

Abstract
Background
Sudden cardiac death often involves arrhythmias triggered by metabolic stress. Loss of mitochondrial function is thought to contribute to the arrhythmogenic substrate, but how mitochondria contribute to uncoordinated electrical activity is poorly understood. It has been proposed that the formation of “metabolic current sinks”, caused by the non-uniform collapse of mitochondrial inner-membrane potential (ΔΨm), contributes to reentrant arrhythmias because ΔΨm depolarization is tightly coupled to the activation of sarcolemmal ATP-sensitive K+ (KATP) channels, hastening action potential repolarization and shortening the refractory period.
Methods and Results
Here we use computational and experimental methods to investigate how ΔΨm instability can induce reentrant arrhythmias. We develop the first tissue level model of cardiac electrical propagation incorporating cellular electrophysiology, excitation-contraction coupling, mitochondrial energetics and reactive oxygen species (ROS) balance. Simulations show that reentry and fibrillation can be initiated by regional ΔΨm loss, due to the disparity of refractory periods inside and outside of the metabolic sink. Computational results are compared with the effects of a metabolic sink generated experimentally by local perfusion of a mitochondrial uncoupler in a monolayer of cardiac myocytes.
Conclusions
The results demonstrate that regional mitochondrial depolarization triggered by oxidative stress activates sarcolemmal KATP currents to form a metabolic sink. Consequent shortening of the action potential inside, but not outside, the sink increases the propensity for reentry. ΔΨm recovery during pacing can lead to novel mechanisms of ectopic activation. The findings highlight the importance of mitochondria as potential therapeutic targets for sudden death associated with cardiovascular disease.
Keywords: arrhythmia (mechanisms), ATP-sensitive potassium channel, mitochondria, reactive oxygen species, reentry
Introduction
Whether the index event is acute coronary occlusion or is associated with chronic progressive heart disease, sudden cardiac death usually involves a paroxysmal, unpredictable event that precipitates ventricular arrhythmias. Among potential mechanisms implicated in promoting electrical instability, dispersion of refractoriness is thought to be a major factor in the susceptibility to reentry and fibrillation. Regional heterogeneity of the effective refractory period (ERP) of the tissue depends on both differences in the action potential duration (APD) of the individual cardiomyocytes and the conduction velocity. In non-ischemic cardiac tissue, unidirectional block occurs when the repolarization gradient exceeds ~3.2msec/mm1,2 and a similar degree of dispersion has been reported to increase the vulnerability to ventricular tachyarrhythmia in intact hearts3. Dispersion of repolarization is also a prominent feature of cardiac ischemia and reperfusion4, concurrent with an increase in the spatiotemporal heterogeneity of ΔΨm5,6. Notably, the timing of ΔΨm depolarization during reperfusion7 and ventricular fibrillation (VF) coincide8.
The mechanisms linking mitochondrial dysfunction to electrical instability in the heart are incompletely understood; however, a large body of evidence indicates that KATP channels are rapidly activated upon energy depletion to cause APD shortening and the concomitant elevation of the ST-segment of the electrocardiogram9. Pharmacological inhibition of sarcolemmal KATP channels blunts APD shortening during ischemia8 and, interestingly, KATP antagonists can also prevent arrhythmias elicited by reperfusion10 or β-adrenergic stress11. In addition, gain12,13 or loss of function14 mutations in atrial KATP channel subunits have been associated with arrhythmias in humans.
The cellular events occurring upstream of KATP channel activation, i.e., the mechanisms that cause an abrupt decrease in energy supply, are crucial to understanding how metabolic stress leads to arrhythmias. In this light, mitochondrial ROS-induced ROS release (RIRR)15 has emerged as a key event that underlies ΔΨm depolarization in cardiac cells. RIRR refers to the autocatalytic production of a burst of ROS by the mitochondrial electron transport chain when mitochondria are subjected to exogenous sources of ROS, or when the antioxidant defenses of the mitochondrion or the cell are compromised16. This phenomenon can be synchronized across the entire mitochondrial network of the cardiomyocyte in a process that depends on local ROS diffusion17 and appears as a propagated ΔΨm depolarization wave17, or as sustained self-organized slow oscillations of ΔΨm (period≈100seconds)18,19. The rapid depolarization of ΔΨm transforms the mitochondria into consumers, rather than producers, of ATP20, causing a drop in the cellular ATP/ADP ratio, activating KATP channels and shortening the APD.
The goal of the present study was to investigate how mitochondrial functional instability alters the dynamics of the electrophysiological substrate to increase the vulnerability to arrhythmias. We utilize a model of the myocardial syncytium that incorporates a ventricular cardiomyocyte model of excitation-contraction coupling, mitochondrial energetics, and RIRR (ECME-RIRR)21 to examine how regional oxidative stress initiates a chain of events that leads to reentry, through the formation of a metabolic current sink. The simulation results are supported by experiments in which a metabolic sink is created in a cardiac cell monolayer. Together, the findings lend credence to the hypothesis that spatiotemporal metabolic instability underlies electrical instability during oxidative stress in the heart.
