

Abstract
Adaptive immune responses often begin with the formation of a molecular complex between a T cell receptor (TCR) and a peptide antigen bound to a major histocompatibility complex (MHC) molecule. These complexes are highly variable, however, due to the polymorphism of MHC genes, the random, inexact recombination of TCR gene segments and the vast array of possible self and pathogen peptide antigens. As a result, it has been very difficult to comprehensively study the TCR repertoire or identify and track more than a few antigen-specific T cells in mice or humans. For mouse studies, this had led to a reliance on model antigens and TCR transgenes. The study of limited human clinical samples, in contrast, requires techniques that can simultaneously survey phenotype, function and reactivity to many T cell epitopes. Thanks to recent advances in single-cell and cytometry methodologies, as well as high-throughput sequencing of the TCR repertoire, we now have or will soon have the tools needed to comprehensively analyze T-cell responses during health and disease.
From the advent of clonal selection theory1,2 to the present day, it has become increasingly clear that the adaptive immune response has, as its central unit, the expression of a single rearranged immunoglobulin or TCR on each B or T cell. And that in general, single cells are the operational units or ‘quanta’ of immunity. With respect to T lymphocytes, this means that understanding their role in immune responses requires comprehensive methods of interrogating the phenotypic and functional characteristics of individual T cells. In this regard, the use of flow cytometry for high-throughput analysis of individual T cells has been the gold standard for many years3. Gradual improvements in flow cytometry allowing simultaneous assessment of expression of surface and intracellular markers4 and the precise temporal patterns of cytokine expression by T cells5-7 have enabled studies on the relationships between T-cell phenotype/function and clinical status in a range of diseases8-14. The study of antigen-specificity, however, is complicated by enormous variability and unpredictability in terms of the epitopes targeted by T cells in any given T-cell response, especially considering the highly polymorphic nature of the MHC, and the fact that intact pathogens typically encode a wide variety of potential T cell epitopes15. Furthermore, as the breadth or number of epitopes targeted by the T cell response can be important, especially in rapidly evolving viral infections16-18, and the phenotypes of T cells targeting different epitopes from the same pathogen can vary significantly19,20, it is important to be able to monitor recognition of numerous epitopes in the response to each pathogen. As a result, the number of parameters analyzed in any given experiment continues to grow beyond the number of colors (12–15) available for fluorescence-based flow cytometry, making the latter type of analysis increasingly arduous or even impossible. Recent developments in methods for analyzing antigen-specific T cells that extend these limits exploit multiplexing and single-cell mass spectrometry-based ‘mass cytometry’20-24. Other emerging technologies that promise to dramatically increase both the speed and depth of information that one can obtain about T-cell responses include techniques allowing the analysis of single-cell mRNA transcripts25,26.
In addition, unlike most mouse models of immunological diseases, wherein the identity of the antigenic epitopes that drive disease initiation and/or progression are known, the instances of human immunological diseases wherein the precise specificities of T cells involved are known remain relatively rare. Therefore, until precise antigenic epitope specificities can be determined, study of these human T cell responses requires alternative approaches; none appear to be more powerful than high-throughput sequencing of TCR repertoires. Data generated by this approach are providing insights into T-cell selection and the nature of repertoire diversity in various T-cell subsets in normal and pathological circumstances27,28. TCR sequencing approaches also allow the identification and tracking of TCR clonotypes or motifs involved in immune responses and various pathologies29-31. Moreover, high-throughput yeast-display approaches represent a way to identify pMHC ligands that bind to these TCR clonotypes or motifs32,33. These approaches hold promise for identifying relevant antigens for immune responses for which relevant antigens are currently completely unknown. For instance, identification of antigens targeted by T cells in patients with auto-inflammatory diseases could facilitate the development of novel treatment options.
In this Review we discuss the advantages, disadvantages and complementarity of these high-dimensional approaches for the study of antigen-specific T cells. Common to each approach is the goal of understanding and/or exploiting the specificity of the T-cell mediated immune response to manipulate or predict outcomes of immunological diseases or vaccine responses. These recent technological advances seem poised to finally make possible the comprehensive analyses of T-cell responses.
Analyzing T-cell phenotypic and functional diversity
Each individual αβ T cell expresses one of as many >1014 different TCR heterodimers34 and each of these TCRs is specific for a very small fraction of possible self or foreign antigens presented in the context of an individual's MHC molecules (Fig. 1a). Thus, in terms of diversity of antigen-specificity alone, T cells are one of the most diverse cell subsets in the body. Several approaches for analyzing this diversity exist, and each has advantages and disadvantages (Table 1).
Figure 1.
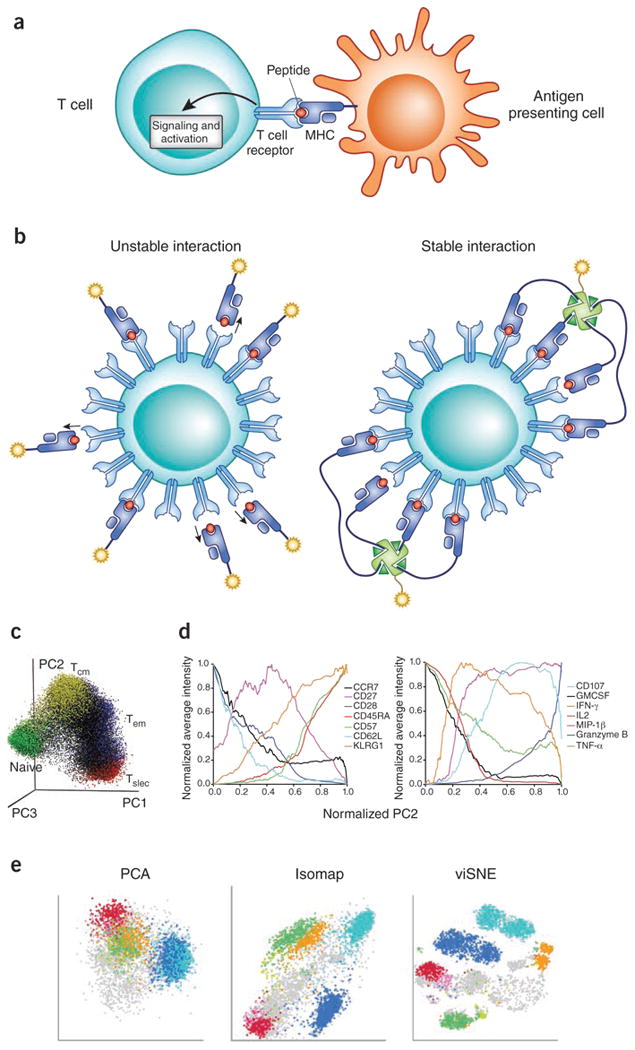
Antigen recognition by T cell receptor and probing antigen specificity with peptide-MHC multimers. (a) Antigen-specific T cell responses are initiated through the interaction of TCR, expressed on T cells, and the corresponding petide-MHC protein complex expressed by antigen-presenting cells. TCR engagement initiates a complex cell signaling cascade the results in T cell activation. (b) Binding of TCR to its specific peptide-MHC ligand is very low (∼1-100 μM) and has very fast dissociation kinetics (t1/2 usually much less than a minute). Thus, monomeric staining reagents are insufficiently stable for the detection of antigen-specific T cells. In contrast, by taking advantage of cooperative binding, multimeric complexes of peptide-MHC allow for remarkably sensitive and accurate detection of antigen-specific T cells57,58. (c) Mass cytometry and dimensionality reduction methods allow integrated analysis of T cell phenotype and function. In the example shown here, to visualize diversity of the peripheral blood CD8+ T cells, 25 parameters were measured for each cell including 16 phenotypic markers and 9 functional markers. Principal component analysis (PCA) was applied to generate three aggregate parameters describing ∼60% of total variance. A representative donor's 3D-PCA plot with cells annotated based on previously defined stringent criteria for naïve, central-memory (Tcm), effector-memory (Tem) and short-lived effector cells (Tslec). Adapted from ref 22. (d) To illustrate the phenotypic and functional meaning of a non-naïve cell progression along the PC2 axis of the PCA plot, average expression of (left) phenotypic and (right) functional parameters were normalized and plotted as a function of normalized PC2 values. This unsupervised analysis provides a hypothetical framework for graded T cell differentiation involving progressive gains and/or losses of surface marker expression and functional capacities. Adapted from ref 22. (e) To illustrate the power of non-linear dimensionality approaches, a linear PCA analysis of bone-marrow derived cells colored by a number of user-defined cell subsets is compared to two different non-linear approaches, Isomap and viSNE. Adapted from ref 38.
