

Abstract
The clinical manifestations of glucocorticoid excess include central obesity, hyperglycaemia, dyslipidaemia, electrolyte abnormalities and hypertension. A century on from Cushing's original case study, these cardinal features are prevalent in industrialized nations. Hypertension is the major modifiable risk factor for cardiovascular and renal disease and reflects underlying abnormalities of Na+ homeostasis. Aldosterone is a master regulator of renal Na+ transport but here we argue that glucocorticoids are also influential, particularly during moderate excess. The hypothalamic–pituitary–adrenal axis can affect renal Na+ homeostasis on multiple levels, systemically by increasing mineralocorticoid synthesis and locally by actions on both the mineralocorticoid and glucocorticoid receptors, both of which are expressed in the kidney. The kidney also expresses both of the 11β-hydroxysteroid dehydrogenase (11βHSD) enzymes. The intrarenal generation of active glucocorticoid by 11βHSD1 stimulates Na+ reabsorption; failure to downregulate the enzyme during adaption to high dietary salt causes salt-sensitive hypertension. The deactivation of glucocorticoid by 11βHSD2 underpins the regulatory dominance for Na+ transport of mineralocorticoids and defines the ‘aldosterone-sensitive distal nephron’. In summary, glucocorticoids can stimulate renal transport processes conventionally attributed to the renin–angiotensin–aldosterone system. Importantly, Na+ and volume homeostasis do not exert negative feedback on the hypothalamic–pituitary–adrenal axis. These actions are therefore clinically relevant and may contribute to the pathogenesis of hypertension in conditions associated with elevated glucocorticoid levels, such as the metabolic syndrome and chronic stress.
Introduction
It is over 100 years since Harvey Cushing described the clinical consequences of severe glucocorticoid excess (Cushing, 1912) and although this syndrome remains rare, the cardinal features of central obesity, dyslipidaemia, impaired glucose metabolism and hypertension are increasingly prevalent in Western society (Batsis et al. 2007). These traits of the ‘metabolic syndrome’ endanger cardiovascular health. Indeed, hypertension is the major modifiable risk factor for both cardiovascular and renal disease and reflects impaired Na+ homeostasis and a diminution of the pressure natriuresis mechanisms (Mullins et al. 2006). Here, we review recent studies that provide a mechanistic framework for regulation of renal Na+ transport by glucocorticoids: two overarching themes are developed. First, that defining glucocorticoid action within the kidney is challenging due to the pleiotropic actions of systemic glucocorticoids. Second, that there are multiple instances where the hypothalamic–pituitary–adrenal axis (HPAA) can influence Na+ transport processes that are classically regulated by the renin–angiotensin–aldosterone system (RAAS). Such ‘crosstalk’ may have a physiological context but there is an implicit capacity for aberrant renal Na+ transport as the HPAA is not regulated by Na+/volume homeostasis.
Integrated responses to glucocorticoids
The renal response to systemic glucocorticoid administration is well characterized (Table 1) but attempts to resolve these actions into specific tubular and vascular components are confounded by two phenomena: the pleiotropic effects of glucocorticoids and the promiscuity of steroid receptor–ligand interactions (Fig. 1).
Table 1.
The integrated renal effects of glucocorticoids. The effects of glucocorticoids on renal haemodynamics and tubular transport function can oppose one another. This is discussed further in the main text
| The effects of glucocorticoids on integrated renal function haemodynamic |
| ↑ renal blood flow |
| variable effect on renal vascular resistance |
| ↑ filtration fraction |
| ↑ glomerular filtration rate |
| water and electrolyte metabolism |
| diuresis |
| natriuresis or antinatriuresis – see main text |
| plasma volume contraction (or sometimes volume expansion – see main text) |
| kaliuresis |
| ↑ renal acid excretion and metabolic alkalosis |
| phosphaturia |
| ↑ amino acid transport |
| ↑ sulphate transport |
| intermediate metabolism within the kidney |
| luconeogenesis |
| ammoniagenesis |
Figure 1. Model of glucocorticoid effects on integrated renal function.
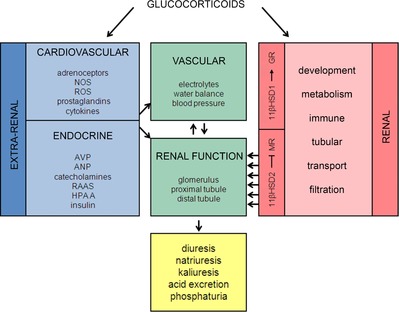
Glucocorticoids act pervasively and therefore their net effect on renal function is determined by extrarenal, haemodynamic and renal tubular factors. The local action of glucocorticoids within the kidney is subject to fine-tuning by the 11βHSD isoforms. The net result is usually a natriuresis, but under some conditions antinatriuresis predominates (see main text). ANP, atrial natriuretic peptide; AVP, arginine vasopressin; GR, glucocorticoid receptor; 11βHSD1/2, 11β-hydroxysteroid dehydrogenase 1/2; HPAA, hypothalamic-pituitary-adrenal axis; NOS, nitric oxide synthase; RAAS, renin–angiotensin–aldosterone system; ROS, reactive oxygen species.
The glucocorticoid receptor (GR) is ubiquitously expressed and systemic administration of glucocorticoids therefore changes many variables, including intermediary metabolism, cardiac output and systemic vascular resistance. This integrated response to glucocorticoids is clinically relevant but it is challenging to identify primary renal events (i.e. those occurring as a direct result of glucocorticoid signalling within the kidney) from secondary responses that are indirect and often countervailing. Thus, in some circumstances glucocorticoids promote renal Na+ retention. This is particularly evident for endogenous glucocorticoids and reflects activation of both GR and mineralocorticoid receptors (MR) in the renal tubule.
In other cases, glucocorticoids – particularly synthetic compounds – induce a powerful natriuresis (Table 1). The conventional explanation for this phenomenon is that the haemodynamic actions of glucocorticoids impair autoregulation, increase glomerular filtration rate (GFR) and, despite the best efforts of glomerulotubular balance, promote natriuresis and kaliuresis. The mechanisms that underpin these haemodynamic effects are not fully defined. Endogenous glucocorticoids certainly exert permissive effects that help maintain both renal blood flow (RBF) and GFR: both are reduced in adrenal insufficiency. Such underperfusion does not relate exclusively to hypotension as RBF is restored by steroid replacement but not by volume replacement alone (Mangos et al. 2003). The effect on renal haemodynamics of exogenous glucocorticoids is more complex and the mechanisms not resolved. Micropuncture evidence in rats found that prednisolone increased single nephron GFR due to dilatation of the glomerular arterioles, with the ultrafiltration coefficient and Starling forces across the glomerular capillary being unaffected (Baylis et al. 1990). Similar data were obtained in dogs (Hall et al. 1980). However, in humans, the glucocorticoid-induced increase in GFR must reflect an increased filtration fraction as RBF remains stable or even falls, causing increased renal vascular resistance (Connell et al. 1987). It is not clear what accounts for these differences but one possibility is differential sensitivity to the catabolic effect of synthetic glucocorticoids as increased renal delivery of amino acids can directly increase RBF (Baylis et al. 1990).
The second confounding phenomenon is the capacity for glucocorticoids to activate MR, exerting effects often antagonistic to their haemodynamic actions. For example, dogs infused with noradrenaline and adrenocorticotrophic hormone (ACTH) become hypertensive and enter a negative Na+ and water balance. However, if the renal perfusion pressure is servo-controlled, hypertension is accompanied by Na+ retention (Woods et al. 1988). Similarly, chronic ACTH infusion into mice increases Na+ excretion (Dunbar et al. 2010) despite the fact that activation of MR and GR promotes Na+ reabsorption in the distal tubule (Bailey et al. 2009). These data underscore the dualistic effect of glucocorticoids, with the direct action on the renal tubules being over-ridden by haemodynamic processes that cause an increase in net urinary Na+ excretion.
