

Abstract
The discovery of activating BRAF V600E mutations in 50% of all cutaneous melanomas has revolutionized the understanding of melanoma biology and provided new strategies for the therapeutic management of this deadly disease. Highly potent small molecule inhibitors of BRAF are now showing great promise as a novel therapeutic strategy for melanomas harboring activating BRAF V600E mutations and are associated with high levels of response. This commentary article discusses the latest data on the role of mutated BRAF in the development and progression of melanoma as the basis for understanding the mechanism of action of BRAF inhibitors in the preclinical and clinical settings. We further address the issue of BRAF inhibitor resistance and outline the latest insights into the mechanisms of therapeutic escape as well as describing approaches to prevent and abrogate the onset of both intrinsic and acquired drug resistance. It is likely that our evolving understanding of melanoma genetics and signaling will allow for the further personalization of melanoma therapy with the goal of improving clinical responses.
Keywords: BRAF, melanoma, vemurafenib, MEK, resistance, Cot, MAPK
Introduction
Melanoma is the most devastating form of skin cancer. Approximately 68,130 new cases of melanoma and 46,770 cases of melanoma in situ, resulting in 8,700 deaths are estimated for the United States in 2010 [1]. Whereas overall rates of cancer death continue to decrease, risk of death from melanoma continues to rise year on year and showed a 7% increase during the period 1990–2006 [1]. For many years disseminated melanoma was assumed to be resistant to all forms of therapeutic intervention. Recent advances in molecular profiling and genome sequencing have shown melanoma to be a heterogeneous group of malignancies whose progression is driven by distinct patterns of oncogenic mutation. Following the successes of targeted therapy agents, such as imatinib in chronic myeloid leukemia (CML) and gastrointestinal stromal (GIST) tumors, there are now hopes that similar advances can be made in melanoma. In this commentary we will review the latest advances in the targeted therapy of melanoma with a special emphasis upon the development of small molecule BRAF inhibitors. As our focus, we will discuss the role of mutant BRAF in the initiation and progression of melanoma and will delineate the mechanisms by which melanoma cells respond to and escape from BRAF inhibitor therapy.
The role of mutated BRAF in melanoma development and progression
The identification of activating mutations in BRAF in ~50% of all cutaneous melanomas in 2002 was a landmark event in the understanding of melanoma biology [2]. Raf (RApidly growing Fibrosarcoma) proteins constitute a 3 member family of Serine/Threonine kinases (ARAF, BRAF and CRAF) with closely overlapping functions that constitute part of the Ras/Raf/MEK/ERK mitogen activated protein kinase (MAPK) signal transduction cascade. Although >50 mutations in BRAF have now been described, the most common BRAF mutation in melanoma, accounting for 80% of all of the BRAF mutations, is a valine to glutamic acid (V600E) substitution [2, 3]. Acquisition of a V600E mutation in BRAF destabilizes the inactive kinase conformation switching the equilibrium towards the active form, leading to constitutive activity [3]. Other BRAF mutations identified from melanoma specimens are the V600K and V600D/V600R variants, which account for 16% and 3% of all BRAF mutations, respectively [4]. A minor sub-group of melanomas were also identified with BRAF mutations in positions other than 600 [5]. These non-V600 position BRAF mutants differ from the position-600 mutants, show impaired intrinsic BRAF kinase activity and require the presence of CRAF to transactivate their MAPK signaling [3]. Analysis of a large panel of melanoma cell lines and tissues revealed that ~1% of melanoma cell lines had either D594G or G469E mutation in BRAF, respectively and that 1% of melanoma specimens harbored a G469A mutation in BRAF [5]. Of the 50% of melanomas that are not BRAF mutant, 15–20% harbor activating Ras mutations and a small percentage are c-KIT mutant. The initiating oncogenic event in the remaining 30–35% of BRAF wild-type melanoma is currently unknown.