Table 1.
Comparison of approaches for analysis of individual T lymphocytes.
| Flow cytometry | Mass cytometry | Single-cell transcriptomics | |
|---|---|---|---|
| Number of parameters per cell | ∼10-20 | ∼40 more possible | Up to full genome (>10,000) |
| Cellular throughput | 10's of thousands per second | Hundreds per second | Very low, Maximum ∼100 cells |
| Sorting possible | Yes | No | Required for most approaches |
| Good antibodies required | Yes | Yes | No |
| Sensitive and quantitative | +++ | ++ | Difficult especially for low abundance transcripts |
| Ability to evaluate antigen-specificity | Up to ∼30 specificities with combinatorial tetramer staining | >100 specificities with combinatorial tetramer staining | No. Possible with TCR sequence. |
| T cell receptor sequence information possible | Yes with sorting | No | Yes |
Flow cytometry
As mentioned above, flow cytometry has been the leading method for measuring this diversity over the past three decades. The latest instruments are remarkably powerful, capable of analyzing and/or sorting cells based on up to ∼18 cellular parameters at >10,000 cells per second4. Such high-dimensional and high-throughput analysis revealed relationships between T-cell phenotype/function and clinical status for a range of diseases. For instance, in several infectious disease settings, a correlation between pathogen control and poly-functionality (that is, the capacity to perform more than one effector function, such as producing more than one cytokine) of the corresponding antigen-specific T cells have been identified8-13. As less simplistic relationships between disease status and antigen-specific T cell phenotypic/functional status must also exist, a great deal of effort is being dedicated to developing computer algorithms that can analyze and extract useful information from this otherwise impossible-to-understand high-dimensional flow cytometry data35-38.
Human T cells are often segregated into four major categories based on surface markers that indicate their proliferative potential, cytotoxicity capacity, and their ability to produce cytokines. They are the following: naïve (CCR7+CD45RA+), central memory (CCR7+CD45RA–), effector memory (CCR7–CD45RA–), and terminal effector (CCR7–CD45RA+)39. Although simple, useful and well accepted, it is clear that much more heterogeneity exists than is captured by these subdivisions. For instance, expression of all possible combinations of CD27, CD28, CD62L and CCR7 are also observed, yet the significance of all of these distinctions is not clear40. Also, even cells fitting the most strict definition of naïve (CD45RA+CD45RO–CD27+CCR7+CD62L+CD28+), yet expressing higher levels of CD95, interleukin 2 receptor subunit-β (IL-2Rβ), CXC-motif chemokine receptor 3 (CXCR3) and leukocyte function-associated antigen-1 (LFA-1), have been designated stem cell-like memory cells with high proliferative capacity41. T cells can also be classified by the their functional capacities (e.g., abilities to produce various cytokines and effector molecules), transcription factor expression profiles42, as well as markers indicative of their tissue trafficking potential43. Combined with antigen-specificity, as discussed below, the number of parameters that can be clearly distinguished by even the latest fluorescent flow cytometers is insufficient for an integrated and comprehensive view of T-cell diversity.
Mass cytometry
Thus, the arrival of mass cytometry has allowed a quantum leap in phenotypic and functional characterization of single T cells. The mass spectrometry-based flow cytometry method (CyTOF) uses isotopically purified heavy metal atoms, instead of fluorophores, as tags 44. Whereas the number of parameters available to fluorescent flow cytometry is limited by broad spectral overlap leading to crosstalk between fluorescent channels, in CyTOF many more parameters (now ∼40 and dozens more should be possible in the near future) can be distinguished. In addition, thanks to reduced crosstalk between channels due to accurate resolution of tags differing by as little as a single atomic mass unit, the analysis and interpretation of CyTOF data is greatly simplified45. In terms of sample throughput, however, mass cytometry is significantly slower (capable of analyzing ∼500 cells per second) than fluorescence flow cytometry (up to tens of thousands of cells per second). Although reduced sample throughput poses a challenge to studies requiring large sample sizes or rare cells, using pre-enrichment strategies such as T cell purification methods or tetramer enrichment approaches as we have applied20,22 make it possible to analyze even extremely rare antigen-specific T cells. Mass-tag barcoding46 can also alleviate the throughput problem by allowing a large number (up to 96 so far) uniquely tagged (barcoded) samples to be analyzed simultaneously. After data acquisition, simple and effective software is available for deconvolution of each barcoded sample.