Why do glucocorticoids exert these countervailing influences? Glucocorticoids induce a catabolic effect on systemic metabolism, promoting the conversion of protein, glycogen and triglyceride stores to amino acids, glucose and free fatty acids. The increase in GFR meets an increased demand for the excretion of waste products and contributes to the stress response. Teleologically, any direct stimulatory effect of glucocorticoids on tubular Na+ reabsorption would stabilize net excretion in the face of increased GFR, preserving salt and water balance in response to physiological stresses that threaten plasma volume. This effect is analogous to that induced by angiotensin II when activated in response to dietary Na+ restriction or hypovolaemia. Angiotensin II constricts the efferent arteriole to stabilize GFR in the face of reduced perfusion pressure and promotes Na+ retention by activation of transport proteins, including the thiazide-sensitive cotransporter (Ashek et al. 2012) and the epithelial Na+ channel (ENaC; Zaika et al. 2013).
However, the full extent of the physiological (and pathophysiological) role of glucocorticoids is both subtle and complex (Fig. 1). Glucocorticoids influence kidney development and the in utero programming of cardiovascular and renal phenotypes (Habib et al. 2011); they influence the pathogenesis of kidney injury (Rafiq et al. 2011) and they contribute to circadian variation in renal function: the glucocorticoid responsive protein, glucocorticoid-induced leucine zipper (GILZ), which features in the regulatory pathways of key Na+ transporters in the distal nephron (see Fig. 5), shows strong circadian oscillations in the kidney (Zuber et al. 2009).
Figure 5. Paradigm of mineralocorticoid signalling: regulation of ENaC in the principal cell.
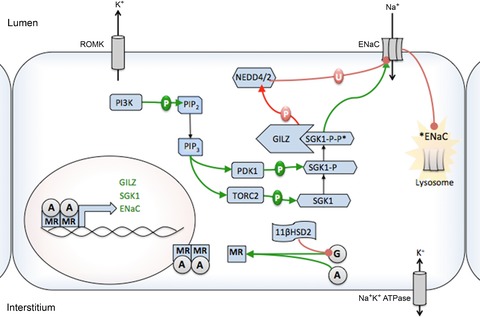
Aldosterone regulates ENaC through MR-binding dependent promotion of SGK-1 expression and ENaCα expression. Two phosphorylation steps, mediated by TORC2 and PDK1, activate SGK1 (SGK1-P-P*). These kinases are activated by the PI3K system. SGK1 phosphorylates Nedd4/2, which is an ubiquitin ligase enzyme that binds ENaC and marks it for withdrawal from the apical membrane and subsequent degradation. SGK1-dependent phosphorylation inhibits Nedd4/2-ENaC binding and promotes the maintenance of ENaC in the membrane. Glucocorticoids are prevented from activating MR in the presence of high levels of 11βHSD2. GILZ expression is also increased upon aldosterone stimulation and is thought to stabilize SGK1. Red connectors represent inhibition through phosphorylation or ubiquitinylation, while green arrows indicate stimulation by phosphorylation or otherwise. Black arrows indicate movement of ions or change to phosphorylated state. A, aldosterone; ENaC, epithelial Na+ channel; G, glucocorticoids; 11βHSD1/2, 11β-hydroxysteroid dehydrogenase 1/2; MR, mineralocorticoid receptor; P, phosphorylation; U, ubiquitinylation.
Interaction between the hypothalamic–pituitary– adrenal axis and the renin–angiotensin–aldosterone system
The HPAA can influence the RAAS at both systemic and local levels. For example, ACTH excess increases the circulating mineralocorticoid ‘load’ in several ways. First, ACTH increases secretion of aldosterone by promoting cholesterol delivery to the mitochondria in the zona glomerulosa cells (Hattangady et al. 2012) and by enhancing CYP11B2 transcription (Takeda et al. 1996). Second, ACTH stimulates production of deoxycorticosterone, a weak mineralocorticoid that is physiologically significant when in excess (Mullins et al. 2009). The stimulatory effect of ACTH on aldosterone seems to be transient, but that on deoxycorticosterone sustained (Dunbar et al. 2010). Finally, ACTH may stimulate renin production in the juxtaglomerular apparatus (Oelkers et al. 1982), although this concept lacks much empirical support.
At a receptor level, glucocorticoids have equal, or perhaps greater, affinity for the MR than does aldosterone (Arriza et al. 1987). This concept is implicated in the hypokalaemia and hypertension of Cushing's syndrome and is discussed in more detail below.
Glucocorticoid signal transduction in the renal tubule
Pre-receptor steroid metabolism governs receptor specificity
GR and MR share a high in vitro affinity for both classes of steroid (Arriza et al. 1987), but differ in their binding kinetics. The renal MR constitutes a high-affinity, low-capacity corticosteroid-binding site (formerly designated ‘type I’), Kd 0.5–3 nm for both aldosterone and cortisol. GRs offer a low-affinity, high-capacity ‘type II’ site, Kd 20–65 nm, for both steroids. The in vivo specificity of the GR and MR for their cognate ligands is, at least in part, a property conferred by the pre-receptor metabolism of glucocorticoids by the 11β-hydroxysteroid dehydrogenase isozymes: type 1 (11βHSD1) converts inactive 11-keto derivatives of glucocorticoids into physiologically active cortisol (corticosterone in rodents), and type 2 (11βHSD2) catalyses the reverse reaction (Chapman et al. 2013). Thus 11βHSD2 confers upon MR specificity for aldosterone that is inherently lacking: cortisol activates MR whereas cortisone does not (Fig. 2). Inhibition of 11βHSD2 activity (using derivatives of glycyrrhetinic acid, the active ingredient of liquorice) promotes Na+ reabsorption and potassium secretion in the distal nephron (Bailey et al. 2001). Genetic ablation of the enzyme, as occurs in apparent mineralocorticoid excess syndrome, causes low renin hypertension and hypokalaemia due in part to increased Na+ reabsorption in the distal nephron (Stewart et al. 1996; Bailey et al. 2008).
Figure 2. Glucocorticoid signal transduction apparatus.
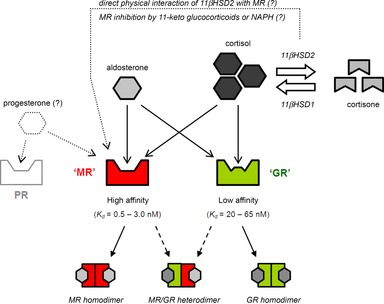
In the kidney, glucocorticoids activate a set of receptors, GR, with low affinity and high capacity. However, they also have the capacity to activate high-affinity MR, but are prevented from doing so in vivo by 11βHSD2, which is expressed in mineralocorticoid-sensitive tissues. GR and MR may form heterodimers but the influence these have on Na+ transport is unknown. Progesterone can influence electrolyte transport by binding MR and/or its cognate receptor. GR, glucocorticoid receptor; 11βHSD1/2, 11β-hydroxysteroid dehydrogenase 1/2; MR, mineralocorticoid receptor; PR, progesterone receptor.