Methods
Model development
The cardiomyocyte model used in this study was the ECME-RIRR21 model, which is based on experimental observations of oxidative stress-induced metabolic oscillations in intact guinea pig cardiomyocytes, in which ΔΨm depolarization is triggered by a ROS-activated inner membrane anion channel18,19. The general scheme of the ECME-RIRR model is shown in Supplemental Fig. 1. To investigate the effects of heterogeneous mitochondrial energetics on cardiac electrical propagation and ventricular arrhythmias at the tissue level, the ECME-RIRR cardiomyocyte model was incorporated into a two-dimensional (2D) finite element model of ventricular tissue (5x5cm2). ECME-RIRR model parameters were identical to those in ref.17 unless indicated otherwise. Electrical activity in the myocardial sheet was described by the monodomain equation22. No-flux conditions on membrane voltage (Vm) were implemented at the model boundaries.
Numerical aspects
The monodomain equation (a partial differential equation, PDE) was discretized at 200μm spatial resolution. Temporal discretization relied on an operator splitting scheme23, whereby a forward Euler method was employed to solve the PDE, and a custom-tailored integration technique22 was used to solve the ordinary differential equations (ODEs) of the ECME-RIRR model. As ECME-RIRR contains both fast (e.g., Ca2+ handling) and slow (e.g, mitochondrial TCA cycle) responses, the numerical scheme used different time steps to integrate the PDE and ODEs. Specifically, the PDE was integrated using a time step of 20μs and the set of ODEs was split into groups of variables that operated at similar time scales so that appropriate time steps could be chosen for each group. This numerical scheme leads to a substantial reduction in execution time22.
Simulation protocol
The model simulation protocol is described in detail in the Supplemental Material. Briefly, we first simulated the effect of a metabolic current sink formed by regional mitochondrial depolarization (Fig. 1A) on electrical wave propagation in the absence or presence of a single-pulse premature stimulus, S2. We then investigated the effect of recovery of ΔΨm in the metabolic sink on electrical wave propagation during the repolarization phase of the mitochondrial oscillations, in the absence of extrastimuli. The effect of lag time between the electrical stimulus and the recovery of mitochondrial energetics (ES-MElag) on the formation of erratic electrical activity was also analyzed.
Figure 1.

Impact of ROS-induced regional mitochondrial depolarization on metabolic sink formation and electrical wave propagation in a 2D tissue model. (A) Regional ΔΨm depolarization in the central region occurs spontaneously when fractional ROS generation by the electron transport chain is increased (ROS shunt increased from the nominal level of 2% to 14% in the central zone); (B) Effects of changing KATP channel density (0, 0.8, 1.8 and 3.8/μm2) throughout the model on the AP at the center of the metabolic sink (action potentials outside of the sink were not affected); (C) Propagation of the electrical wave through the metabolic sink at 90ms after the S1 stimulus (1Hz stimulus applied at lower left corner); and (D) sodium channel availability (jNa; inactivation gate parameter) with different KATP channel densities. 2D tissue model size: 5x5cm2, 63,000 nodes; Sink zone radius, r=1cm; Tissue conductivity: 0.1S/m. ECME-RIRR model parameters were otherwise identical to those in ref.17.
Experimental protocol
Neonatal rat ventricular myocytes (NRVM) were isolated from ventricles of 2-day-old neonatal Sprague-Dawley rats (Harlan Laboratories), as previously described24. The procedure conformed to the protocols in the National Institutes of Health, Guide for the Care and Use of Animals (NIH publication No. 85-23, Revised 1996). Cells were resuspended in Medium 199 (Invitrogen) and supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen). After two steps of preplating, 850,000 cells were plated on each of plastic coverslips (D=2.1cm) coated with fibronectin (25μg/mL). After one day, serum was reduced to 2%. Monolayers were used for experiments after 5–7 days in culture.
To observe changes in sarcolemmal membrane potential, monolayers were loaded with 5μM di-4-ANEPPS (Invitrogen) for 15min, placed in the optical mapping setup, and continuously superfused with Tyrode’s solution consisting of (in mM) 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 5 HEPES, and 5 glucose. To create a metabolic sink, a custom-made local perfusion device25 was used to expose the center part of the monolayer to Tyrode’s solution containing the mitochondrial uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 1μM; Sigma-Aldrich); the rest of the monolayer received only normal Tyrode’s solution. FCCP was intentionally used as a tool to generate a metabolic sink without inducing extensive oxidative modifications of ion channels and transporters so that experimental and model conditions were comparable. Due to the shorter action potential (AP) of NRVM as compared to the AP model, the sizes of the monolayer and central sink region in the experiments were smaller, so that spatial relationships in the experiment corresponded to those in the simulations. Changes in the membrane voltage were recorded with a 464-element photodiode array (WuTech) and analyzed using software written in LabVIEW (Texas Instruments) and MATLAB (MathWorks). Glibenclamide, a selective blocker of KATP channels, was used at a concentration of 10μM. Experiments were performed at 37°C.
Regional ΔΨm depolarization by local FCCP perfusion was recorded using the potentiometric fluorescent indicator tetramethylrhodamine methyl ester (TMRM, 2μM) in the dequench mode26. After TMRM was loaded for 1hour, the monolayer was imaged from above by means of a cooled CCD camera (MicroMax 1300Y; Princeton Instruments).