There is now a wealth of evidence demonstrating that mutated BRAF is a bona fide melanoma oncogene. Mechanistically, mutated BRAF exerts most of its oncogenic effects through the activation of the MAPK pathway [6]. MAPK activity drives the uncontrolled growth of melanoma cells by upregulating the expression of cyclin D1 and through the suppression of the cyclin dependent kinase inhibitor p27KIP1. Pre-clinical studies have shown that introduction of mutated BRAF into immortalized melanocytes leads to anchorage independent growth and tumor formation in immunocompromised mice [6]. Conversely, downregulation of mutated BRAF using RNAi causes cell cycle arrest and apoptosis in both in vitro and in vivo BRAF V600E mutant melanoma models [6]. Although it has been suggested that the acquisition of the BRAF V600E mutation is an early event in melanoma development, with 80% of all benign nevi shown to be BRAF mutant, the available evidence indicates that mutant BRAF alone cannot initiate melanoma [7, 8]. The introduction of V600E mutated BRAF into primary human melanocytes does not lead to oncogenic transformation and is instead associated with the onset of senescence [8]. Likewise, an immunohistochemical analysis of a large cohort of melanocytic nevi revealed positive staining for senescence associated beta galactosidase as well as histological markers of growth arrest [8].
Instead, melanoma development seems to require both BRAF/MAPK and phospho-inositide 3-kinase (PI3K)/AKT pathway activity. In BRAF mutant melanoma cells this can arise through the loss of expression or functional inactivation of the tumor suppressor phosphatase and tensin homolog (PTEN) which is lost in 10–30% of melanoma cell lines and 10% of human tumor material [9, 10]. Activation of AKT signaling in BRAF mutant melanoma also occurs as the result of increased AKT3 expression and also rarely through the acquisition of activating E17K mutations in AKT3 [6]. The requirement for both mutant BRAF and activation of the PI3K/AKT signaling pathway in melanoma initiation and progression is supported by transgenic mouse studies showing that introduction of the BRAF-V600E mutation in concert with the suppression of PTEN expression is required for full melanoma development [11].
In addition to its well-characterized effects upon growth, there is emerging evidence that aberrant BRAF signaling also regulates the survival of melanoma cells. A number of studies have shown that siRNA knockdown of BRAF and small molecule BRAF inhibitors induce apoptosis in BRAF V600E mutant melanoma cells through the regulation of the pro-apoptotic proteins BIM, BMF, BAD and Mcl-1 [12–15]. The best studied of these molecules is the BH3-only protein BIM which exerts its cytotoxic activity by binding to and antagonizing the anti-apoptotic proteins Bcl-2, Bcl-w, Bcl-XL and Mcl-1 [16, 17]. Expression of BIM is regulated both transcriptionally and post-transcriptionally by a number of signaling pathways, including BRAF/MEK/ERK, JNK, p38 MAPK and PI3K/AKT [18]. BIM exists as three isoforms BIM-EL (extra long), BIM-L (long) and BIM-S (short) that are generated by alternate splicing. Of the three splice forms BIM-S is thought to be the most important for apoptosis induction. It is known that the BRAF V600E mutation regulates BIM expression through the MEK/ERK pathway-mediated phosphorylation of the extra-long form of BIM (BIM-EL) at Serine 69, leading to its subsequent degradation by the proteasome [12, 19]. Inhibition of BRAF also regulates BIM splicing and leads to the selective upregulation of BIM-S expression [20]. The essential role of the BIM-S splice form for BRAF inhibitor mediated apoptosis was demonstrated by the siRNA knockdown of BIM-S and the fact that the introduction of BRAF V600E into BRAF wild-type melanoma cells and melanocytes downregulated basal levels of BIM-S expression [20]. In these instances, the increase in BIM-S expression observed was associated with an upregulation of the splicing factor SRp55 [20].