Nevertheless, the physiological role of 11βHSD2 in the kidney is more complex. The pre-receptor metabolism of glucocorticoids by the enzyme will protect MR but cells of the distal tubule also express GR. It is unlikely that these receptors are physiologically redundant and their activation will be influenced by 11βHSD2-mediated metabolism of cortisol. In heterologous expression systems, 11βHSD2 appears able to influence the subcellular localization of MR, possibly through a direct physical interaction. Furthermore, cortisone blocks the interaction between aldosterone and MR, suggesting that the ‘inactive’ 11-keto-glucocorticoids generated by 11βHSD2 may act as autocrine or paracrine MR antagonists (Odermatt et al. 2001). NAPH generation by 11βHSD2 can also alter the intracellular redox potential, locking MR–cortisol complexes in an inactive state (Funder, 2010). Thus, glucocorticoids can bind MR but the receptor is not physiologically activated unless excess reactive oxygen species are present. This adverse redox environment is generated in the kidney of salt-sensitive animals after a period of high Na+ intake: glucocorticoid-induced activation of MR promotes inflammation and fibrosis (Luther et al. 2012).
There is also the potential for interaction with sex steroids, as progesterone acts as a partial agonist for MR and GR (Arriza et al. 1987). Moreover, bona fide progesterone receptors are also expressed in the distal nephron, where they probably participate in the regulation of solute transport. Progesterone derived from the adrenal gland promotes renal potassium retention in male and ovariectomized female potassium-depleted mice (Elabida et al. 2011). This probably reflects direct signalling via the progesterone receptor as activation of GR or MR would be kaliuretic. Moreover, the potassium retention was blocked by RU486, an antagonist of the progesterone receptor. RU486 also antagonizes GR but the progesterone-induced potassium retention was not associated with induction of classic GR response genes.
Renal expression of glucocorticoid receptor, mineralocorticoid receptor and 11β-hydroxysteroid dehydrogenase isozymes
The expression patterns of MR and 11βHSD within the kidney are depicted in Fig. 3: GR is widely expressed in the kidney, with mRNA being detected in most cells. In contrast, MR and 11βHSD2 have a more restricted distribution: co-localization of these in the connecting tubule and principal cells of the collecting duct defines the ‘aldosterone-sensitive distal nephron’ (ASDN). The expression of 11βHSD2 in the distal convoluted tubule (DCT) is less certain and it is probable that the enzyme is not expressed at high levels, if at all (Bostanjoglo et al. 1998; Campean et al. 2001). Although DCT cells express both GR and MR and are responsive to both corticosteroids, the conventional model of the ASDN does not apply in this segment. This is an important concept as the DCT reabsorbs more of the filtered Na+ load (∼7%) than does the collecting duct (<5%). Abnormal glucocorticoid status could, via activation of the thiazide-sensitive transporter in the DCT, imperil Na+ and blood pressure homeostasis.
Figure 3. Renal sites of MR and 11βHSD expression.
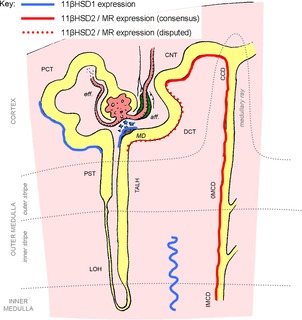
Gluocorticoid receptors are expressed throughout the renal tubule and in the glomerulus and are not shown on this figure. See main text for details. aff., afferent; CCD, cortical collecting duct; CNT, connecting tubule; DCT, distal convoluted tubule; eff., efferent; 11βHSD1/2, 11β-hydroxysteroid dehydrogenase 1/2; IMCD, inner medullary collecting duct; LOH, loop of Henle; MD, macula densa; MR, mineralocorticoid receptor; OMCD, outer medullary collecting duct; PCT, proximal convoluted tubule; PST, proximal straight tubule; TALH, thick ascending limb of Henle's loop.
Several groups have proposed that 11βHSD2 abundance decreases along a gradient as one moves proximally from the cortical collecting duct, through the connecting tubule, to DCT. This has led to speculation that there may be an ‘ASDN proper’ in which aldosterone dominates regulation of Na+ transport through MR, and an intermediate segment expressing low levels of 11βHSD2 in which MR is activated by aldosterone under basal conditions and/or by glucocorticoids if they are present in excess, during activation of the HPAA or at certain periods during the circadian cycle (Gaeggeler et al. 2005). It is also possible that 11βHSD2 expression/activity is physiologically regulated by Na+ and K+ status (Thompson et al. 2000): DCT and the connecting tubule are plastic, homeostatically responsive epithelia capable of rapid remodelling of the molecular apparatus for Na+ transport.
11β-Hydroxysteroid dehydrogenase 1 and the glucocorticoid-amplified proximal nephron
11βHSD1 is located in the S3 proximal tubule (Fig. 3) and macula densa (Gong et al. 2008; Odermatt & Kratschmar, 2012). It has also been detected in the interstitial cells of the renal medulla (Castello et al. 1989; Rundle et al. 1989). As these cells lack the hexose-6-phosphate dehydrogenase and cannot generate NADPH, 11βHSD1 may therefore act as a dehydrogenase here, catalysing the same reaction as 11βHSD2 (Gomez-Sanchez et al. 2008). The urinary steroid profile following siRNA knockdown of medullary 11βHSD1 suggests that this is not correct and 11βHSD1 acts predominantly as a reductase (Liu et al. 2008). This leads to the concept of a ‘glucocorticoid-amplified proximal nephron’ and detailed physiological examination of 11βHSD1 activity in the renal tubule is required. Data are surprisingly scarce but downregulation of renal 11βHSD1 is an adaptive response to either salt loading or increased blood pressure (Dunbar et al. 2010). Failure to transcriptionally repress the encoding gene, hsd11b1, contributes to the pathogenesis of salt-sensitive hypertension. This phenomenon was demonstrated in innovative studies using the Dahl salt-sensitive (DSS) rat and a consomic control strain (DSS-13BN) with attenuated salt sensitivity of blood pressure. Renal medullary expression of 11βHSD1 was downregulated in response to high dietary salt in DSS-13BN but not in DSS rats. The authors hypothesized that failure to downregulate 11βHSD1 contributed to renal Na+ retention and hypertension. To test this hypothesis, knockdown of 11βHSD1 expression/activity was induced by injecting siRNA into the renal medulla in vivo: hsd11b1 knockdown attenuated salt-sensitive hypertension in the DSS rats (Liu et al. 2008). The molecular mechanism underpinning the rescue of salt-sensitive blood pressure was not determined. It may be that 11βHSD1 knockdown had a direct impact on Na+ transport processes in the medullary tubular epithelium (i.e. on NKCC2 function in the thick ascending limb of Henle's loop). There is no consensus in the literature concerning the effect of glucocorticoids on NKCC2 activity, with both transcriptional repression of slc12a1 (Bailey et al. 2009) and increased abundance of the protein (Frindt & Palmer, 2012) being reported. Nevertheless, hsd11b1 knockdown in the renal medulla resulted in a reduction in the concentration of corticosterone in the urine. This raises the possibility that 11βHSD1 in the interstitial cells of the medulla exerts a paracrine effect on transport processes in the distal nephron by altering the concentration of active glucocorticoids in the downstream tubular fluid and/or peritubular capillaries.