Results
Regional ΔΨm depolarization forms a metabolic sink
Our first objective was to determine, using modeling studies, whether regional mitochondrial depolarization can form a metabolic current sink and, if so, how the sink affects electrical wave propagation. As expected from prior single cell simulations21, increasing the fraction of ROS production in the mitochondria of myocytes in the central region resulted in abrupt spontaneous ΔΨm depolarization confined to that region (Fig. 1A). The effect of regional mitochondrial depolarization on electrical wave propagation was highly dependent on the density of KATP channels (σKATP). In the absence of KATP current (IK,ATP), regional mitochondrial depolarization had no effect on the AP or electrical wave propagation in this simulation. With moderate KATP channel density (e.g., σKATP=0.8 or 1.8/μm2), the collapse of ΔΨm caused significant shortening of APD and reduction of AP amplitude (APA) in the central region (Fig. 1B). The effect on wave propagation was to dramatically shorten the wavelength in the central zone, with little effect on the wavefront (Fig. 1C). When σKATP was increased to 3.8/μm2, APD decreased dramatically and APA was further reduced (Fig. 1B), resulting in a very thin propagating wave (Fig. 1C) and a very short refractory period within the sink, as demonstrated by the recovery of the j-gate of the Na channel (Fig. 1D). In fact, the Na+ channels in the metabolic sink recovered from inactivation even before the wave outside the sink had passed through the region, making the sink vulnerable to early re-excitation. The voltage gradient at the border of the sink could, in theory, have provided a source of re-excitation of the metabolic sink; however, in this simulation, the current was not sufficient to reach the threshold for re-excitation. Increasing σKATP to values higher than 3.8/μm2 resulted in the metabolic sink becoming completely inexcitable, owing to a very high threshold for excitation conferred by the high background K+ conductance. Changing KATP channel density alone had no effect on ΔΨm.
Regional mitochondrial depolarization forms a substrate for arrhythmias
The susceptibility to reentry in the metabolic current sink model was explored by applying a premature S2 at or near the border of the central zone (1.8/μm2≥σKATP≥3.8/μm2). Our simulations showed that there was an S1–S2 coupling interval window (~150–205ms) within which S2 induced reentrant activity. For example, as shown in Fig. 2A (σKATP=3.8/μm2; see also Movie 1), when the S1–S2 interval was 170ms, the S2-induced excitation propagated immediately into the metabolic sink as a thin wave that broke up as it emerged from the sink region, spiraling back on itself to reenter the sink and spawning multiple wavelets at several points near the border. This fibrillatory activity was sustained for 600ms before dying out, terminating as a result of the no-flux boundary conditions of the model. The role of the metabolic current sink in establishing fibrillation propensity was supported by the fact that the phase singularities sustaining the turbulent electrical behavior arose initially at the border of the central zone (Fig. 2B and Movie 2). Similar behavior was observed for larger sizes of the metabolic sink; however, reentry did not take place for a metabolic sink of smaller radius, e.g. 0.5cm, since by the time the S2-elicited wave propagated through the sink, the surrounding normal tissue was still refractory after the S1 propagation.
Figure 2.

Sustained fibrillation in the tissue model (1Hz-pacing) evoked by a second stimulus (S2) near the border of the metabolic sink (A). The lower panel (B) shows maps of AP phase with the calculated phase singularities (magenta dots). The S1-S2 time interval is 170ms. σKATP=3.8/μm^2, r=1cm.
Recovery of mitochondrial energetics induces spontaneous arrhythmias
Next, we conducted simulations to investigate the effect of recovery of ΔΨm during the repolarization phase of the mitochondrial oscillations on electrical activity in the paced tissue (1.8/μm2≥σKATP≥3.8/μm2). Recovery of ΔΨm in the metabolic sink always resulted in spontaneous wavefront generation from the back of the S1 wave, which we will call waveback breakthrough, which propagated as a rebound wave through the metabolic sink. This rebound wave affected the electrical activity in the tissue in a sink size-dependent fashion. As shown in Fig. 3 (σKATP=3.8/μm2), when the sink size was relatively small (r=0.5cm), the wave was confined within the sink zone without breaking out (Fig. 3A, upper panel). When the sink was larger (r=1cm), the rebound wave propagated through the sink and entered normal tissue, forming spiral waves (Fig. 3A, middle panel, and Movie 3). A further increase in sink size (r=2cm) resulted in wavebreaks and turbulent electrical activity (Fig. 3A, lower panel and Movie 4). The relationship between sink size and the induction of arrhythmias for these σKATP values is summarized in Fig. 3B.
Figure 3.

Effect of recovery of ΔΨm on electrical propagation. (A) ΔΨm repolarization in the metabolic sink induced either a rebound excitation contained within the sink, reentry, or fibrillation in the 2D tissue model, depending on the sink size (r=0.5, 1 and 2cm, respectively); (B) Scheme of the effect of sink size on the genesis of abnormal electrical activity. σKATP=3.8/μm^2.
