodeling by virtue of its effects on endothelial cell survival and proliferation.8,9 This and other complementary mechanisms are proposed to mediate fibrogenic effects of VEGF during liver injury through the VEGFR2 receptor.6 Indeed, in vivo inhibition of VEGFR2 reduced fibrogenesis and portal hypertension, and VEGF overexpression increased hepatic collagen content during fibrosis development.13 We confirmed these observations by showing that an anti-VEGF antibody that selectively blocks VEGF from activating VEGFR2 but not VEGFR131 impairs BDL-induced fibrogenesis. Accordingly, we predicted that the neutralizing antibody would also accelerate hepatic tissue repair. Unexpectedly, tissue repair was impaired by VEGF inhibition, indicating that VEGF might promote fibrosis resolution. These dual effects of VEGF during fibrogenesis and fibrosis resolution are reminiscent of the dual effects of macrophages that contribute to both fibrogenesis and fibrosis resolution.4 We therefore explored the possible relationship between VEGF and monocyte lineage cells during fibrosis resolution. Indeed, VEGF is a monocyte chemoattractant and promotes vascular permeability, with both activities potentially contributing to the observed effects of VEGF on organ remodeling.12,32 This concept is supported by the present studies in which macrophage function, as well as the sinusoidal permeability that can contribute to monocyte infiltration, were significantly impaired by VEGF inhibition. Macrophages are essential, not only for injury-associated fibrogenesis but also for fibrosis resolution.4 After we genetically depleted macrophages, fibrosis regression was delayed in a manner similar to VEGF inhibition. In addition, both these interventions, VEGF blockade and macrophage depletion, were associated with impaired CXCL9 and MMP13 production.
CXCL9 is a CXCR3 ligand that reduces recruitment of Th1-regulatory T cells29,33,34 and impairs angiogenesis.13,35,36 Recent reports show that CXCL9 has additional antifibrotic effects directly on HSC.13,29 MMPs are increased during both fibrosis progression and resolution in temporal and cell-type–specific manners.3,15,37 Among various MMPs, MMP13 has received particular attention as a macrophage-derived MMP that drives fibrosis resolution.3,22,24 We therefore explored the relationship of VEGF, CXCL9, and MMP13 further in vitro. VEGF promoted CXCL9 production in Kupffer cells consistent with recent observations that CXCL9 levels were elevated in a VEGF-overexpressing transgenic mouse.13 Direct effects of VEGF on HSC were less significant in our experimental conditions (Supplementary Figure 12). The mechanism by which VEGF promotes fibrosis regression involves the CXCL9 antifibrotic pathway.
Next, to evaluate how Kupffer cell–derived CXCL9 promotes repair of fibrotic tissue, we used a combination of conditioned media and recombinant protein experiments that revealed an important role for MMP13. Our in vitro studies showed that CXCL9 stimulates MMP13 production from HSC and Kupffer cells. Autocrine and paracrine loops between macrophages and HSC involving CXCL9 and MMP13 are critical for VEGF-mediated fibrosis resolution.
To evaluate our observations in an additional fibrosis reversal model, we investigated the role of VEGF in a CCl4 withdrawal model. In this model, VEGF neutralization attenuated liver tissue repair based on Sirius Red staining. In addition, adenoviral overexpression of VEGF increased tissue repair as well as levels of CXCL9 and MMP13. Similar findings were observed with adenoviral overexpression of CXCL9 in this model. Interestingly, in the CCl4 withdrawal model, although AdVEGF and AdCXCL9 increased MMP13 levels, no basal increase in MMP13 was observed, which is in contradistinction to the BDL/CJ model. We speculate that this might be due to different mechanisms of injury and repair in hepatocyte- and cholangiocyte-based liver insult.38 However, others have noted an increase of MMP13 at early time points in CCl4 fibrosis resolution,24 and thus MMP13 induction, can be influenced by the precise temporal kinetics of CCl4 administration and withdrawal in this model, which might have varied in our study compared with earlier studies.
In summary, the data uncover a dual and opposing role of VEGF on fibrogenesis and fibrosis resolution through critical effects of VEGF on sinusoidal permeability, monocyte infiltration, and SAM function. Mechanistically, we identify a VEGF-driven CXCL9–MMP13 axis that mediates fibrosis resolution (Figure 7). In total, our findings identify new mechanisms and potential targets to promote hepatic tissue repair and fibrosis resolution.
Figure 7.