Glucocorticoid receptor in the distal nephron
Classical studies of receptor–ligand interactions in collecting duct cells in vitro demonstrate that mineralocorticoids can bind to the GR. Indeed, binding assays indicate that physiological concentrations of aldosterone would induce a low level of GR occupancy, but the biological significance of this is not clear (Gaeggeler et al. 2005). An interaction between aldosterone status and the GR has been demonstrated in mouse and rat kidneys (Ackermann et al. 2010), using nuclear translocation as a proxy for receptor activation. Contrary to our understanding of the ASDN, suppression of aldosterone by dietary NaCl loading resulted in a reduction in the nuclear localization of GR in the ASDN whereas MR localization was not affected. Conversely, adrenalectomy resulted in the loss of nuclear MR and GR in all nephron segments. The nuclear MR signal was restored in all nephron segments by physiological doses of corticosterone. The GR signal was restored in most nephron segments but not in the ASDN. Furthermore, in a colonic cell line expressing both receptors (an unusual phenomenon in epithelial cell lines), GR activation did not itself induce ENaC but was a prerequisite for the full MR-mediated response to aldosterone (Bergann et al. 2011). Similar findings have been reported in neuronal cell lines (Tsugita et al. 2009). The interaction between the receptors is not understood but GR may be sine qua non for formation of the MR/MR homodimer or may even form a heterodimer with MR (Fig. 2), as is suggested by FRET microscopy (Nishi et al. 2004).
These data challenge our conventional view of the steroid control of Na+ transport in the ASDN and the consequences for Na+ transport are not clear. If glucocorticoids, via GR, physiologically regulate MR, an important homeostatic role for 11βHSD2 may be to control intracellular glucocorticoid concentration and thereby govern GR activation.
Signal transduction downstream of steroid receptor activation
Activated steroid receptors translocate to the nucleus, where they act as transcription factors. There is considerable overlap in the GR and MR response genes, providing an additional mechanism through which both classes of steroid can activate a common set of biological effector pathways.
Genomic responses to glucocorticoids: well-characterized pathways
GR activation in the collecting duct stimulates the transcription of Sgk1 and GILZ (Muller et al. 2003; Nguyen Dinh Cat et al. 2009). However, the role of Sgk1 in the renal response to glucocorticoids in vivo remains obscure. Dexamethasone increases the abundance of Sgk1 transcripts in whole kidney homogenates. Our data indicate that Sgk1 is physiologically active: dexamethasone increases the phosphorylation of the Sgk1 target NDRG1 (Fig. 4). Dexamethasone does not, however, increase Sgk1 expression in isolated cortical collecting ducts (Muller et al. 2003); Sgk1 expression in the distal renal tubule was not altered in response to overexpression of GR in the collecting duct (Nguyen Dinh Cat et al. 2009). These observations suggest that whereas Sgk1 participates in the response to glucocorticoids in some kidney cells, it does not do so in the ASDN, where some (yet unknown) mechanisms preserve Sgk1 as an aldosterone-responsive gene. Dexamethasone upregulates NHE3 activity in cultured renal cells in an Sgk1-dependent fashion, providing in vitro evidence that Sgk1-dependent pathways may participate in glucocorticoid-regulated solute transport in the proximal nephron (Wang et al. 2007).
Figure 4. Dexamethasone increases the abundance of phosphorylated NDRG1 (P-NDRG1-Thr346,356,366) in whole mouse kidney, indicative of increased SgK1 activity.
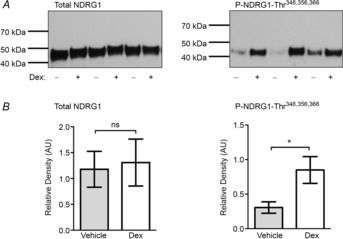
C57BL6 mice were treated with dexamethasone (1 mg kg−1) or vehicle (0.9% saline) and kidneys collected after 6 h. A, kidneys were probed with antibodies to the phosphorylated form of NDRG1 (P-NDRG1-Thr346,356,366) or total NDRG1 (T-NDRG1). NDRG1 is a substrate for SGK1 and this phosphoprotein is a surrogate indicator of SGK1 activity. B, densitometry analysis indicated P-NDRG1 was significantly increased in the dexamethasone-treated group but there was no change in the T-NDRG1. Data are means ± s.e.m., n = 6. *P < 0.05, by Student's t test. Dex, dexamethasone.
Both Sgk1 and GILZ are also classic MR response genes (Fig. 5). The Sgk1-Nedd4–2-ENaC pathway provides the canonical mechanism whereby aldosterone stimulates Na+ reabsorption in the principal cell (Snyder et al. 2002); the Sgk1-Nedd4–2 pathway also operates in the DCT to stimulate NCC (Arroyo et al. 2011). In cultured collecting duct (mpkCCD) cells, GILZ participates in the regulation of ENaC activity by aldosterone (Soundararajan et al. 2005).
Genomic responses to glucocorticoids: an unbiased approach
We have made a systematic attempt to identify the renal transcriptional response to glucocorticoids in mice exposed to 12 days of exogenous ACTH. mRNA prepared from whole kidneys was hybridized with an Affymetrix GeneChip (Santa Clara, CA, USA), and the results subjected to a pathway analysis (Dunbar et al. 2010). This model predominantly reflects glucocorticoid-mediated signalling, the stimulation by ACTH of aldosterone production (vide supra) being transient (Dunbar et al. 2010). A large number of genes were differentially regulated, including known targets such as Sgk1 as well as novel gene pathways concerned with organic anion/cation transport, vitamin D, calcium and xenobiotic metabolism.
The WNK-SPAK cascade
The WNK-SPAK kinase network acts as a master regulator of electrolyte transport in the distal renal tubule, and can transduce mineralocorticoid signals (Hoorn et al. 2011). Sgk1 phosphorylates WNK4 at ser1169, thus relieving inhibition by WNK4 of downstream targets, including NCC, ENaC and ROMK (Ring et al. 2007; Rozansky et al. 2009).
However, GR can also regulate WNK signalling. WNK4 expression is reduced when GR is overexpressed in the collecting duct (Nguyen Dinh Cat et al. 2009) and GR negatively regulates WNK4 transcription in mpkDCT cells and in the murine DCT in vivo (Mu et al. 2011) (vide infra). Basal WNK4 mRNA expression is higher in mice lacking GR in the distal nephron, suggesting that endogenous glucocorticoids exert a tonic antinatriuretic effect through their effects on WNK signalling (Mu et al. 2011).
Glucocorticoid effects on tubule Na+ transport
The systemic administration of glucocorticoids induces effects on solute transport processes along the length of the renal tubule. For example, dexamethasone treatment in rats causes an increase in the abundance of NHE3, NCC, NKCC2, the full-length isoform of α-ENaC and the cleaved isoform of γ-ENaC in whole kidney homogenates (Frindt & Palmer, 2012). Glucocorticoids influence cellular morphology and proliferation in the distal renal tubule, causing amplification of the basolateral membranes of the principal cells in the cortical collecting tubules in the rabbit (Wade et al. 1979). However, these studies are unable to discriminate between the specific effects of glucocorticoid signalling in renal tubular cells and a ‘passive’ response to glucocorticoid-induced changes in systemic haemodynamics and/or intermediate metabolism.
Glucocorticoid effects in the proximal tubule
Regulation of NHE3 and Na+-Pi cotransporter 2
In the rat glucocorticoids stimulate Na+ bicarbonate reabsorption by activating NHE3 (Zallocchi et al. 2003) and suppress sodium phosphate cotransport by Na+-Pi cotransporter 2 (Loffing et al. 1998). These events contribute to glucocorticoid-induced increases in the renal excretion of acid and phosphate (Table 1).