Coupling between mitochondrial energetics and cellular electrical activity in the model is primarily through the ATP-sensitive potassium channel, hence, we examined the behavior of IK,ATP during recovery of the metabolic sink to better understand the mechanism of waveback breakthrough. As shown in Fig. 4A for the 2cm radius case, when the S1 wave, initiated from the left-lower corner of the model, reached the edge of the sink (at 58ms, point a, top row), mitochondria within the sink were partially repolarized (ΔΨm=37mV, Fig. 4B), but IK,ATP was still activated, at 22μA/cm2 (the spatial distribution of IK,ATP in the sheet at the same instant of time is presented in Fig. 4A middle row, while the temporal traces of IK,ATP at points a–d are depicted in Fig. 4C). While the S1 wave propagated through the sink zone, mitochondria continued to recover (Fig. 4A middle row), resulting in a diminution of IK,ATP. At 132ms, when ΔΨm had recovered to 90% of its maximal value (121mV), IK,ATP was almost completely inactivated (2.4μA/cm2 at point c in Fig. 4A, and Fig. 4C). The rapid inactivation of KATP current reversed the current dissipation by the sink, thus lowering the threshold for re-excitation. The combination of short ERP and lowered threshold for firing permitted waveback breakthrough of activation (point d), leading to a rebound wave.
Figure 4.

Electrical wave, ΔΨm recovery, and IK,ATP during waveback breakthrough. (A) Propagation of the electrical wave (upper panel), recovery of ΔΨm (middle panel), and KATP current (lower panel). (B) The time course of ΔΨm recovery at the sink center. (C) Inactivation of KATP currents accompanying mitochondrial repolarization. σKATP=3.8/μm^2, r=2cm.
To dissect the spatiotemporal determinants of the aberrant electrical behavior induced by ΔΨm changes, we examined how the timing of metabolic sink recovery (relative to S1) affects the induction of spontaneous arrhythmias. Fig. 5A shows the energetic state of the sink when the wavefront reaches the edge of the sink (58ms) for various intervals between electrical stimulation and mitochondrial recovery, ES-MElag, while Fig. 5B shows the time course of ΔΨm repolarization at the center of the sink. We found that when the lag was too large, e.g., 154ms, or too small, e.g., 23ms, no wavebreak or fibrillation was observed (Fig. 5C). When the lag was in the range of 45 to 150ms, irregular electrical activity was induced (Fig. 5C). Particularly, two types of “arrhythmias” were observed: rebound waves which exited the sink when the lag was between 45 and 103ms, and fibrillation when the lag was between 103 and 150ms. This relationship is summarized in Fig. 5D.
Figure 5.

Effect of electrical stimulus-mitochondrial energetics lag time (ES-MElag) on reentry induced by the metabolic sink. (A) ΔΨm distribution at the time when S1 reaches the edge of the metabolic sink as ES-MElag is varied. (B) Dynamics of ΔΨm recovery at the metabolic sink center with different lag times. (C) Recovery of ΔΨm induces rebound propagation or fibrillation depending on ES-MElag. (D) Summary of the effect of lag times on the induction of arrhythmias. σKATP=3.8/μm2, r=2cm.
Spontaneous arrhythmias due to metabolic recovery did not always originate directly from the waveback. In simulations where the conductivity in the central zone was decreased to reflect the possible gap junction sensitivity to the level of ATP, the stimulus-induced wavefront could not penetrate the sink zone because additional excitatory current was required, as shown in Fig. 6 (σKATP=3.8/μm2 and r=2cm, sink zone conductivity 0.03S/m). Instead, rebound waves were initiated at the edge of the sink at sites of high transmembrane voltage gradient when the excitation threshold decreased in the sink during mitochondrial recovery. In this example, conduction velocity was lower in the sink as compared to that in Fig. 3; therefore, the rebound wave took longer to propagate through it, allowing for the surrounding normal tissue to recover from the S1 wave, resulting in reentry even though the radius was smaller than that shown to be resistant in Fig. 3B. Thus, a decrease in gap junctional conductance, which was incorporated into the model to reproduce the magnitude of wavefront slowing observed in the experiments (Fig. 7B), also has the effect of decreasing the minimum size of the metabolic sink supporting reentry.
Figure 6.
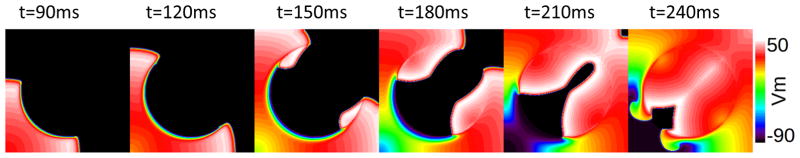
Induction of reentry during recovery of ΔΨm under the conditions of reduced tissue conductivity (gap junction uncoupling) in the metabolic sink (conductivity decreased from 0.1S/m in normal tissue to 0.03S/m in the central region). σKATP=3.8/μm2, r=2cm. Similar behavior can be achieved with tissue conductivities in the interval 0.1–0.03S/m and σKATP above 3.8/μm2.
Figure 7.