Malignantly transformed cells are highly invasive and there is good evidence that oncogenic BRAF plays a key role in this process. Early studies, that predated the discovery of BRAF mutations, showed constitutive MAPK signaling activity to drive the invasion of melanoma cells through the increased expression of the pro-migratory β3 integrin receptor and the upregulation of matrix metalloproteinase (MMP) expression [21]. It has since been shown that activation of the BRAF/MEK/ERK pathway aids the motile phenotype of melanoma through reorganization of the cytoskeleton. Two recent studies demonstrated a role for mutant BRAF in regulating the expression of RND3/RhoE/Rho8, a regulator of the crosstalk between the BRAF/MEK/ERK and Rho/Rock/LIM kinase/Cofilin pathways [22]. Silencing of BRAF using siRNA or inhibition of MEK downregulated RND3 expression, which in turn increased stress fiber formation and enhanced focal adhesion stability. Depletion of RND3 by siRNA was found to prevent the invasion of melanoma cells in a 3D collagen implanted spheroid cell culture model [22].
Other recent work showed mutated BRAF to induce the invasion of melanoma cells through a novel pathway involving the release of cytosolic calcium [23]. This discovery came from a microarray screen that identified the cyclic GMP phosphodiesterase PDE5A as a novel gene that was downregulated by oncogenic BRAF. Although the re-introduction of PDE5A did not confer a growth advantage to BRAF V600E melanoma cells it did significantly suppress cell invasion [23]. A mechanistic analysis showed that downregulation of PDE5A by mutant BRAF increased levels of intracellular cGMP leading to cytosolic calcium release. The increased intracellular calcium then led to phosphorylation of myosin light chain 2 (MLC2), which enhanced cell contractility and led to an increase in the invasive capacity. Of clinical relevance, the authors observed that a number of commonly used PDE inhibitors such as sildenafil (more commonly known as Viagra) and tadalafil blocked the activity of PDE5A and enhanced the contractility and invasion of the melanoma cells [23]. It was suggested that the use of these PDE inhibitors could be deleterious in patients with BRAF mutant melanoma.
In addition to the direct effects upon melanoma cell behavior described above, the presence of a BRAF mutation also regulates the interaction of melanoma cells and the host microenvironment, in particular by allowing the tumor cells to escape immune surveillance. Inhibition of BRAF/MAPK signaling in BRAF V600E mutant melanoma cells is known to reduce the release of immunosuppressive cytokines and reverses the suppressive effects of melanoma cell culture supernatants upon dendritic cell activation [24]. There is also evidence that the presence of a BRAF mutation allows melanoma cells to escape T-cell recognition. Two recent studies have shown that increased BRAF/MEK/ERK signaling suppresses the expression of highly immunogenic differentiation antigens from melanoma cell lines [25, 26]. These effects were noted to be dependent upon continuous BRAF/MAPK signaling and the expression of the pigmentations antigens could be restored following the inhibition of either BRAF or MEK. There seemed to be some benefit of inhibiting the MAPK pathway using BRAF rather than MEK inhibitors, with BRAF inhibition shown to restore the antigen specific function of T-cells, whereas MEK inhibition actually suppressed T-cell activity [26]. Given the current interest in combining BRAF and MEK inhibitors with immunotherapies such as ipilimumab, these results may also be of clinical relevance.