Glucocorticoid effects in the distal convoluted tubule
Glucocorticoids stimulate NaCl reabsorption in the DCT. The underlying molecular mechanisms show the DCT to be a site at which several key natritropic signals interact. In a mouse model of salt-sensitive hypertension with sympathetic activation, an epigenetic interaction between adrenergic and glucocorticoid signalling is indicated. This effect is mediated by the WNK kinases and results in the regulation of NCC expression, phosphorylation and transport activity (Mu et al. 2011). β-adrenergic stimulation activated NCC by suppressing WNK4 expression through a GR-dependent mechanism. Activated β2-adrenoreceptors induced histone acetylation at negative glucocorticoid response elements in the WNK4 promoter, enhancing GR binding. There was corresponding negative regulation of Wnk4 by GR in the DCT in vivo: adrenalectomy or a GR antagonist abolished the inhibitory effect of noradrenaline on Wnk4 mRNA expression (and this was restored by dexamethasone in the case of adrenalectomy).
As WNK4 is itself a negative regulator of NCC activity, these findings support a model in which GR activation in the DCT tonically stimulates NaCl reabsorption. Consistent with this, basal Wnk4 mRNA abundance was higher in mice lacking GR in the distal nephron.
Glucocorticoid effects in the aldosterone-sensitive distal nephron: in vitro and in vivo data
The ability of glucocorticoids to stimulate electrogenic Na+ transport in the collecting ducts has been demonstrated in vitro using cultured cell lines that faithfully maintain many of the in vivo characteristics of principal cells (Naray-Fejes-Toth & Fejes-Toth, 1990; Laplace et al. 1992; Bens et al. 1999; Gaeggeler et al. 2005). In mCCDcl1 cells, corticosterone stimulates amiloride-sensitive transport even in the presence of intact 11βHSD2 (Gaeggeler et al. 2005), albeit at higher concentrations than aldosterone (K½ = 18 nm for corticosterone; 0.52 nm for aldosterone). High concentrations of dexamethasone stimulate this current via an RU486-sensitive pathway, suggesting that this is a consequence – at least in part – of GR activation. The mechanisms whereby GR stimulates Na+ transport in the collecting duct remain to be established: regulation of endothelin-1 expression has been implicated in vitro (Stow et al. 2012).
Glucocorticoid effects on Na+ transport in the ‘aldosterone-sensitive distal nephron’ in vivo
Despite the robust in vitro data, a direct demonstration that glucocorticoids physiologically activate ENaC in vivo has been lacking. Seven days of treatment with dexamethasone increased the abundance of the full-length isoform of α-ENaC in rat kidney, but had no effect on electrogenic Na+ transport in split open collecting ducts (Frindt & Palmer, 2012). Mice heterozygotes for a null mutation in Hsd11b2 (the gene encoding 11βHSD2) have salt-sensitive blood pressure associated with elevated levels of circulating glucocorticoids (Bailey et al. 2011). Salt loading induced hypokalaemia in hsd11b2+/− mice, and the trans-tubular potassium gradient >7 indicated enhanced ‘mineralocorticoid’ activity in the distal nephron. The salt sensitivity and hypokalaemia were both corrected by GR antagonism but not by MR antagonism, suggesting that GR activation might exert a pathophysiological role in the ASDN.
The MR knockout mouse provides further evidence of glucocorticoids exerting mineralocorticoid effects in the distal nephron. Global constitutive knockout in MR is fatal; the animals die from excessive urinary solute losses at about day 10, indicating that GR is not able to completely compensate for the lack of MR (Berger et al. 1998). However, glucocorticoids are capable of a partial compensation: triamcinolone treatment enhanced ENaC expression and activity in salt-supplemented MR knockout mice (Schulz-Baldes et al. 2001).
The direct effects of GR signalling in the distal nephron has been investigated using two transgenic mouse models: Ksp-Cre+ GRloxP/loxP mice, in which GR was specifically and constitutively deleted in the AQP2-positive distal nephron (Goodwin et al. 2010), and Hoxb7-tetON2-hGR mice in which GR was conditionally overexpressed in cells of collecting duct lineage (Nguyen Dinh Cat et al. 2009). Loss of GR from the distal nephron had no effect on the response to chronic dexamethasone, which induced a rise in blood pressure and a natriuresis that was indistinguishable from that in wild-type mice (Goodwin et al. 2010). However, basal blood pressure was higher in Ksp-Cre+ GRloxP/loxP mice, raising the possibility that GR activity in the ASDN participates in blood pressure homeostasis in the ‘normal healthy’ adult. This being a constitutive knockout, a developmental effect cannot be discounted. Overexpression of GR in the distal nephron had no effect on net urinary Na+ or K+ excretion, although there was a reduction in urinary aldosterone indicative of compensatory RAAS inhibition (Nguyen Dinh Cat et al. 2009). There was a clear transcriptional response to GR overexpression (discussed above). Taken together, these data suggest that GR-mediated signalling may modulate Na+ transport in the collecting duct, but the regulation of net renal Na+ excretion is physiologically dominated by the RAAS. Moreover, it is possible that some of the effects of mineralocorticoids in the distal nephron may be mediated independently of MR and GR. In rats chronically infused with aldosterone, combined MR and GR blockade did not prevent the increase in ENaC protein abundance and trafficking to the apical cell membrane (Nielsen et al. 2007). The physiological effect of this is uncertain: spironolactone did rescue the hypokalaemia and prevented the increase in Na+/K+-ATPase abundance, suggesting that electrogenic Na+ transport in the principal cell was reduced, despite the continued presence of ENaC in the apical membrane.
Implications for hypertension and salt sensitivity in humans
The renal tubules possess the molecular apparatus requisite for glucocorticoid-responsive Na+ transport. The expression of 11βHSD1 and 11βHSD2 will influence these processes by regulating local glucocorticoid concentrations. Several lines of evidence challenge the conventional role of renal 11βHSD2 as a mere enzymatic guardian of the MR. Equally compelling, is evidence showing stimulation by glucocorticoids of Na+ reabsorption at several sites along the nephron. More provocatively, experiments in cell lines and in native kidney indicate that GR has an important permissive role for aldosterone signalling at MR. The HPAA is thus able to influence renal Na+ excretion at multiple levels (Fig. 6) – and such regulation escapes the negative feedback mechanisms inherent within the RAAS.
Figure 6. Interactions between the RAAS and the HPAA.
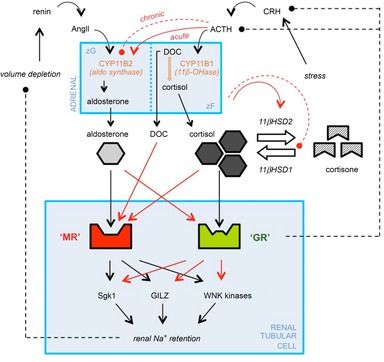
Dashed arrows (with filled circles at the head) represent negative feedback loops. Red arrows are potential sites of crosstalk between the RAAS and the HPAA. Interactions arise at multiple levels: pre-receptor (changes in ligand availability), receptor (receptor–ligand promiscuity) and post-receptor (common second messenger systems). HPAA activation can increase the activity of 11βHSD2 and inhibit 11βHSD1. The other potential routes of crosstalk are discussed in the main text. ACTH, adrenocorticotrophic hormone; AngII, angiotensin II; CRH, corticotrophin releasing hormone; DOC, deoxycorticosterone; HPAA, hypothalamic–pituitary–adrenal axis; 11βHSD1/2, 11β-hydroxysteroid dehydrogenase 1/2; MR, mineralocorticoid receptor; RAAS, renin–angiotensin–aldosterone system.