Effect of regional ΔΨm depolarization on electrical propagation in cardiac cell (NRVM) monolayers. (A) Regional ΔΨm depolarization induced by perfusion in a circular zone (D=0.5cm) with FCCP (1μM). (B) Top: Loss of ΔΨm decreases APA and APD, resulting in wavefront slowing and a shorter wavelength within the metabolic sink. Bottom: Simulations that closely match the experimental results (σKATP=0.6/μm2, sink zone tissue conductivity 0.03S/m). (C, D) Changes in optically measured AP amplitude and duration in the sink with or without glibenclamide (10μM), which prevented rapid APD shortening and blunted the APA decrease. Perfusion with FCCP starts at time 0. (E) Sustained reentry during perfusion with FCCP. (F) Transient reentry during washout of FCCP. Dashed circles in panels E and F show the border of the metabolic sink. Bipolar point stimulation: 1Hz in B-D, 4Hz in E,F; frame rate: 50Hz in B, 33Hz in E, 25Hz in F.
Regional ΔΨm depolarization induces abnormal electrical activity in NVRM monolayers
Local perfusion25 of the center part (D=0.5cm) of the monolayer with FCCP induced regional ΔΨm depolarization (Fig. 7A), and resulted in a fast decrease in both APA and APD50 (Fig. 7C and Fig. 7D; Supplemental Fig. 2). As a consequence of ΔΨm depolarization, wavelength shortened, and wave slowing was also observed (Fig. 7B-top). Simulations including both KATP–dependent coupling and decreased gap junctional conductance closely matched these results (Fig. 7B-bottom). The heterogeneity of refractoriness and slower conduction observed in the NRVM monolayers after induction of the metabolic sink occasionally led to reentry. Movie 5 and Fig. 7E show such an event in a monolayer paced at 4Hz. Spiral waves continued for several minutes after pacing was turned off. Also, heterogeneity in refractoriness and conduction due to reperfusion of the metabolic sink with normal physiological solution to restore ΔΨm caused transient or sustained reentry. Fig. 7F shows formation of a transient reentrant wave in a monolayer as the sink started to regain excitability upon FCCP washout. Overall, reentry occurred in 6 of 10 monolayers paced at 1–4Hz (4 monolayers during FCCP perfusion and 4 during washout).
KATP channels played a key role in the electrophysiological changes induced by the metabolic sink. Glibenclamide (10μM) blunted the APA decrease and largely prevented the APD shortening (Fig. 7C and Fig. 7D). Consequently, in contrast to FCCP perfusion alone, glibenclamide preserved excitability of the sink area. Transient reentry occurred for 1–2 minutes in only 2 out of 6 monolayers (1 monolayer during FCCP perfusion and 1 during washout). The effect of glibenclamide was even more pronounced at room temperature (Supplemental Fig. 3).
To study the effect of sink size on the occurrence of reentry, in another set of experiments (21 monolayers), a larger area (D=1.2cm) of the NRVM monolayer was perfused with FCCP and thus a larger metabolic sink was induced. In this case, reentry occurred during FCCP perfusion in response to an S2 stimulus (S1–S2=200ms) on the border of the large sink in 3 out of 3 monolayers tested, while extrastimuli at the border of the smaller sink did not cause reentry in any monolayer. In the absence of an S2 stimulus, spontaneous activity was observed to be initiated close to the edge of the sink and led to reentry in 2 of 18 monolayers even at the low pacing rate of 1Hz (Movie 6).
Discussion
In this study, 2D simulations and experiments in cardiac cell monolayers demonstrated that the formation of a metabolic sink, induced by regional mitochondrial depolarization, profoundly affects electrical activity in the tissue. The overall influence of energetic collapse is to decrease APA and APD, decreasing wavelength and introducing regions of very short refractory period that facilitate reentry. In both computational and experimental models, premature beats at locations surrounding the metabolic sink resulted in spiral wave reentry and fibrillation in a sink size-dependent manner. Furthermore, our simulations showed that metabolic recovery could result in spontaneous electrical instability through a novel mechanism involving waveback breakthrough, depending on the size of the metabolic sink and the timing of ΔΨm recovery with respect to wavefront arrival.
Sarcolemmal KATP channels are activated by a decrease in cytosolic ATP and mediate a weakly inwardly rectifying background K+ current27,28. Mitochondrial ΔΨm depolarization can potentiate KATP channel opening because uncoupling of oxidative phosphorylation results in a reversal of the mitochondrial ATP synthase, accelerating the depletion of intracellular ATP. Increased background K+ conductance through KATP channels pulls the resting membrane potential (Em) close to the equilibrium potential for K+ (EK). After 10min of ischemia, resting membrane potential is, in fact, equal to EK29. KATP current activation accounts for most of the AP shortening during the early phase of ischemia, as evidenced by the ability of KATP channel inhibitors such as glibenclamide to prevent shortening of the AP duration over the first 10min of ischemia8. Concomitant with AP shortening, there is a monotonic decrease in APA and upstroke velocity during the first 10min of ischemia, which can be partially prevented by glibenclamide treatment8, suggesting that KATP channels may contribute to both the AP shortening and early conduction slowing in the ischemic heart. Our experiments supported this idea by showing that blocking sarcolemmal KATP channels blunted the loss of APA and APD during mitochondrial depolarization and lowered the chance of reentry in monolayers of cardiomyocytes. The effect of glibenclamide was even more evident at room temperature (Supplemental Fig. 3), likely due to temperature-dependent alterations in metabolic flux. In the intact heart, with longer durations of mitochondrial dysfunction, however, other mechanisms besides KATP activation come into play, including a further decrease in conduction velocity, gap junctional uncoupling, catecholamine release (at ~15–20min), increases in Ca2+i (2–3 fold) and Mg2+ (>3 fold), and a second phase of ATP decline.