Although mass cytometry is far from a comprehensive ‘proteomics’ method, extending cellular analysis into 40 dimensions means that each cell can be parsed into one of 240 (∼1 trillion) possible bins, allowing cells to be classified in unprecedented detail. It also means that a wide variety of T-cell markers can be can be assessed simultaneously providing a view of the overall diversity of a sample of cells. We have used mass cytometry to broadly probe the relationship between CD8+ T cell phenotype, function and specificity by simultaneously assessing several markers of each. For example, after using some of the then-available parameters to isolate single CD8+ T cells, we probed six different antigen-specificities using peptide–MHC tetramers (discussed below, see Fig. 1b), and used the remaining 25 parameters to evaluate expression of 16 surface and nine functional markers22. In that study, we chose the parameters and the timing of assessment to maximize the amount of information obtained form each cell and to reveal variation. Nonetheless, a caveat of this approach is that only a snapshot of information from a single time-point taken from a single blood sample is available for each cell. Accurately tracking the kinetics of cellular responses requires an ability to continuously monitor single cells (as described below for the kinetics of cytokine secretion and in references5-7).
As there is a great deal of interest in T-cell poly-functionality as an index of the strength of an immune response, in the same study we took advantage of the large number cellular parameters available to mass cytometry by assessing nine different functional capacities (eight cytokines and a marker of granular release, CD107), in populations of activated human CD8+ T cells. This gave us 512 (29 ) possible combinations and we were able to observe at least 242 of these, indicating that a vast number of possible functional combinations can be expressed. Furthermore, by using MHC tetramers bearing influenza, cytomegalovirus (CMV) and Epstein-Barr virus (EBV) epitopes together with these nine functional markers, we showed that T cells specific for each of the different viruses expressed partially overlapping but distinct combinations of cytokines, ranging from 50 to 100 different combinations of cytokines observed. Although only a few antigen-specificities have been analyzed in this manner so far, these results indicate that T-cell responses are far more complex than previously thought, at least for CD8+ T cells, and that they seem tailored to the particular pathogen22.
To integrate both phenotypic and functional capacities of the cells, we also combined all the measured information about each cell and performed dimensionality reduction using principal component analysis (PCA). PCA uses weighted combinations of each of the measurements to create composite parameters that maximally represent the data with a minimal number of new parameters. This approach provides an unbiased composite representation of the cellular diversity that incorporates both phenotypic and functional information. It also provides insights about how these markers, and different cell subsets, are related to each other (Fig. 1c, d)22. However, the represented pattern depends heavily on the cellular markers chosen for the analysis and on the composition of the cells being analyzed. We anticipate that this and other challenges of analyzing high-dimensional cellular data will improve as new computational methods are developed and their utilities compared for various applications35. For example, more sophisticated analogs of principal component analysis that combine the measured parameters in a non-linear fashion are capable explaining more variation in fewer dimensions and should enable better resolution of T-cell diversity38 (Fig. 1e).
Advantages of both fluorescence and mass cytometry include their ability to accurately and sensitively quantify protein levels at the individual cell level in rapid succession (hundreds to tens of thousands per second). However, their dependence on the availability of reliable antibodies specific for each protein severely limits their utility for discovery of novel proteins of interest. Furthermore, though the number of channels continues to increase, even for mass cytometry the number of isotopes in the periodic table represents a hard cap on the number probes possible by this approach.
Single T cell phenotyping and temporal analysis using microwells
Another important new technique was developed by Love and colleagues, who spread T cells onto slides engineered to contain 100,000 microwells, such that many wells are occupied by only a single cell5-7. These cells can then be stimulated and many potential cytokines analyzed using antibody capture. This system revealed something unique, which is that the individual T cells typically make multiple cytokines, but not necessarily at the same time and over a several day timespan14. This temporal complexity is not captured in other systems and takes advantage of the unique ability of this system to analyze the output of the same cells over time, and in a very high-throughput manner.
Single cell transcription profiling
Going forward, many of the limitations of flow cytometry and mass cytometry will be overcome through the use of single cell gene transcription profiling. However, because copy numbers of mRNA can be as low as just a few copies per cell, single cell gene expression analysis remains a challenge and the data need to be interpreted carefully48. Nonetheless, after careful validation, higher-throughput (up to 96 cells at a time) single-cell quantitative reverse transcriptase PCR (RT-PCR) systems are already allowing up to 96 mRNA transcripts to be analyzed in each cell in a quantitative fashion25,37,49 (Fig. 2). Highlighting diversity in cellular mRNA transcript levels, this approach proved to be useful in distinguishing gene expression profiles of tetramer-stained and sorted antigen-specific T cells responding to the same antigen in the context of different gene-based vaccination49. Though it is a dramatic improvement over bulk analysis without single-cell resolution (Fig. 2), the analysis of only 96 cells at a time severely limits the current applicability of this approach. Here we anticipate that implementation of barcoding-based technologies (as discussed below) will gradually increase throughput and broaden the applicability of single-cell transcriptional profiling.
Figure 2.
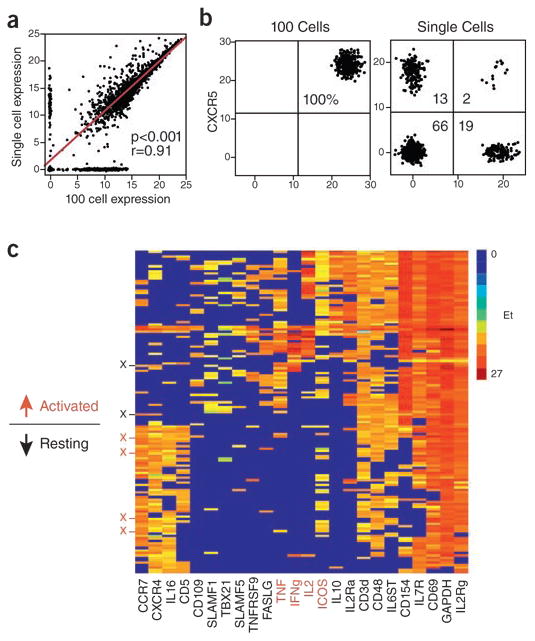
Single cell analysis can reveal heterogeneity in gene expression among T cells. The data shown here were generated using high-throughput BioMark™ microfluidics for multiplex quantitative RT-PCR analysis25. (a) After some careful optimization relative single cell gene expression can be validated by plotting compared to relative expression levels obtained from ‘bulk’ analysis of 100 cells. Good correspondence indicates reliable single cell gene expression measurements. (b) The value of single cell analysis is illustrated by comparing expression of CXCR5 and CCL5 mRNA in single T cells and in ‘bulk’ populations of 100 T cells. Averaging expression of genes across 100 cells masks diversity revealed at the single-cell level. (c) Further illustrating the power of the single-cell approach, expression levels of 24 genes (columns) in 163 cells (rows) is shown. In this example, half of the cells were stimulated prior to analysis (noted as ‘Activated’); the other half were unstimulated (‘Resting’). Unsupervised clustering discriminated all but six cells (noted with X on y-axis) into activated vs. resting categories based on gene expression. Adapted from ref 25.