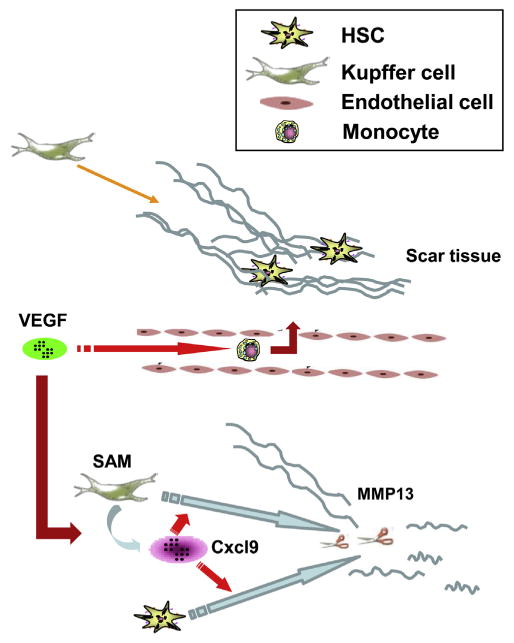
Proposed model of VEGF during liver fibrosis resolution. During fibrosis resolution, VEGF promotes monocyte infiltration and SAM accumulation in scar tissue by increasing sinusoidal endothelial cell permeability and stimulating monocyte migration. SAM in turn up-regulates CXCL9 expression. CXCL9 promotes Kupffer and activated stellate cells to produce MMP13 to remodel scar and promote fibrosis resolution.
Supplementary Material
Acknowledgments
Funding: Supported by National Institutes of Health (NIH) grants DK59015 and AA021171 (to VS); NIH 1R21A1 DK076873 and NIH U19 AI066313 (to DS).
Abbreviations used in this paper
- AdCXCL9
adenovirus expressing murine CXCL9
- AdVEGF
adenovirus expressing murine VEGF
- BDL
bile duct ligation
- CCl4
carbon tetrachloride
- CJ
cholecystojejunostomy
- CSF1R
colony-stimulating factor 1 receptor
- CXCL9
chemokine (C-X-C motif) ligand 9
- GFP
green fluorescent protein
- HSC
hepatic stellate cell
- HUVEC
human umbilical vein endothelial cell
- IP
intraperitoneally
- MAFIA
macrophage fas-induced apoptosis
- MMP13
matrix metalloproteinase 13
- mRNA
messenger RNA
- NCF1
neutrophil cytosolic factor 1
- SAM
scar-associated macrophage
- VEGF
vascular endothelial growth factor
Footnotes
Conflicts of interest: This author discloses the following: Rolf A. Brekken received research funding from Affitech AS, a company developing r84 for clinical use. The remaining authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://dx.doi.org/10.1053/j.gastro.2014.01.061.
References
Author names in bold designate shared co-first authorship.
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taura K, De Minicis S, Seki E, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–1738. doi: 10.1053/j.gastro.2008.07.065. [DOI] [PubMed] [Google Scholar]
- 6.Yoshiji H, Kuriyama S, Yoshii J, et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut. 2003;52:1347–1354. doi: 10.1136/gut.52.9.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thabut D, Routray C, Lomberk G, et al. Complementary vascular and matrix regulatory pathways underlie the beneficial mechanism of action of sorafenib in liver fibrosis. Hepatology. 2011;54:573–585. doi: 10.1002/hep.24427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 9.Clauss M. Molecular biology of the VEGF and the VEGF receptor family. Semin Thromb Hemost. 2000;26:561–569. doi: 10.1055/s-2000-13213. [DOI] [PubMed] [Google Scholar]
- 10.Senger DR, Van de Water L, Brown LF, et al. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastasis Rev. 1993;12:303–324. doi: 10.1007/BF00665960. [DOI] [PubMed] [Google Scholar]
- 11.Lee CG, Link H, Baluk P, et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10:1095–10103. doi: 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cursiefen C, Chen L, Borges LP, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahin H, Borkham-Kamphorst E, Kuppe C, et al. Chemokine Cxcl9 attenuates liver fibrosis-associated angiogenesis in mice. Hepatology. 2012;55:1610–1619. doi: 10.1002/hep.25545. [DOI] [PubMed] [Google Scholar]
- 14.Cao S, Yaqoob U, Das A, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest. 2010;120:2379–2394. doi: 10.1172/JCI41203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popov Y, Sverdlov DY, Bhaskar KR, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G323–G334. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinha S, Cao Y, Dutta S, et al. VEGF neutralizing antibody increases the therapeutic efficacy of vinorelbine for renal cell carcinoma. J Cell Mol Med. 2010;14:647–658. doi: 10.1111/j.1582-4934.2008.00578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnett SH, Kershen EJ, Zhang J, et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. 2004;75:612–623. doi: 10.1189/jlb.0903442. [DOI] [PubMed] [Google Scholar]