The present simulations focused primarily on how KATP channel activation by mitochondrial depolarization alters the excitable substrate and can lead to arrhythmias. The model was designed to represent the effects of mitochondrial dysfunction during periods of high oxidative stress, such as reperfusion. Previously, we showed that in guinea pig hearts, reperfusion after 30minutes of global ischemia evokes reentrant arrhythmias, and that treatment with a compound that prevents or reverses RIRR-mediated mitochondrial depolarization (4′-chlorodiazepam) eliminates post-ischemic VF and improves AP recovery8. Similarly, we have shown that significant spatiotemporal heterogeneity of ΔΨm is present during ischemia-reperfusion in isolated hearts5,6. Moreover, we have demonstrated that in normoxic hearts exposed to oxidative stress (reduced glutathione depletion with diamide), an increase in the incidence of ventricular arrhythmias occurs in parallel with heterogeneous ΔΨm loss in clusters of myocytes. The arrhythmias and the mitochondrial collapse of ΔΨm were both inhibited by 4′-chlorodiazepam30.
Because mitochondrial RIRR activates KATP current and shortens APD19, a major assumption of the metabolic sink hypothesis was that heterogeneous repolarization was responsible for the enhanced susceptibility to reentry in these studies. The computational and experimental tests described in the present work demonstrate that regional mitochondrial depolarization can indeed increase the susceptibility to reentry in response to an extrastimulus. Furthermore, simulations uncovered a novel ectopic trigger mechanism of fibrillation, occurring spontaneously during ΔΨm recovery. The latter mechanism is a model prediction that remains to be demonstrated in experimental systems or intact hearts. This will require high speed, high resolution imaging of both mitochondrial and sarcolemmal membrane voltages simultaneously. Nevertheless, in addition to more well established mechanisms of spontaneous automaticity (i.e., EAD and DAD), the mechanisms of waveback breakthrough and rebound waves could contribute to the high rate of reentrant arrhythmias observed when mitochondria are recovering and the spatiotemporal heterogeneity of ΔΨm is high during early reperfusion5.
The effect of KATP channel inhibition on ischemia-reperfusion injury and arrhythmogenesis is variable and very species-dependent. For example, in mice treated with the KATP channel blocker HMR1098, ischemia causes a much more rapid contracture of the isolated perfused heart (within 5 minutes of the onset of ischemia) compared to untreated hearts31. In this species, APs are already very short and the heart rate is extremely high (~600bpm), which could potentially explain why KATP channel activation during ischemia is essential to prevent a tetanus-like depolarization of the membrane potential under metabolic stress. In contrast, in larger animals with slower heart rates and longer AP plateau potentials (e.g. guinea pig, dogs, humans), glibenclamide treatment does not accelerate ischemic contracture, although it does prevent the protective effects of ischemic or chemical preconditioning32. In fact, KATP channel inhibition has been shown to prevent VF after acute myocardial infarction in non-insulin-dependent diabetic patients33. Furthermore, KATP channel inhibition effectively prevented arrhythmias induced by the combination of acute myocardial ischemia and exercise in dogs with healed myocardial infarctions9,10,34. Notably, the antiarrhythmic effect was observed with compounds (HMR1883 and congeners) that were designed to selectively inhibit the heart sarcolemmal KATP channel without affecting pancreatic insulin release or mitochondrial KATP channels35. Thus, consistent with the present findings, available evidence supports the arrhythmogenic role of KATP in association with ischemia-reperfusion injury in large animals. Moreover, enhanced arrhythmogenicity related to KATP activation was confirmed in a recent study in explanted failing and non-failing human hearts36.
Previous work, from the Jalife group, has emphasized how increased inward rectifier37,38, delayed rectifier39, and more recently, hERG40 K+ current expression can increase the susceptibility to reentry in structurally normal, well perfused, hearts. This arrhythmogenic effect is primarily caused by AP shortening and increased rotor frequency. Conditions of ischemia-reperfusion are an even more extreme example of an acute and dramatic increase in a weak inward rectifier background current (IK,ATP). Thus, the general principles of rotor stabilization and increased fibrillation risk apply, although many other factors also come into play, including changes in transmembrane K+, Na+, and Ca2+ gradients, pH, ATP, Mg2+, as well as tissue conductivity. In fact, as the experiments with the mitochondrial uncoupler show, mitochondrial ΔΨm collapse impacts the AP and the propagating wavefront in a manner that is more complex than expected simply from KATP channel activation, even though this mitochondrial intervention induces less oxidative stress than ischemia-reperfusion per se (data not shown). More pronounced effects on conduction velocity and APA in the experiments than in the simulations are likely to be due to metabolic regulation of targets not yet represented in the computational model. Importantly, although the FCCP-induced metabolic sink evokes less oxidative stress than other metabolic or oxidative challenges, and in that sense behaves more like the present computational model, this does not mean that multiple targets of oxidative stress would not play a role in arrhythmogenesis when ROS-induced ROS release underlies the formation of metabolic sinks in the heart. We continue to define these additional targets in ongoing studies.