Also exciting is the emergence of single-cell RNA sequencing methods that allow genome wide mRNA transcript quantification26,48,50. The use of barcoded primers used to multiplex the analysis of a large number cells in a single DNA sequencing reaction will surely broaden the utility of the single-cell transcriptomics approach48. Although it will always be important to evaluate cellular protein levels instead of relying strictly on mRNA levels, accurate genome-wide transcriptomic information from single cells, if of sufficient throughput and sensitivity, will add insight to that gained using the proteomic methods discussed above; it might also enable selection of markers to analyze using those methods. That said, cytometry methods able to directly evaluate T cell antigen-specificity by using peptide-MHC multimers, as discussed in the next section, will remain essential.
Characterizing T-cell antigen specificity
Studies of human immunology are limited by the availability of cellular material, which is usually restricted to small volumes of blood. Furthermore, to varying extents, depending on the immune response of interest, responsive T cells often represent a very small minority of total blood lymphocytes. Thus, to achieve sufficient signal above the inherent noise of complex blood samples, it is imperative to restrict analysis to the relevant cells—ideally through identification of T cells specific for the antigen of interest. However, as mentioned above, the precise antigen-specificities of T cells are only known for a handful of immune responses. Without knowledge of precise antigenic epitopes, only indirect methods relying on a cellular response (e.g., proliferation, cytokine production, surface marker expression changes8,51-56) to stimulation with whole-antigen can be used. However, in these approaches, the analysis is ‘single-plex’ in that each sample of cells can only be interrogated with a single antigen (or single mixture of antigens). Thus, epitope mapping is either very low resolution or requires large volumes of sample. Detecting rare antigen-specific cells is also problematic especially if any other cells in the sample are activated and contribute to background signal. Lastly, depending on the method of detection (unless high-dimensional approaches are used, see previous section), the phenotypic and/or functional profile of the antigen-specific cells can be limited and influenced in unpredictable ways by the stimulation used to identity them.
Improvements to methods enabling direct detection antigen-specific cells are overcoming each of these limitations. As a result of their cooperative binding and ability to specifically label antigen-specific T cells (Fig. 1), peptide–MHC tetramers (or multimers) are now widely used for directly identifying and characterizing antigen-specific T cells57,58. Several recent technical advances have made this approach especially practical, including higher-throughput production of hundreds or thousands of tetramers from one batch of prepared MHC class I protein-loaded ultraviolet (UV)-cleavable and exchangeable peptides59,60 or cleavable and exchangeable class II-associated Ii peptide (CLIP)-loaded MHC class II proteins61; detection of rare antigen-specific T-cell populations using magnetic bead-based enrichment of the peptide-MHC tetramer labelled antigen-specific cells allowing the characterization of very rare T cells, even those in the naïve T cell repertoire61,62; and an ability to probe larger numbers of T-cell antigen specificities in a single sample using combinatorial staining approaches23,24,63.
Most recently, we demonstrated an approach that incorporates each of these improvements and exploits mass cytometry20. In this approach >100 antigen specificities and >20 phenotypic or functional markers can be probed in a single blood or tissue sample (Fig. 3a-c). We used this approach to simultaneously screen for, identify and phenotypically characterize rotavirus-specific cells in blood and intestinal lymphocyte samples from healthy human donors. Parallel analysis of T cells specific for a range of previously identified viral (and other) antigens provided ‘landmarks’ for classifying and defining the phenotypic profiles the identified cells. Through this parallel analysis, and consistent with what had previously observed for EBV19, we identified significant differences in the phenotypic properties of T cells targeting different epitopes derived from the same virus20 (Fig. 3d, e). Assuming that it will be possible to increase the number T-cell specificities that can be probed simultaneously (reconstruction experiments show that even a thousand different specificities may be possible20) using this approach, and apply it to study CD4+ T cells and MHC-class II restricted antigens (McGuire, Davis, unpublished results), we anticipate that this method will enable rapid identification of antigens related to a wide range of pathologies currently characterized by poorly defined T cell responses.
Figure 3.
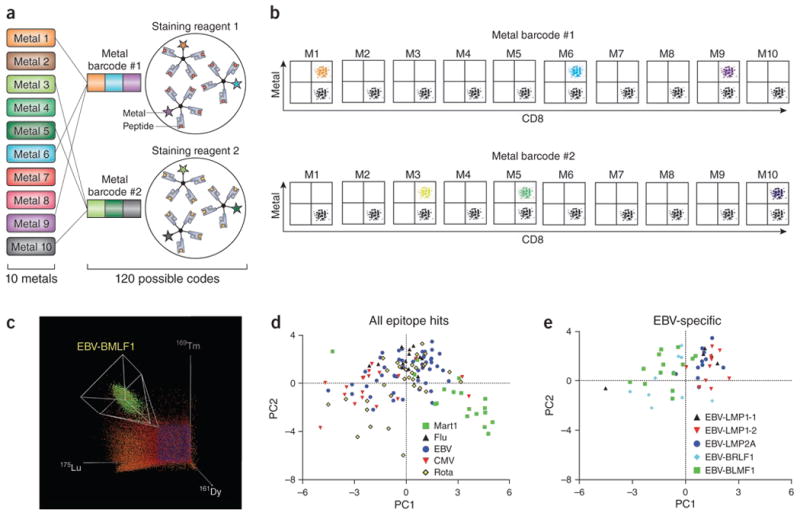
Highly mulitplexed analysis of T–cell antigen-specificity using mass cytometry based combinatorial peptide–MHC tetramer staining. (a-b) By coding each T cell antigen specificity (each pMHC tetramer) by a unique combination of three out ten possible metal tags, up to 120 different antigen specificities can be simultaneously measured in a single sample. Cells staining positive with three and only three of the metals comprising each code can be identified as specific for the pMHC epitope corresponding to that code. Adapted from NBT 31:609. (c) This example illustrates the identification of EBV-BRLF-1-specific T cells (colored green) by virtue of their staining positive for 161Dy, 169Tm, and 175Lu after incubation with a pMHC tetramer barcoded with these metals. This panel shows just one of the 120 possible 3D dot-plots for each sample. Adapted from ref 20. (d) If T cells are also stained with antibodies specific for phenotypic and functional markers, PCA dimensionality reduction can then be used to further summarize the phenotype of T cells specific for each epitope. In this example, each dot represents T cells specific for the indicated epitope from one of the 17 different donors analyzed. T cells displaying previously defined phenotypes and specific for well-characterized antigens can be used as ‘landmarks’ on these plots to help classify the composite phenotype of T cells specific for previously uncharacterized antigens. Adapted from ref 20. (e) This example illustrates the power of this approach to accurately define the status of antigen-specific T cells. Here the phenotypic status of EBV-specific cells targeting lytic- (BRLF1, BLMF1) versus latency-associated (LMP1, LMP2) antigens are delineated suggesting that T cells specific for lytic-cycle antigens have encountered antigen more recently20. Adapted from ref 20.