However, is this relevant for human health? Many of the mechanisms whereby glucocorticoids cause hypertension reside in the vasculature, and central and autonomic nervous systems, but there is a clear contribution from an antinatriuretic effect in the renal tubules. This is clinically important: salt-sensitive humans have an enhanced stress-induced activation of the HPAA (Weber et al. 2008) and attenuated glucocorticoid clearance (Kerstens et al. 2003). Glucocorticoids stimulate electrogenic Na+ transport in humans when present in excess, as in Cushing's syndrome. We do not know, however, if this contributes to hypertension in states of moderate glucocorticoid excess or in the metabolic syndrome, where the tissue availability of active glucocorticoid is enhanced (Pereira et al. 2012). Hsd11b2 gene polymorphisms are associated with salt sensitivity in blood pressure in normotensive and hypertensive subjects, suggesting that glucocorticoids can breach the 11βHSD2 barrier even when they are not present in vast excess.
The molecular pathways whereby glucocorticoids contribute to salt-sensitive hypertension have been elucidated in mice. These studies provide a mechanistic explanation for the long-recognized ability of combined glucocorticoid and adrenergic stimulation to exert tubular antinatriuretic effects. Mechanisms in the distal nephron predominate: the NCC is activated during sympathetic stimulation (Mu et al. 2011) and ENaC is increased in 11βHSD2 heterozygotes (Craigie et al. 2012). These pathways are attractive therapeutic targets to ameliorate the salt-sensitive hypertension associated with chronic stress and other states of sympathetic and HPAA activation in humans. There is also a pressing clinical need to develop a mechanistic understanding of the effects of renal 11βHSD1 activity on Na+ transport. Systemic 11βHSD1 inhibitors are in development for use as modifiers of cardiovascular risk in obesity, type 2 diabetes mellitus and the metabolic syndrome (Hughes et al. 2008; Hadoke et al. 2009), conditions in which salt-sensitive hypertension is prevalent (Hall, 2003; Fujita, 2010). One potential side-benefit of 11βHSD1 inhibition in such cases might be an improvement in salt sensitivity because of diminished active glucocorticoid generation in the renal medulla. An antihypertensive effect of 11βHSD1 inhibitors has recently been reported in a clinical trial and in the spontaneously hypertensive rat (Bauman et al. 2013).
Conclusion
Conditions associated with modest elevations in circulating glucocorticoids are common. Moreover, the widespread therapeutic use of GR agonists may flatten the dynamic regulation of the HPAA with deleterious consequences for renal Na+ homeostasis. Glucocorticoids regulate Na+ transport in the proximal and distal renal tubule and, in particular, can stimulate Na+ reabsorption in the post-macular segments. 11βHSD2 activity is low in the DCT, which is emerging as a critical site for the regulation of renal Na+ excretion by various signalling pathways, including glucocorticoids. These effects have implications for human health and disease, with the potential to contribute to the pathogenesis of Na+-sensitive hypertension in the metabolic syndrome and in chronic stress.
Acknowledgments
We thank Dr Chris Kenyon for his critical appraisal of the manuscript and for Fig. 1, Prof. Stuart Wilson for discussion of data, The British Heart Foundation Centre of Research Excellence Award and Kidney Research UK for research funding. RWH was supported by a Clinical Research Fellowship from The Wellcome Trust; JRI by a British Heart Foundation 4-year PhD studentship.
Biography
Robert W. Hunter is a Clinical Lecturer on the Edinburgh Clinical Academic Track (ECAT) and a Speciality Registrar in Nephrology and General Internal Medicine. He received a BA in Physiology from the University of Cambridge and a BM BCh from the University of Oxford. His PhD, from the University of Edinburgh, focused on structure/function relationships in the distal renal tubule. Jessica Ivy is a BritishHeart Foundation PhD student at the University of Edinburgh. Jessica has a First Class degree in Pharmacology and an MSc with Distinction in Cardiovascular Biology, both from the University of Edinburgh. Her PhD combines in vivo approaches for cardiovascular and renal physiology with molecular techniques to explore the contribution of glucocorticoid excess to salt-sensitive hypertension. Matt Bailey is a renal physiologist with a PhD from the University of London. He had postdoctoral training at UCL and CNRS in Saclay, France. A Wellcome Trust Fellowship took him to Yale University to work with Gerhard Giebisch and Steve Hebert and he moved to Edinburgh in 2003, where he is currently a Senior Lecturer. Matt started his career as a single nephron micropuncturist, developing electrophysiological approaches to assess transepithelial electrolyte flux in vivo. At Edinburgh, he became interested in human disease and currently investigates renal function in mouse models of hypertension.
Additional information
Competing interests
None declared.
Author contributions
All authors approved the final version for publication.
References
- Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol. 2010;299:F1473–F1485. doi: 10.1152/ajprenal.00437.2010. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- Arroyo JP, Lagnaz D, Ronzaud C, Vazquez N, Ko BS, Moddes L, Ruffieux-Daidie D, Hausel P, Koesters R, Yang B, Stokes JB, Hoover RS, Gamba G, Staub O. Nedd4–2 modulates renal Na+-Cl− cotransporter via the aldosterone-SGK1-Nedd4–2 pathway. J Am Soc Nephrol. 2011;22:1707–1719. doi: 10.1681/ASN.2011020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashek A, Menzies RI, Mullins LJ, Bellamy CO, Harmar AJ, Kenyon CJ, Flatman PW, Mullins JJ, Bailey MA. Activation of thiazide-sensitive co-transport by angiotensin II in the cyp1a1-Ren2 hypertensive rat. PLoS One. 2012;7:e36311. doi: 10.1371/journal.pone.0036311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MA, Unwin RJ, Shirley DG. In vivo inhibition of renal 11β-hydroxysteroid dehydrogenase in the rat stimulates collecting duct sodium reabsorption. Clin Sci (Lond) 2001;101:195–198. [PubMed] [Google Scholar]
- Bailey MA, Paterson JM, Hadoke PW, Wrobel N, Bellamy CO, Brownstein DG, Seckl JR, Mullins JJ. A switch in the mechanism of hypertension in the syndrome of apparent mineralocorticoid excess. J Am Soc Nephrol. 2008;19:47–58. doi: 10.1681/ASN.2007040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MA, Mullins JJ, Kenyon CJ. Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of Cushing syndrome. Hypertension. 2009;54:890–896. doi: 10.1161/HYPERTENSIONAHA.109.134973. [DOI] [PubMed] [Google Scholar]
- Bailey MA, Craigie E, Livingstone DE, Kotelevtsev YV, Al-Dujaili EA, Kenyon CJ, Mullins JJ. Hsd11b2 haploinsufficiency in mice causes salt sensitivity of blood pressure. Hypertension. 2011;57:515–520. doi: 10.1161/HYPERTENSIONAHA.110.163782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsis JA, Nieto-Martinez RE, Lopez-Jimenez F. Metabolic syndrome: from global epidemiology to individualized medicine. Clin Pharmacol Ther. 2007;82:509–524. doi: 10.1038/sj.clpt.6100355. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Whitehead A, Contino LC, Cui J, Garcia-Calvo M, Gu X, Kevin N, Ma X, Pai LY, Shah K, Shen X, Stribling S, Zokian HJ, Metzger J, Shevell DE, Waddell ST. Evaluation of selective inhibitors of 11β-HSD1 for the treatment of hypertension. Bioorg Med Chem Lett. 2013;23:3650–3653. doi: 10.1016/j.bmcl.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Baylis C, Handa RK, Sorkin M. Glucocorticoids and control of glomerular filtration rate. Semin Nephrol. 1990;10:320–329. [PubMed] [Google Scholar]
- Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999;10:923–934. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- Bergann T, Fromm A, Borden SA, Fromm M, Schulzke JD. Glucocorticoid receptor is indispensable for physiological responses to aldosterone in epithelial Na+ channel induction via the mineralocorticoid receptor in a human colonic cell line. Eur J Cell Biol. 2011;90:432–439. doi: 10.1016/j.ejcb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schutz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9429. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostanjoglo M, Reeves WB, Reilly RF, Velazquez H, Robertson N, Litwack G, Morsing P, Dorup J, Bachmann S, Ellison DH. 11β-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9:1347–1358. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- Campean V, Kricke J, Ellison D, Luft FC, Bachmann S. Localization of thiazide-sensitive Na+-Cl− cotransport and associated gene products in mouse DCT. Am J Physiol Renal Physiol. 2001;281:F1028–F1035. doi: 10.1152/ajprenal.0148.2001. [DOI] [PubMed] [Google Scholar]
- Castello R, Schwarting R, Muller C, Hierholzer K. Immunohistochemical localization of 11β-hydroxysteroid dehydrogenase in rat kidney with monoclonal antibody. Ren Physiol Biochem. 1989;12:320–327. doi: 10.1159/000173209. [DOI] [PubMed] [Google Scholar]
- Chapman K, Holmes M, Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–1206. doi: 10.1152/physrev.00020.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens. 1987;5:425–433. [PubMed] [Google Scholar]
- Craigie E, Evans LC, Mullins JJ, Bailey MA. Failure to downregulate the epithelial sodium channel causes salt sensitivity in Hsd11b2 heterozygote mice. Hypertension. 2012;60:684–690. doi: 10.1161/HYPERTENSIONAHA.112.196410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing H. The Pituitary Body and its Disorders: clinical states produced by disorders of the hypophysis cerebri. J.B. Lippincott Company; 1912. [Google Scholar]
- Dunbar DR, Khaled H, Evans LC, Al-Dujaili EA, Mullins LJ, Mullins JJ, Kenyon CJ, Bailey MA. Transcriptional and physiological responses to chronic ACTH treatment by the mouse kidney. Physiol Genomics. 2010;40:158–166. doi: 10.1152/physiolgenomics.00088.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elabida B, Edwards A, Salhi A, Azroyan A, Fodstad H, Meneton P, Doucet A, Bloch-Faure M, Crambert G. Chronic potassium depletion increases adrenal progesterone production that is necessary for efficient renal retention of potassium. Kidney Int. 2011;80:256–262. doi: 10.1038/ki.2011.15. [DOI] [PubMed] [Google Scholar]
- Frindt G, Palmer LG. Regulation of epithelial Na+ channels by adrenal steroids: mineralocorticoid and glucocorticoid effects. Am J Physiol Renal Physiol. 2012;302:F20–F26. doi: 10.1152/ajprenal.00480.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension. 2010;55:813–818. doi: 10.1161/HYPERTENSIONAHA.109.149062. [DOI] [PubMed] [Google Scholar]
- Funder JW. Aldosterone and mineralocorticoid receptors in the cardiovascular system. Prog Cardiovasc Dis. 2010;52:393–400. doi: 10.1016/j.pcad.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger JD, Rossier BC. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16:878–891. doi: 10.1681/ASN.2004121110. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez EP, Romero DG, Rodriguez de AF, Warden MP, Krozowski Z, Gomez-Sanchez CE. Hexose-6-phosphate dehydrogenase and 11β-hydroxysteroid dehydrogenase-1 tissue distribution in the rat. Endocrinology. 2008;149:525–533. doi: 10.1210/en.2007-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Morris DJ, Brem AS. Human renal 11β-hydroxysteroid dehydrogenase 1 functions and co-localizes with COX-2. Life Sci. 2008;82:631–637. doi: 10.1016/j.lfs.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Goodwin JE, Zhang J, Velazquez H, Geller DS. The glucocorticoid receptor in the distal nephron is not necessary for the development or maintenance of dexamethasone-induced hypertension. Biochem Biophys Res Commun. 2010;394:266–271. doi: 10.1016/j.bbrc.2010.02.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib S, Gattineni J, Twombley K, Baum M. Evidence that prenatal programming of hypertension by dietary protein deprivation is mediated by fetal glucocorticoid exposure. Am J Hypertens. 2011;24:96–101. doi: 10.1038/ajh.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadoke PWF, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br J Pharmacol. 2009;156:689–712. doi: 10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41:625–633. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- Hall JE, Morse CL, Smith MJ, Jr, Young DB, Guyton AC. Control of arterial pressure and renal function during glucocorticoid excess in dogs. Hypertension. 1980;2:139–148. doi: 10.1161/01.hyp.2.2.139. [DOI] [PubMed] [Google Scholar]
- Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol. 2012;350:151–162. doi: 10.1016/j.mce.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoorn EJ, Nelson JH, McCormick JA, Ellison DH. The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol. 2011;22:605–614. doi: 10.1681/ASN.2010080827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes KA, Webster SP, Walker BR. 11-Beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors in type 2 diabetes mellitus and obesity. Expert Opin Investig Drugs. 2008;17:481–496. doi: 10.1517/13543784.17.4.481. [DOI] [PubMed] [Google Scholar]
- Kerstens MN, Kleij van der FG, Boonstra AH, Sluiter WJ, Koerts J, Navis G, Dullaart RP. Salt loading affects cortisol metabolism in normotensive subjects: relationships with salt sensitivity. J Clin Endocrinol Metab. 2003;88:4180–4185. doi: 10.1210/jc.2002-021625. [DOI] [PubMed] [Google Scholar]
- Laplace JR, Husted RF, Stokes JB. Cellular responses to steroids in the enhancement of Na+ transport by rat collecting duct cells in culture. Differences between glucocorticoid and mineralocorticoid hormones. J Clin Invest. 1992;90:1370–1378. doi: 10.1172/JCI116003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Singh RJ, Usa K, Netzel BC, Liang M. Renal medullary 11β-hydroxysteroid dehydrogenase type 1 in Dahl salt-sensitive hypertension. Physiol Genomics. 2008;36:52–58. doi: 10.1152/physiolgenomics.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffing J, Lotscher M, Kaissling B, Biber J, Murer H, Seikaly M, Alpern RJ, Levi M, Baum M, Moe OW. Renal Na/H exchanger NHE-3 and Na-PO4 cotransporter NaPi-2 protein expression in glucocorticoid excess and deficient states. J Am Soc Nephrol. 1998;9:1560–1567. doi: 10.1681/asn.v991560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JM, Luo P, Wang Z, Cohen SE, Kim HS, Fogo AB, Brown NJ. Aldosterone deficiency and mineralocorticoid receptor antagonism prevent angiotensin II-induced cardiac, renal, and vascular injury. Kidney Int. 2012;82:643–651. doi: 10.1038/ki.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangos GJ, Whitworth JA, Williamson PM, Kelly JJ. Glucocorticoids and the kidney. Nephrology. 2003;8:267–273. doi: 10.1111/j.1440-1797.2003.00215.x. [DOI] [PubMed] [Google Scholar]
- Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between alphaENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+ J Am Soc Nephrol. 2003;14:1107–1115. doi: 10.1097/01.asn.0000061777.67332.77. [DOI] [PubMed] [Google Scholar]
- Mullins LJ, Bailey MA, Mullins JJ. Hypertension, kidney, and transgenics: a fresh perspective. Physiol Rev. 2006;86:709–746. doi: 10.1152/physrev.00016.2005. [DOI] [PubMed] [Google Scholar]
- Mullins LJ, Peter A, Wrobel N, McNeilly JR, McNeilly AS, Al-Dujaili EA, Brownstein DG, Mullins JJ, Kenyon CJ. Cyp11b1 null mouse, a model of congenital adrenal hyperplasia. J Biol Chem. 2009;284:3925–3934. doi: 10.1074/jbc.M805081200. [DOI] [PubMed] [Google Scholar]
- Naray-Fejes-Toth A, Fejes-Toth G. Glucocorticoid receptors mediate mineralocorticoid-like effects in cultured collecting duct cells. Am J Physiol Renal Physiol. 1990;259:F672–F678. doi: 10.1152/ajprenal.1990.259.4.F672. [DOI] [PubMed] [Google Scholar]
- Nguyen Dinh Cat A, Ouvrard-Pascaud A, Tronche F, Clemessy M, Gonzalez-Nunez D, Farman N, Jaisser F. Conditional transgenic mice for studying the role of the glucocorticoid receptor in the renal collecting duct. Endocrinology. 2009;150:2202–2210. doi: 10.1210/en.2008-1531. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Kwon TH, Frokiaer J, Knepper MA, Nielsen S. Maintained ENaC trafficking in aldosterone-infused rats during mineralocorticoid and glucocorticoid receptor blockade. Am J Physiol Renal Physiol. 2007;292:F382–F394. doi: 10.1152/ajprenal.00212.2005. [DOI] [PubMed] [Google Scholar]
- Nishi M, Tanaka M, Matsuda K, Sunaguchi M, Kawata M. Visualization of glucocorticoid receptor and mineralocorticoid receptor interactions in living cells with GFP-based fluorescence resonance energy transfer. J Neurosci. 2004;24:4918–4927. doi: 10.1523/JNEUROSCI.5495-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odermatt A, Kratschmar DV. Tissue-specific modulation of mineralocorticoid receptor function by 11β-hydroxysteroid dehydrogenases: an overview. Mol Cell Endocrinol. 2012;350:168–186. doi: 10.1016/j.mce.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Odermatt A, Arnold P, Frey FJ. The intracellular localization of the mineralocorticoid receptor is regulated by 11β-hydroxysteroid dehydrogenase type 2. J Biol Chem. 2001;276:28484–28492. doi: 10.1074/jbc.M100374200. [DOI] [PubMed] [Google Scholar]
- Oelkers W, Kohler A, Belkien L, Fuchs-Hammoser R, Maiga M, Scherer B, Weber PC. Studies on the mechanism by which ACTH stimulates renin activity and angiotensin II formation in man. Acta Endocrinol. 1982;100:573–580. doi: 10.1530/acta.0.1000573. [DOI] [PubMed] [Google Scholar]
- Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:869–881. doi: 10.1111/j.1463-1326.2012.01582.x. [DOI] [PubMed] [Google Scholar]
- Rafiq K, Nakano D, Ihara G, Hitomi H, Fujisawa Y, Ohashi N, Kobori H, Nagai Y, Kiyomoto H, Kohno M, Nishiyama A. Effects of mineralocorticoid receptor blockade on glucocorticoid-induced renal injury in adrenalectomized rats. J Hypertens. 2011;29:290–298. doi: 10.1097/hjh.0b013e32834103a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci U S A. 2007;104:4025–4029. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signalling pathway. J Clin Invest. 2009;119:2601–2612. doi: 10.1172/JCI38323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundle SE, Funder JW, Lakshmi V, Monder C. The intrarenal localization of mineralocorticoid receptors and 11β-dehydrogenase: immunocytochemical studies. Endocrinology. 1989;125:1700–1704. doi: 10.1210/endo-125-3-1700. [DOI] [PubMed] [Google Scholar]
- Schulz-Baldes A, Berger S, Grahammer F, Warth R, Goldschmidt I, Peters J, Schutz G, Greger R, Bleich M. Induction of the epithelial Na+ channel via glucocorticoids in mineralocorticoid receptor knockout mice. Pflugers Arch. 2001;443:297–305. doi: 10.1007/s004240100694. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4–2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8. doi: 10.1074/jbc.C100623200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem. 2005;280:39970–39981. doi: 10.1074/jbc.M508658200. [DOI] [PubMed] [Google Scholar]
- Stewart PM, Krozowski ZS, Gupta A, Milford DV, Howie AJ, Sheppard MC, Whorwood CB. Hypertension in the syndrome of apparent mineralocorticoid excess due to mutation of the 11β-hydroxysteroid dehydrogenase type 2 gene. Lancet. 1996;347:88–91. doi: 10.1016/s0140-6736(96)90211-1. [DOI] [PubMed] [Google Scholar]
- Stow LR, Voren GE, Gumz ML, Wingo CS, Cain BD. Dexamethasone stimulates endothelin-1 gene expression in renal collecting duct cells. Steroids. 2012;77:360–366. doi: 10.1016/j.steroids.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Miyamori I, Yoneda T, Hatakeyama H, Inaba S, Furukawa K, Mabuchi H, Takeda R. Regulation of aldosterone synthase in human vascular endothelial cells by angiotensin II and adrenocorticotropin. J Clin Endocrinol Metab. 1996;81:2797–2800. doi: 10.1210/jcem.81.8.8768832. [DOI] [PubMed] [Google Scholar]
- Thompson A, Bailey MA, Michael AE, Unwin RJ. Effects of changes in dietary intake of sodium and potassium and of metabolic acidosis on 11β-hydroxysteroid dehydrogenase activities in rat kidney. Exp Nephrol. 2000;8:44–51. doi: 10.1159/000020647. [DOI] [PubMed] [Google Scholar]
- Tsugita M, Iwasaki Y, Nishiyama M, Taguchi T, Shinahara M, Taniguchi Y, Kambayashi M, Nishiyama A, Gomez-Sanchez CE, Terada Y, Hashimoto K. Glucocorticoid receptor plays an indispensable role in mineralocorticoid receptor-dependent transcription in GR-deficient BE(2)C and T84 cells in vitro. Mol Cell Endocrinol. 2009;302:18–25. doi: 10.1016/j.mce.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Wade JB, O'Neil RG, Pryor JL, Boulpaep EL. Modulation of cell membrane area in renal collecting tubules by corticosteroid hormones. J Cell Biol. 1979;81:439–445. doi: 10.1083/jcb.81.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang H, Lang F, Yun CC. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am J Physiol Cell Physiol. 2007;292:C396–C404. doi: 10.1152/ajpcell.00345.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CS, Thayer JF, Rudat M, Sharma AM, Perschel FH, Buchholz K, Deter HC. Salt-sensitive men show reduced heart rate variability, lower norepinephrine and enhanced cortisol during mental stress. J Hum Hypertens. 2008;22:423–431. doi: 10.1038/jhh.2008.11. [DOI] [PubMed] [Google Scholar]
- Woods LL, Mizelle HL, Hall JE. Control of sodium excretion in NE-ACTH hypertension: role of pressure natriuresis. Am J Physiol. 1988;255:R894–R900. doi: 10.1152/ajpregu.1988.255.6.R894. [DOI] [PubMed] [Google Scholar]
- Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: recent advances and new insights. Curr Hypertens Rep. 2013;15:17–24. doi: 10.1007/s11906-012-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallocchi M, Igarreta P, Calvo JC, Reboucas NA, Damasco MC. The mechanisms of brush border Na+/H+ exchanger activation by corticosteroids. Med Sci Monit. 2003;9:BR85–90. [PubMed] [Google Scholar]
- Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, Bonny O, Firsov D. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A. 2009;106:16523–16528. doi: 10.1073/pnas.0904890106. [DOI] [PMC free article] [PubMed] [Google Scholar]