The epidemiological, pathological and prognostic characteristics of BRAF mutant melanoma
Although mutations in BRAF are not classic ultraviolet (UV) radiation signature mutations, UV exposure does seem to play a role in their acquisition. BRAF V600E mutant melanomas tend to occur on sun exposed skin and their incidence correlates well with skin phenotypes that have poor UV protection (such as pale skin, poor tanning response, red hair coloration, freckling) [27, 28]. There is also good epidemiological evidence that individuals with a poor tanning response associated with polymorphisms in the melanocortin receptor 1 (MC1R) have an increased risk of developing melanomas that harbor BRAF V600E mutations [29]. The duration of sun exposure also seems to predict for the incidence of BRAF V600E mutant melanoma. Younger melanoma patients (<55 yrs), with a lower cumulative UV exposure are more likely to develop melanomas that are BRAF V600E mutant [30]. In contrast, NRAS mutant melanomas are more often observed in older patients with a more sustained history of sun exposure. The presence of a BRAF mutation may also dictate the biological behavior of the melanoma. Careful pathological examination of large numbers of BRAF, NRAS and c-KIT mutant melanoma specimens showed BRAF-mutated melanoma cells to have an increased upward migration into the epidermis and a greater propensity for nest formation [30]. BRAF mutant melanoma cells also tended to be larger, rounded and more pigmented than those harboring activating mutations in NRAS [30]. New data also suggests that the presence of the BRAF mutation has prognostic influence for melanoma patients. A prospective analysis of a large cohort of Australian melanoma patients (n=197) revealed that although there were no associations between BRAF mutation status, the site of metastatic disease, serum LDH levels and ECOG performance status, there was a strong association between the presence of a BRAF mutation and inferior survival in the metastatic setting (8.5 months in BRAF wild-type patients vs 5.7 months for BRAF mutant patients) [4].
In vitro targeting of BRAF
The identification of BRAF mutations in melanoma led to the development of a number of small molecule BRAF kinase inhibitors that are now undergoing intensive preclinical and clinical investigation. The first putative BRAF inhibitor to be thoroughly investigated in melanoma was the multi-kinase inhibitor sorafenib (BAY43-9006, Nexxavar) [31]. Although sorafenib was originally developed as a CRAF inhibitor, it also had some activity against BRAF and was the first kinase inhibitor available for evaluation in BRAF mutant melanoma [32]. In animal xenograft studies, sorafenib treatment led to minor levels of regression in BRAF V600E mutated melanoma and induced limited levels of apoptosis [5, 32]. Subsequent pre-clinical investigations showed sorafenib to be a relatively weak inhibitor of BRAF, with many off-target effects (including inhibition of VEGFR, PDGFR, FLT-3 and p38 MAP kinase [31, 33]), it was therefore concluded that any anti-melanoma activity seen to sorafenib was independent of its effects upon BRAF inhibition [34]. Following the evaluation of sorafenib, a new generation of highly specific and potent BRAF inhibitors has been developed. These drugs show a greater selectivity for mutant BRAF and have fewer off-target effects; the list of those currently under pre-clinical investigation includes: SB590885, GSK2118436, PLX4032 (RG704, vemurafenib), AZ628, XL281 and GDC-0879. Of these, the two that have been most comprehensively investigated are vemurafenib and its analogue PLX4720 [35–37].
Consistent with the role of BRAF/MAPK signaling in the regulation of cell growth, treatment of BRAF V600E mutated melanoma cell lines with pharmacological inhibitors of BRAF leads to a profound G1 phase cell cycle arrest. Indeed, the BRAF inhibitors SB590885 [38], AZ628 [39] and PLX4720 [35] all have cytostatic effects upon melanoma cell lines harboring the BRAF V600E mutation. Significantly, the more potent BRAF inhibitors, such as vemurafenib /PLX4720, are also pro-apoptotic in a large proportion of BRAF mutant melanoma cell lines – an effect that was well correlated with the ability of the drug to induce BIM expression [10, 20]. The effects of vemurafenib were noted to be BRAF mutation specific, and equivalent responses were seen in melanoma models with both heterozygous and homozygous BRAF mutations [36]. Little effect was observed in cell lines and xenografts if both BRAF alleles were wild-type [36]. Not all BRAF mutated melanoma cell lines were similarly sensitive to vemurafenib and PLX4720, with some cell lines exhibiting intrinsic resistance [40–42]. Responses to vemurafenib and PLX4720 in human melanoma xenograft models were impressive; with either partial or complete responses observed in all cases, with a close relationship observed between drug exposure and response within individual xenograft models [36, 43]. The structure and kinase selectivity of vemurafenib was recently published, and showed the drug to be a pan-Raf inhibitor (IC50: BRAF V600E: 31 nM, wild-type BRAF: 100nM, CRAF 48: nM) with significant inhibitory activity (<100nM) against a number of other kinases (ACK1, MAP4K5 and SRMS) [36]. The importance of these other kinases for the melanoma specific effects of vemurafenib remains to be determined [36]. Another BRAF inhibitor currently exciting much interest in both the pre-clinical and clinical arenas is GSK2118436, an ATP-competitive inhibitor of BRAF V600E/D/K, wild-type BRAF and CRAF [37].