Further model development will be required to more completely describe the changes in the electrophysiological substrate induced by ischemia-reperfusion. Nevertheless, by incorporating mitochondrial energetics into the tissue electrical model, the first step has been taken to begin to correlate metabolic dysfunction with downstream effects on whole heart electrophysiology. This will be vital for understanding how metabolic remodeling, which is present in metabolic syndrome, hypertrophy, heart failure, and diabetes, influences the electrophysiological substrate and potentially contributes to sudden cardiac death. Moreover, the findings underscore the importance of considering mitochondria as targets of therapeutic intervention, not only for preserving healthy cardiac muscle during stress, but also for the arrhythmias associated with cardiovascular disease.
Supplementary Material
Ischemia-reperfusion injury is known to trigger potentially fatal arrhythmias involving reentrant electrical activity, but the mechanisms leading from energetic impairment to arrhythmias are incompletely understood. Here, using a combined computational and experimental approach, we examine how regional depolarization of mitochondrial inner membrane potential (ΔΨm) can promote reentry in a cardiac cell monolayer through the formation of a “metabolic current sink.” A 2D cardiac tissue computational model revealed that oxidative stress-mediated collapse of ΔΨm, and the consequent activation of ATP-sensitive K+ channels in part of the monolayer, increases the vulnerability to reentry by increasing the dispersion of repolarization of the electrical substrate. Similar effects were observed in a cardiac cell monolayer exposed to a chemical uncoupler of mitochondrial oxidative phosphorylation. The findings implicate mitochondria as potential targets for antiarrhythmic therapy.
Acknowledgments
The authors thank Mr. Robert Blake for technical support.
Funding Sources: This work was supported by NIH grants P01-HL081427, R01HL105216, R37-HL54598 (to B.O’R.), R01HL103428, R01HL105216 (to N.T.), R00HL095648 (to L.Z.), and HL108557 (to B.M.).
Footnotes
Journal Subject Codes: [132] Arrhythmias - basic studies, [4] Acute myocardial infarction, [91] Oxidant stress, [107] Biochemistry and metabolism, [7] Chronic ischemic heart disease
Conflict of Interest Disclosures: None.
References
- 1.Laurita KR, Rosenbaum DS. Interdependence of modulated dispersion and tissue structure in the mechanism of unidirectional block. Circ Res. 2000;87:922–928. doi: 10.1161/01.res.87.10.922. [DOI] [PubMed] [Google Scholar]
- 2.Qu Z, Garfinkel A, Weiss JN. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells, 1: Single extrasystoles. Biophys J. 2006;91:793–804. doi: 10.1529/biophysj.106.080945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo CS, Munakata K, Reddy CP, Surawicz B. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation. 1983;67:1356–1367. doi: 10.1161/01.cir.67.6.1356. [DOI] [PubMed] [Google Scholar]
- 4.Levites R, Banka VS, Helfant RH. Electrophysiologic effects of coronary occlusion and reperfusion. Observations of dispersion of refractoriness and ventricular automaticity. Circulation. 1975;52:760–765. doi: 10.1161/01.cir.52.5.760. [DOI] [PubMed] [Google Scholar]
- 5.Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O’Rourke B, Akar FG. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol. 2010;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–660. doi: 10.1016/j.yjmcc.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berkich DA, Salama G, LaNoue KF. Mitochondrial membrane potentials in ischemic hearts. Arch Biochem Biophys. 2003;420:279–286. doi: 10.1016/j.abb.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 8.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Therap. 2008;120:54–70. doi: 10.1016/j.pharmthera.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Billman GE, Englert HC, Scholkens BA. HMR 1883, a novel cardioselective inhibitor of the ATP-sensitive potassium channel. Part ii: Effects on susceptibility to ventricular fibrillation induced by myocardial ischemia in conscious dogs. J Pharmacol Exp Ther. 1998;286:1465–1473. [PubMed] [Google Scholar]
- 11.Kim SJ, Zhang H, Khaliulin I, Choisy SC, Bond R, Lin H, El Haou S, Milnes JT, Hancox JC, Suleiman MS, James AF. Activation of glibenclamide-sensitive ATP-sensitive K+ channels during beta-adrenergically induced metabolic stress produces a substrate for atrial tachyarrhythmia. Circ Arrhythm Electrophysiol. 2012;5:1184–1192. doi: 10.1161/CIRCEP.112.975425. [DOI] [PubMed] [Google Scholar]