As described above, it is currently impossible to predict a priori which of the multitude of possible T-cell epitopes will be targeted by a T cell-mediated immune response15,64. An ability to probe >100 or possibly >1000 candidate epitopes is helpful but alone will not solve this problem, especially for diseases where the range of possible antigens is too large. As we and others have demonstrated for rotavirus and several other viral pathogens (e.g., influenza virus, Dengue virus and human immunodeficiency virus)20,65-68 with relatively small genomes, epitope or MHC-binding prediction algorithms69,70 provide a means of narrowing the possibilities and have been applied with some success. However, even for these small-genome pathogens, the number of candidate epitopes can be quite large when prediction stringencies are relaxed in effort to prevent missing epitopes or when considering viruses with high mutation rates and significant epitope variation that also need to be covered. Furthermore, binding prediction algorithms perform especially poorly for MHC alleles for which there is much less peptide binding data with which to train the algorithms71. Sometimes it can help to broadly map epitopes through cellular stimulation-based approaches or in the case of MHC class II-restricted cells, tetramer guided epitope mapping72 before narrowing down on the precise epitopes using peptide-MHC multimers. Another solution is to use careful transcript sequence analysis as has been done for the identification of tumor-specific mutations encoding candidate tumor antigens based on whole-exome sequencing73. For example, in one recent study, >400 candidate antigens were screened using combinatorial tetramer staining of tumor infiltrating lymphocytes in a lymph-node metastases to identify a single dominant epitope from a melanoma patient74. Lastly, direct identification of MHC-bound peptides through peptide elution75,76 and ever-improving mass spectrometry/proteomics-based peptide sequencing should provide more accurate means of identifying candidate epitopes for a range of applications77,78. Although aided by the development of more sensitive and sophisticated peptide-identifying mass spectrometers79, this approach is still currently limited by the large number antigen-presenting cells required and the associated bias against low abundance and low affinity peptide ligands. Thus, despite the ongoing improvements being made to T cell epitope discovery methods, identification of antigens for a number of diseases, such as autoimmune and auto-inflammatory diseases remain elusive. In these cases, TCR sequencing-based approaches hold promise as a reliable means of tracking and understanding antigen-specific T cell responses, even when the identities of the antigens are unknown.
TCR repertoire sequencing
The advent of inexpensive and massively parallel DNA sequencing has started to impact T-cell analysis, starting with the pioneering work of Robins and colleagues80 to understand the scope of the human TCRβ repertoire. From these studies, it was found that natural biases in VDJ recombination lead to an overrepresentation of common rearrangement events and a higher than expected frequency of such sequences common between different donors81,82. However, the vast majority of unique TCR sequences are present at low frequencies, as would be expected from early estimates of possible TCR diversity34. Considering these data, estimates of TCRβ repertoire diversity in an individual are difficult to calculate , yet lower bounds for total TCRβ diversity in a healthy adult27,80 are ∼3–4 × 106 approximately consistent with previous estimates83. Single TCR chain deep sequencing studies have also shown that the clonal diversity of memory cells is greater than might be expected, with only a minority of memory T cell clones clearly expanded84. TCR sequencing has also been applied to accurately measure the TCR repertoire diversity of responding T-cell populations in cancer31, during human herpes virus infection30, during CMV or polyomavirus BK (BKV) reactivation or during acute allograft rejection85. This technique also holds promise for investigating the extent of changes in TCR repertoire during aging86. This approach has also proven to be a very sensitive means of accurately detecting the presence of rare malignant T cells, such as in minimal residual disease in acute T lymphoblastic leukemia87.
Extending this approach, several higher-throughput means of obtaining endogenous pairs of TCRα and β sequences from individual cells are emerging (Fig. 4a). These include single-cell mRNA capture and emulsion linkage RT-PCR, as recently described for simultaneous sequencing of heavy and light chains of immunoglobulin in single B cells88, single cell barcoding as has been used for single cell mRNA sequencing28,48 and direct cellular emulsion linkage PCR89. Each of these methods allows the sequencing of α and β or γ and δ TCR sequences from individual cells much more efficiently than current practice.
Figure 4.
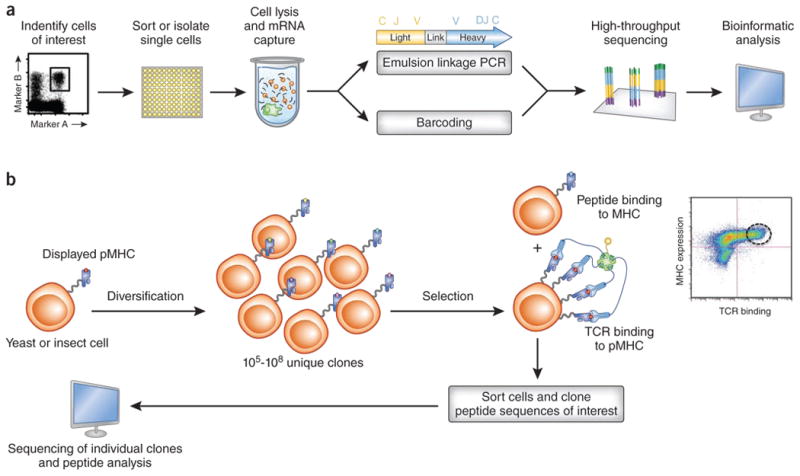
Strategies for high-throughput single-cell analysis of TCR sequences and identification of TCR ligands. (a) As demonstrated for B cell immunologlobulin genes88, sequencing of V regions of endogenously paired TCRa and TCRb genes can be performed in high-throughput by mRNA capture and emulsion linkage RT-PCR, or by direct cellular emulsion linkage PCR89. Cell-specific barcoded tags can also be introduced at the single-cell stage as a general means of increasing the throughput of this type of approach48. (b) Libraries of yeast clones displaying 105-108 unique peptides tethered to MHC molecules enables high-throughput screening for peptides capable of binding MHC (because these peptides result in proper folding and surface expression of pMHC complexes) and for peptides capable of binding to a TCR of interest (here the TCR is used as a tetrameric staining reagent). After multiple rounds of selection, hits are sorted, cloned, and sequenced; the peptide sequences can then be analyzed32,33.