Clinical targeting of BRAF: phase I, II and III trials
Sorafenib was the first RAF inhibitor to enter clinical development in patients with melanoma [44]. In the initial series of studies patients were not selected on the basis of genotype and although some responses were seen, these were not correlated with BRAF mutational status [44]. Large phase III randomized studies of sorafenib in combination with chemotherapy were associated with low response rates and there was little evidence that the effects of sorafenib observed were mediated through BRAF inhibition [33, 45].
Clinically, the most highly studied of the new class of BRAF specific inhibitors is vemurafenib. In the recent phase I clinical trial, 80% of melanoma patients (n=32) selected for the presence of the BRAF V600E mutation responded to PLX4032 (dosed at 240mg/kg – 960 mg/kg BID) and showed significant levels of tumor regression [46]. Pharmacodynamic studies (inhibition of Ki67 and pERK staining in pre- and post-treatment paired biopsies) suggested that >80% BRAF inhibition was required for clinical activity to be observed. It was further noted that inhibition of cytoplasmic pERK levels, but not inhibition of nuclear pERK levels, correlated well with tumor response [36]. In line with preclinical studies showing the importance of mutated BRAF for the metabolic activity of melanoma cells, vemurafenib treatment was also observed to significantly diminish tumor fluoro-deoxy glucose (FDG) uptake as measured by positron emission tomography (PET) imaging [36, 46, 47]. Vemurafenib was generally well tolerated with the most common side effects being rash, arthralgia, photosensitivity and fatigue. Intriguingly, >23% of patients rapidly (mostly <12 weeks of treatment) developed squamous cell carcinomas (SCC) of the keratoacanthoma (KA) type on areas of sun exposed skin [46]. These tumors were removed surgically and did not recur. In the phase II BRAF In Melanoma (BRIM)-2 trial, 132 patients received 960mg of vemurafenib BID. The primary endpoint was best overall response, with duration of response, progression free survival, overall response and safety as the secondary endpoints. In this trial, 52.3% (n=69) of patients had a complete (2.3%) or partial response (50%), 29.5% (n=39) had stable disease and 13.6% (n=18) had progressive disease. Average duration of response was 6.8 months and progression free survival was 6.2 months [48]. Reported side effects were similar to those from the phase I trial, with 24% of patients developing KA. The phase III trial of vemurafenib (BRIM-3) in which nearly 680 patients were randomized 1:1 against dacarbazine has now closed. Data from this trial have been submitted to the FDA for possible regulatory approval.
GSK2118436 is a highly potent small molecule BRAF inhibitor (In vitro kinase selectivity: BRAF; V600E – 0.6 nM, V600K – 0.5 nM, V600D – 1.9 nM, wild-type BRAF – 12 nM, CRAF – 5nM) being evaluated clinically in BRAF mutant melanoma. The recent phase I/II clinical trial of GSK2118436 differed from that of the vemurafenib study by including melanoma patients with non-V600E BRAF mutations (V600K and V600D) and individuals with brain metastases [48]. In the study population 77% harbored V600E BRAF mutations and 19% harbored V600K BRAF mutations. Like vemurafenib, response rates to GSK2118436 were very impressive. In the BRAF V600E mutated melanoma cohort, the overall response rate was 77%, and 44% of BRAF V600K mutated melanoma patients (4/9) also showed a response [48]. Progression free survival was 8.3 months. Significantly, GSK2118436 was found to be active in melanoma patients with untreated brain metastases (n=10), with magnetic resonance imaging (MRI) studies confirming partial responses in 3 out of 10 patients. Overall, 9 out of 10 of the patients with brain metastases showed some level of response, with the responses in the brain matching those achieved at other organ sites [48]. The drug was generally well-tolerated and side-effects were mild. Like vemurafenib, the development of SCC of the KA type was noted in patients treated with GSK2118436 (>70 mg BID). Pharmacodynamic analysis showed 150mg of GSK2118436 BID twice daily to inhibit intratumoral phospho-ERK by >90%, reduce expression of Ki67, induce expression of the cell cycle inhibitor p27 and decrease FDG-PET uptake. Interestingly, it was noted that increased dosing of GSK2118436 up to 300–600mg did not result in proportional increases in plasma drug levels suggesting that hepatic metabolism was being induced. The drug is known to have a number of metabolites, at least three of which are highly active. Enrollment for the phase II trial of single-agent GSK2118436 in BRAF V600 melanoma is already completed and a phase III trial is currently underway. A phase II trial of GSK2118436 in patients with untreated brain metastases is also accruing.