- 12.Antzelevitch C, Barajas-Martinez H. A gain-of-function I(K-ATP) mutation and its role in sudden cardiac death associated with J-wave syndromes. Heart Rhythm. 2010;7:1472–1474. doi: 10.1016/j.hrthm.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medeiros-Domingo A, Tan BH, Crotti L, Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp D, Schwartz PJ, Makielski JC, Ackerman MJ. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7:1466–1471. doi: 10.1016/j.hrthm.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tester DJ, Tan BH, Medeiros-Domingo A, Song C, Makielski JC, Ackerman MJ. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 K(ATP) channel and sudden infant death syndrome. Circulation Cardiovasc Genet. 2011;4:510–515. doi: 10.1161/CIRCGENETICS.111.960195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010;6:e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki N, Sato T, Marban E, O’Rourke B. ATP consumption by uncoupled mitochondria activates sarcolemmal K(ATP) channels in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2001;280:H1882–1888. doi: 10.1152/ajpheart.2001.280.4.H1882. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Cortassa S, Wei AC, Aon MA, Winslow RL, O’Rourke B. Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J. 2009;97:1843–1852. doi: 10.1016/j.bpj.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plank G, Zhou L, Greenstein JL, Cortassa S, Winslow RL, O’Rourke B, Trayanova NA. From mitochondrial ion channels to arrhythmias in the heart: Computational techniques to bridge the spatio-temporal scales. Philos Transact A Math Phys Eng Sci. 2008;366:3381–3409. doi: 10.1098/rsta.2008.0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu Z, Garfinkel A. An advanced algorithm for solving partial differential equation in cardiac conduction. IEEE Trans Biomed Eng. 1999;46:1166–1168. doi: 10.1109/10.784149. [DOI] [PubMed] [Google Scholar]
- 24.Abraham MR, Henrikson CA, Tung L, Chang MG, Aon M, Xue T, Li RA, BOR, Marban E. Antiarrhythmic engineering of skeletal myoblasts for cardiac transplantation. Circ Res. 2005;97:159–167. doi: 10.1161/01.RES.0000174794.22491.a0. [DOI] [PubMed] [Google Scholar]
- 25.Lin JW, Garber L, Qi YR, Chang MG, Cysyk J, Tung L. Region of slowed conduction acts as core for spiral wave reentry in cardiac cell monolayers. Am J Physiol Heart Circ Physiol. 2008;294:H58–65. doi: 10.1152/ajpheart.00631.2007. [DOI] [PubMed] [Google Scholar]
- 26.Davidson SM, Yellon D, Duchen MR. Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol Biol. 2007;372:421–430. doi: 10.1007/978-1-59745-365-3_30. [DOI] [PubMed] [Google Scholar]
- 27.Lederer WJ, Nichols CG, Smith GL. The mechanism of early contractile failure of isolated rat ventricular myocytes subjected to complete metabolic inhibition. J Physiol. 1989;413:329–349. doi: 10.1113/jphysiol.1989.sp017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 29.Kleber AG. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ Res. 1983;52:442–450. doi: 10.1161/01.res.52.4.442. [DOI] [PubMed] [Google Scholar]
- 30.Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki M, Saito T, Sato T, Tamagawa M, Miki T, Seino S, Nakaya H. Cardioprotective effect of diazoxide is mediated by activation of sarcolemmal but not mitochondrial ATP-sensitive potassium channels in mice. Circulation. 2003;107:682–685. doi: 10.1161/01.cir.0000055187.67365.81. [DOI] [PubMed] [Google Scholar]
- 32.O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–432. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomuscio A, Vergani D, Marano L, Castagnone M, Fiorentini C. Effects of glibenclamide on ventricular fibrillation in non-insulin-dependent diabetics with acute myocardial infarction. Coron Artery Dis. 1994;5:767–771. [PubMed] [Google Scholar]
- 34.Billman GE, Houle MS, Englert HC, Gogelein H. Effects of a novel cardioselective ATP-sensitive potassium channel antagonist, 1-[[5-[2-(5-chloro-o-anisamido)ethyl]-beta-methoxyethoxyphenyl]sulfonyl]-3-methylthiourea, sodium salt (HMR 1402), on susceptibility to ventricular fibrillation induced by myocardial ischemia: In vitro and in vivo studies. J Pharmacol Exp Ther. 2004;309:182–192. doi: 10.1124/jpet.103.061416. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- 36.Fedorov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, Schuessler RB, Moazami N, Nichols CG, Efimov IR. Effects of KATP channel openers diazoxide and pinacidil in coronary-perfused atria and ventricles from failing and non-failing human hearts. J Mol Cell Cardiol. 2011;51:215–225. doi: 10.1016/j.yjmcc.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578:315–326. doi: 10.1113/jphysiol.2006.121475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jalife J. Inward rectifier potassium channels control rotor frequency in ventricular fibrillation. Heart Rhythm. 2009;6:S44–48. doi: 10.1016/j.hrthm.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munoz V, Grzeda KR, Desplantez T, Pandit SV, Mironov S, Taffet SM, Rohr S, Kleber AG, Jalife J. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007;101:475–483. doi: 10.1161/CIRCRESAHA.107.149617. [DOI] [PubMed] [Google Scholar]
- 40.Hou L, Deo M, Furspan P, Pandit SV, Mironov S, Auerbach DS, Gong Q, Zhou Z, Berenfeld O, Jalife J. A major role for HERG in determining frequency of reentry in neonatal rat ventricular myocyte monolayer. Circ Res. 2010;107:1503–1511. doi: 10.1161/CIRCRESAHA.110.232470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.