In terms of applications, an especially promising approach would be to use single-cell methodologies and barcoding to analyze antigen-specific T-cell responses in the many cases where the major epitopes are unknown. A glimpse of the potential insights that can be gained with this approach is found in our recently published study of T-cell responses in patients with celiac disease29. In this disease, patients have a strong autoimmune response to gluten, which inflames the gut and causes serious discomfort and damage. In most cases, eating a gluten-free diet eliminates the symptoms. We monitored the peripheral blood lymphocytes of celiac patients who had ingested gluten for any unusual responses. What was expected based on the work of Sollid and colleagues90 was that gliadin (a subset of gluten proteins) specific CD4+ T cells would be activated and then enter the circulatory system approximately six days following the first gluten exposure. However, we found that populations of CD8+ T cells and γδ T cells appeared (and disappeared) with the same kinetics, all expressing gut homing receptors. We then employed a single-cell TCR amplification method using a large panel of primers to show that both the CD8+ and γδ T cell populations were highly enriched for particular sequences and that complementarity-determining region 3 (CDR3) encoded by these sequences were shared between many of the patients. This implies that all three of these different types of T cells are recognizing specific ligands in this disease and somehow coordinating their responses. It also shows that it is possible to analyze T-cell responses directly after particular immunological events (albeit with a time lag) from a blood sample and assess the clonality of the response. In this study and in an analogous study that identifyied oligoclonal skin resident T-cell populations responding to herpes virus infection30, one chain from each T cell population (TCRα, β, γ or δ) was analyzed. However, it is also possible to amplify both chains from single T cells as described above (which we have done, Han et al. unpublished data). Adapting this methodology to a high-throughput system of single cell analysis, as has been done by Georgiou88 and Robinson (personal communication) for plasmablasts, one could envisage a very powerful way to analyze T-cell responses to disease or vaccination that is entirely independent of other methods and requires no knowledge of the major antigens, but can lead to them.
Antigen specificity from TCR sequence?
As introduced above, it would be ideal if high-throughput sequencing of enough TCR sequences could be used to computationally predict T-cell specificity from a given TCR heterodimer sequence. This would solve many of the problems and challenges discussed in this Review. However, unfortunately, we do not anticipate this to be possible anytime soon. The reasons for this are several-fold but stem from the highly variable nature of each of the components of the TCR–peptide–MHC complex. Unlike the binding of short linear peptides to MHC, which is mediated by relatively simple motifs and mostly predictable anchor residues91, the energetics of TCR binding to peptide–MHC rely on highly unpredictable and numerous contacts between the TCR and both the MHC and the antigenic peptide92, although peptide residues generally contribute more than the MHC residues93. The challenges associated with such a problem are highlighted by difficulties in the field of predicting protein-protein interactions in general, even when there are high-resolution structures for each94,95. Furthermore, it is clear that such a problem has far from a unique solution for a given TCR. This is because TCRs generally have a great deal of flexibility in one or both of the CDR3 loops which are the principal determinants of peptide specificity96; this flexibility allows them to recognize multiple different peptides which may not share any sequence homology. For example, Garcia and colleagues solved the structures of one TCR binding to four structurally distinct peptide-MHC complexes; in each case the CDR3s of the TCR adopted a different conformation32. Considering these observations together with the relatively ubiquitous cross-reactivity of TCRs to homologous peptides 97,98, one can conclude that most TCRs can likely recognize many different peptides, and with very low affinities (∼10,000 lower than most affinity matured antibodies, for example). This means the “fit” between a TCR and a ligand is much less stringent and therefore significantly harder to model than the fit between an antibody and its cognate antigen. Thus, we are not optimistic about the possibility of being able to computationally predict TCR antigen-specificity solely from sequence information, at least by some form of structural prediction. An alternative may be to identify specific sequence motifs that can be associated with particular antigen responses (as found for antibody sequences in Dengue fever virus infection99). But for now we will have to rely on experimental approaches for identifying T-cell antigens based on cloned TCRs. Fortunately, some such approaches are already proving to be useful.
One general strategy for identifying antigens bound by a particular cloned TCR is to create libraries of cells (insect/baculovirus or yeast), each expressing a single peptide–MHC antigen that can be selected by staining with tetramerized soluble TCR proteins. For this approach, one needs to know the restricting element (e.g., the appropriate MHC allele) before addressing the even larger challenge of identifying the corresponding peptide epitope. The power of this approach can be illustrated by the challenge of identifying the pMHC recognized by the TCR expressed on the murine diabetogenic T-cell clone, BDC-2.5. Insect cell display was used to identify several mimotopes (presumably irrelevant peptides also capable of stimulating these cells) 100,101 that eventually aided in the identification of the true endogenous antigen. This case was made especially difficult due to the abnormal MHC binding register of the pathogenic epitope, which was not found in any of the insect cell libraries101. A possibly faster approach is to generate yeast display libraries restricted to sequences derived from the pathogen of interest, as was demonstrated for influenza virus–specific human leukocyte antigen DR (HLA-DR)-restricted CD4+ T cells102. Extending this to large random libraries89, 90 one can construct yeast display libraries in which a large number (106–107) different peptides can be expressed bound to a give MHC molecule and that these libraries can then be screened with a particular TCR to identify ligands32,33 (Fig. 4b). This system enabled identification of a number of interesting H-2Ld-restricted ligands of well-described mouse T cell receptor clones. These included an epitope with a sequence totally unrelated to the sequence to the original antigen; although having a high affinity for the TCR, apparently due to an abnormal docking orientation this epitope failed to trigger T-cell activation32. Hits like this and other mimotopes may be difficult to distinguish from immunologically relevant epitopes by this approach. However, as demonstrated101, identification of a consensus sequence from the hits searched against sequence databases containing candidate antigens should help tremendously to narrow down the possibilities especially in cases where the source of antigen is totally unknown. From this, shortlisted candidates can be used as input for a tetramer panel (as described above) or stimulation-based studies of polyclonal T cell populations from which the TCRs of interest had been identified. Thus, as large-scale yeast-display screening approaches continue to improve in throughput and depth, we are optimistic that many more elusive disease-associated T-cell antigens will be identified, possibly leading to novel therapeutics and/or diagnostics.
Concluding remarks
The complexity of T-cell responses to pathogens in outbred populations has, in most cases, severely limited their assessment. This has forced a reliance on highly constrained animal models, which cannot give us a complete picture of how the different types of T cells function during an actual disease or vaccine challenge. It has also greatly inhibited attempts to find T-cell correlates of protection in vaccine or epidemiological work. Fortunately, the technologies discussed here represent a quantum leap in our ability to capture that complexity, and thus we may finally have the tools we need to broadly assess T-cell responses in most situations and to understand their contributions to immunity in much more depth. We anticipate that these approaches will give us much better ways of evaluating new vaccines and immunotherapies in the very near future.