- 18.Chinnery HR, Carlson EC, Sun Y, et al. Bone marrow chimeras and c-fms conditional ablation (Mafia) mice reveal an essential role for resident myeloid cells in lipopolysaccharide/TLR4-induced corneal inflammation. J Immunol. 2009;182:2738–2744. doi: 10.4049/jimmunol.0803505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Chan CC, Kwon OS, et al. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 20.Melgar-Lesmes P, Tugues S, Ros J, et al. Vascular endothelial growth factor and angiopoietin-2 play a major role in the pathogenesis of vascular leakage in cirrhotic rats. Gut. 2009;58:285–292. doi: 10.1136/gut.2008.155028. [DOI] [PubMed] [Google Scholar]
- 21.Popov Y, Sverdlov DY, Sharma AK, et al. Tissue transglutaminase does not affect fibrotic matrix stability or regression of liver fibrosis in mice. Gastroenterology. 2011;140:1642–1652. doi: 10.1053/j.gastro.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 23.Sivak JM, Ostriker AC, Woolfenden A, et al. Pharmacologic uncoupling of angiogenesis and inflammation during initiation of pathological corneal neovascularization. J Biol Chem. 2011;286:44965–44975. doi: 10.1074/jbc.M111.294967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashiyama R, Inagaki Y, Hong YY, et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45:213–222. doi: 10.1002/hep.21477. [DOI] [PubMed] [Google Scholar]
- 25.Barleon B, Sozzani S, Zhou D, et al. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 26.Zhu C, Xiong Z, Chen X, et al. Soluble vascular endothelial growth factor (VEGF) receptor-1 inhibits migration of human monocytic THP-1 cells in response to VEGF. Inflammation Res. 2011;60:769–774. doi: 10.1007/s00011-011-0332-7. [DOI] [PubMed] [Google Scholar]
- 27.Popov Y, Patsenker E, Bauer M, et al. Halofuginone induces matrix metalloproteinases in rat hepatic stellate cells via activation of p38 and NFkappaB. J Biol Chem. 2006;281:15090–15098. doi: 10.1074/jbc.M600030200. [DOI] [PubMed] [Google Scholar]
- 28.Gao B, Seki E, Brenner DA, et al. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G225. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wasmuth HE, Lammert F, Zaldivar MM, et al. Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology. 2009;137:309–319. 319 e1–e3. doi: 10.1053/j.gastro.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee WY, Moriarty TJ, Wong CH, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol. 2010;11:295–302. doi: 10.1038/ni.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan LA, Carbon JG, Roland CL, et al. r84, a novel therapeutic antibody against mouse and human VEGF with potent anti-tumor activity and limited toxicity induction. PLoS One. 2010;5:e12031. doi: 10.1371/journal.pone.0012031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May D, Djonov V, Zamir G, et al. A transgenic model for conditional induction and rescue of portal hypertension reveals a role of VEGF-mediated regulation of sinusoidal fenestrations. PloS One. 2011;6:e21478. doi: 10.1371/journal.pone.0021478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santodomingo-Garzon T, Han J, Le T, et al. Natural killer T cells regulate the homing of chemokine CXC receptor 3-positive regulatory T cells to the liver in mice. Hepatology. 2009;49:1267–1276. doi: 10.1002/hep.22761. [DOI] [PubMed] [Google Scholar]
- 34.Erhardt A, Wegscheid C, Claass B, et al. CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J Immunol. 2011;186:5284–5293. doi: 10.4049/jimmunol.1003750. [DOI] [PubMed] [Google Scholar]
- 35.Strieter RM, Burdick MD, Gomperts BN, et al. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16:593–609. doi: 10.1016/j.cytogfr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Campanella GS, Colvin RA, Luster AD. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PloS one. 2010;5:e12700. doi: 10.1371/journal.pone.0012700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim Biophys Acta. 2013;1832:876–883. doi: 10.1016/j.bbadis.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Iwaisako K, Brenner DA, Kisseleva T. What's new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J Gastroenterol Hepatol. 2012;27(Suppl 2):65–68. doi: 10.1111/j.1440-1746.2011.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.