References
- 1.Burnet FM. A Modification of Jerne's Theory of Antibody Production using the Concept of Clonal Selection. The Australian Journal of Science. 1957;20:67–69. doi: 10.3322/canjclin.26.2.119. [DOI] [PubMed] [Google Scholar]
- 2.Burnet FM. The clonal selection theory of acquired immunity. Nashville: Vanderbilt University Press. 1959;3 [Google Scholar]
- 3.Hulett HR, Bonner WA, Barrett J, Herzenberg LA. Cell sorting: automated separation of mammalian cells as a function of intracellular fluorescence. Science. 1969;166:747–749. doi: 10.1126/science.166.3906.747. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay PK, Roederer M. Cytometry: today's technology and tomorrow's horizons. Methods. 2012;57:251–258. doi: 10.1016/j.ymeth.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han Q, Bradshaw EM, Nilsson B, Hafler DA, Love JC. Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab on a chip. 2010;10:1391–1400. doi: 10.1039/b926849a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han Q, et al. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci U S A. 2012;109:1607–1612. doi: 10.1073/pnas.1117194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varadarajan N, et al. Rapid, efficient functional characterization and recovery of HIV-specific human CD8+ T cells using microengraving. Proc Natl Acad Sci U S A. 2012;109:3885–3890. doi: 10.1073/pnas.1111205109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Betts MR, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 10.Makedonas G, Betts MR. Living in a house of cards: re-evaluating CD8+ T-cell immune correlates against HIV. Immunol Rev. 2011;239:109–124. doi: 10.1111/j.1600-065X.2010.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan J, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A. 2008;105:20410–20415. doi: 10.1073/pnas.0810114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker BD, Yu XG. Unravelling the mechanisms of durable control of HIV-1. Nature reviews Immunology. 2013;13:487–498. doi: 10.1038/nri3478. [DOI] [PubMed] [Google Scholar]
- 13.Precopio ML, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. The Journal of experimental medicine. 2007;204:1405–1416. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamanaka YJ, Gierahn TM, Love JC. The dynamic lives of T cells: new approaches and themes. Trends Immunol. 2013;34:59–66. doi: 10.1016/j.it.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akram A, Inman RD. Immunodominance: a pivotal principle in host response to viral infections. Clinical immunology. 2012;143:99–115. doi: 10.1016/j.clim.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Kiepiela P, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 17.Pereyra F, et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis. 2008;197:563–571. doi: 10.1086/526786. [DOI] [PubMed] [Google Scholar]
- 18.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 19.Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. The Journal of experimental medicine. 2002;195:893–905. doi: 10.1084/jem.20011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newell EW, et al. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nature biotechnology. 2013;31:623–629. doi: 10.1038/nbt.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadrup SR, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–526. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- 24.Newell EW, Klein LO, Yu W, Davis MM. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat Methods. 2009;6:497–499. doi: 10.1038/nmeth.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dominguez MH, et al. Highly multiplexed quantitation of gene expression on single cells. Journal of Immunological Methods. 2013;391:133–145. doi: 10.1016/j.jim.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren EH, Matsen F.A.t, Chou J. High-throughput sequencing of B- and T-lymphocyte antigen receptors in hematology. Blood. 2013;122:19–22. doi: 10.1182/blood-2013-03-453142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.La Gruta NL, Thomas PG. Interrogating the relationship between naive and immune antiviral T cell repertoires. Curr Opin Virol. 2013;3:447–451. doi: 10.1016/j.coviro.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han A, et al. Dietary gluten triggers concomitant activation of CD4+ and CD8+ alphabeta T cells and gammadelta T cells in celiac disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13073–13078. doi: 10.1073/pnas.1311861110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature. 2013;497:494–497. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emerson RO, et al. High-throughput sequencing of T cell receptors reveals a homogeneous repertoire of tumor-infiltrating lymphocytes in ovarian cancer. J Pathol. 2013 doi: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams JJ, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 2011;35:681–693. doi: 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Birnbaum ME, Dong S, Garcia KC. Diversity-oriented approaches for interrogating T-cell receptor repertoire, ligand recognition, and function. Immunological Reviews. 2012;250:82–101. doi: 10.1111/imr.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 35.Aghaeepour N, et al. Critical assessment of automated flow cytometry data analysis techniques. Nature methods. 2013;10:228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finak G, et al. Mixture models for single-cell assays with applications to vaccine studies. Biostatistics. 2013 doi: 10.1093/biostatistics/kxt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amir EA, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature biotechnology. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 40.Romero P, et al. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J Immunol. 2007;178:4112–4119. doi: 10.4049/jimmunol.178.7.4112. [DOI] [PubMed] [Google Scholar]
- 41.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nature medicine. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nature reviews Immunology. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 44.Ornatsky O, Baranov VI, Bandura DR, Tanner SD, Dick J. Multiple cellular antigen detection by ICP-MS. J Immunol Methods. 2006;308:68–76. doi: 10.1016/j.jim.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 45.Bjornson ZB, Nolan GP, Fantl WJ. Single-cell mass cytometry for analysis of immune system functional states. Curr Opin Immunol. 2013;25:484–494. doi: 10.1016/j.coi.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nature biotechnology. 2012 doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Claassen M. Shooting Movies of Signaling Network Dynamics with Multiparametric Cytometry. Curr Top Microbiol Immunol. 2013 doi: 10.1007/82_2013_350. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nature reviews Genetics. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 49.Flatz L, et al. Single-cell gene-expression profiling reveals qualitatively distinct CD8 T cells elicited by different gene-based vaccines. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5724–5729. doi: 10.1073/pnas.1013084108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 51.Brunner KT, Mauel J, Cerottini JC, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. 1968;14:181–196. [PMC free article] [PubMed] [Google Scholar]
- 52.Peters PJ, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. The Journal of experimental medicine. 1991;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Betts MR, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 54.Waldrop SL, Pitcher CJ, Peterson DM, Maino VC, Picker LJ. Determination of antigen-specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen-specific homeostatic mechanism in HIV-associated immunodeficiency. J Clin Invest. 1997;99:1739–1750. doi: 10.1172/JCI119338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Rosa SC, et al. Vaccination in humans generates broad T cell cytokine responses. J Immunol. 2004;173:5372–5380. doi: 10.4049/jimmunol.173.9.5372. [DOI] [PubMed] [Google Scholar]
- 56.Frentsch M, et al. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nature medicine. 2005;11:1118–1124. doi: 10.1038/nm1292. [DOI] [PubMed] [Google Scholar]
- 57.Altman JD, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 58.Davis MM, Altman JD, Newell EW. Interrogating the repertoire: broadening the scope of peptide-MHC multimer analysis. Nature reviews Immunology. 2011;11:551–558. doi: 10.1038/nri3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toebes M, et al. Design and use of conditional MHC class I ligands. Nat Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 60.Grotenbreg GM, et al. Discovery of CD8+ T cell epitopes in Chlamydia trachomatis infection through use of caged class I MHC tetramers. Proc Natl Acad Sci U S A. 2008;105:3831–3836. doi: 10.1073/pnas.0711504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Day CL, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112:831–842. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moon JJ, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen RS, et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nature protocols. 2012;7:891–902. doi: 10.1038/nprot.2012.037. [DOI] [PubMed] [Google Scholar]
- 64.Chang CX, et al. Sources of diversity in T cell epitope discovery. Frontiers in bioscience : a journal and virtual library. 2011;16:3014–3035. doi: 10.2741/3895. [DOI] [PubMed] [Google Scholar]
- 65.Assarsson E, et al. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J Virol. 2008;82:12241–12251. doi: 10.1128/JVI.01563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maciel M, Jr, et al. Comprehensive analysis of T cell epitope discovery strategies using 17DD yellow fever virus structural proteins and BALB/c (H2d) mice model. Virology. 2008;378:105–117. doi: 10.1016/j.virol.2008.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weiskopf D, et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2046–2053. doi: 10.1073/pnas.1305227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoof I, et al. Interdisciplinary analysis of HIV-specific CD8+ T cell responses against variant epitopes reveals restricted TCR promiscuity. Journal of immunology. 2010;184:5383–5391. doi: 10.4049/jimmunol.0903516. [DOI] [PubMed] [Google Scholar]
- 69.Lundegaard C, et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36:W509–512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wee LJ, Lim SJ, Ng LF, Tong JC. Immunoinformatics: how in silico methods are re-shaping the investigation of peptide immune specificity. Front Biosci (Elite Ed) 2012;4:311–319. doi: 10.2741/e378. [DOI] [PubMed] [Google Scholar]
- 71.Rivino L, et al. Defining CD8+ T Cell Determinants during Human Viral Infection in Populations of Asian Ethnicity. Journal of immunology. 2013;191:4010–4019. doi: 10.4049/jimmunol.1301507. [DOI] [PubMed] [Google Scholar]
- 72.Yang J, et al. Multiplex mapping of CD4 T cell epitopes using class II tetramers. Clin Immunol. 2006;120:21–32. doi: 10.1016/j.clim.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. The EMBO journal. 2013;32:194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Rooij N, et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013 doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rotzschke O, et al. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature. 1990;348:252–254. doi: 10.1038/348252a0. [DOI] [PubMed] [Google Scholar]
- 76.Marrack P, Ignatowicz L, Kappler JW, Boymel J, Freed JH. Comparison of peptides bound to spleen and thymus class II. The Journal of experimental medicine. 1993;178:2173–2183. doi: 10.1084/jem.178.6.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fortier MH, et al. The MHC class I peptide repertoire is molded by the transcriptome. The Journal of experimental medicine. 2008;205:595–610. doi: 10.1084/jem.20071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kasuga K. Comprehensive analysis of MHC ligands in clinical material by immunoaffinity-mass spectrometry. Methods in molecular biology. 2013;1023:203–218. doi: 10.1007/978-1-4614-7209-4_14. [DOI] [PubMed] [Google Scholar]
- 79.Baker ES, et al. Mass spectrometry for translational proteomics: progress and clinical implications. Genome Med. 2012;4:63. doi: 10.1186/gm364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Robins HS, et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Venturi V, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. Journal of immunology. 2011;186:4285–4294. doi: 10.4049/jimmunol.1003898. [DOI] [PubMed] [Google Scholar]
- 83.Arstila TP, et al. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 84.Klarenbeek PL, et al. Human T-cell memory consists mainly of unexpanded clones. Immunology letters. 2010;133:42–48. doi: 10.1016/j.imlet.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 85.Dziubianau M, et al. TCR Repertoire Analysis by Next Generation Sequencing Allows Complex Differential Diagnosis of T Cell-Related Pathology. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2013 doi: 10.1111/ajt.12431. [DOI] [PubMed] [Google Scholar]
- 86.Boyd SD, Liu Y, Wang C, Martin V, Dunn-Walters DK. Human lymphocyte repertoires in ageing. Curr Opin Immunol. 2013;25:511–515. doi: 10.1016/j.coi.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu D, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4:134ra163. doi: 10.1126/scitranslmed.3003656. [DOI] [PubMed] [Google Scholar]
- 88.DeKosky BJ, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nature biotechnology. 2013;31:166–169. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Turchaninova MA, et al. Pairing of T-cell receptor chains via emulsion PCR. European Journal of Immunology. 2013;43:2507–2515. doi: 10.1002/eji.201343453. [DOI] [PubMed] [Google Scholar]
- 90.Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nature reviews Immunology. 2013;13:294–302. doi: 10.1038/nri3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reay PA, Kantor RM, Davis MM. Use of global amino acid replacements to define the requirements for MHC binding and T cell recognition of moth cytochrome c (93-103) J Immunol. 1994;152:3946–3957. [PubMed] [Google Scholar]
- 92.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- 93.Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 2002;418:552–556. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- 94.Janin J. Protein-protein docking tested in blind predictions: the CAPRI experiment. Mol Biosyst. 2010;6:2351–2362. doi: 10.1039/c005060c. [DOI] [PubMed] [Google Scholar]
- 95.Ritchie DW. Recent progress and future directions in protein-protein docking. Curr Protein Pept Sci. 2008;9:1–15. doi: 10.2174/138920308783565741. [DOI] [PubMed] [Google Scholar]
- 96.Reiser JB, et al. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- 97.Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity. 2013;38:373–383. doi: 10.1016/j.immuni.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Su LF, Davis MM. Antiviral memory phenotype T cells in unexposed adults. Immunological Reviews. 2013;255:95–109. doi: 10.1111/imr.12095. [DOI] [PubMed] [Google Scholar]
- 99.Parameswaran P, et al. Convergent antibody signatures in human dengue. Cell host & microbe. 2013;13:691–700. doi: 10.1016/j.chom.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Crawford F, et al. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol Rev. 2006;210:156–170. doi: 10.1111/j.0105-2896.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 101.Stadinski BD, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nature Immunology. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wen F, Esteban O, Zhao H. Rapid identification of CD4+ T-cell epitopes using yeast displaying pathogen-derived peptide library. Journal of Immunological Methods. 2008;336:37–44. doi: 10.1016/j.jim.2008.03.008. [DOI] [PubMed] [Google Scholar]