All of the BRAF inhibitors evaluated so far, including sorafenib, vemurafenib, GSK2118436 and XL281 have induced proliferative squamous lesions in the skin [36, 46]. These lesions occur at sun-exposed skin sites, are frequently rapidly growing and can be managed with surgery or other local control measures. Although the mechanisms underlying the development of these SCC remains to be fully determined, there is now strong preclinical evidence that BRAF targeted agents may have direct growth promoting effects upon initiated, but not fully transformed cells. Whereas BRAF inhibitors such as PLX4720 and GDC-0879 inhibit the activation of BRAF/MEK/ERK in BRAF mutant cell lines, they are known to increase MEK/ERK signaling in cell lines with RAS mutations and constitutive activity in receptor tyrosine kinases such as HER2 [49–51]. From a mechanistic standpoint it has been shown that wild-type Raf kinase activation induces Raf dimerization. The paradoxical increase in MAPK signaling that occurs when BRAF is inhibited in tumor cells that are BRAF wild-type arises as a result of increased CRAF-CRAF dimer formation that in turn activates MEK [52, 53]. In addition to this, preclinical studies have also shown vemurafenib to enhance FAK signaling in NRAS mutant melanoma cells, which together with increased MAPK activity, increases invasive potential [49]. There is also evidence that BRAF inhibition increases the survival of NRAS mutant tumor cells, in part by modulating Mcl-1 expression [51]. Taken together, these results all suggest a need for the careful screening of melanoma patients for the BRAF mutation prior to the initiation of BRAF inhibitor therapy.
Mechanisms of intrinsic BRAF inhibitor resistance
Although the presence of an activating BRAF mutation generally predicts for a response to BRAF inhibitors, a significant proportion of BRAF V600E mutated melanoma cell lines show signs of intrinsic drug resistance [10, 41, 42]. Similar findings were observed in the phase I clinical trial of vemurafenib, where ~20% of the patients whose melanomas harbored the BRAF V600E mutation did not meet the RECIST criteria threshold for a response [46]. Melanomas are known to have complex mutational profiles and harbor concurrent alterations in many genes including CDK2, CDK4, MITF and AKT3. How these genes and possibly others impact upon the biological behavior of melanoma cells and modulate the response to BRAF inhibitors is not yet understood.
In melanoma cells, constitutive BRAF/MEK/ERK signaling drives cell cycle entry and uncontrolled growth by increasing cyclin D1 expression. It is now well established that inhibition of BRAF in BRAF V600E mutant melanoma cell lines leads to both inhibition of cyclin D1 expression and cell cycle arrest. A recent array comparative genomic (aCGH) analysis of a large panel of melanoma cell lines and tumor specimens showed 17% to harbor a BRAF V600E mutation in conjunction with amplification of cyclin D1 [54]. In Western Blot experiments, the amplified cell lines had increased cyclin D1 protein expression and showed intrinsic resistance to SB590885 [54]. Overexpression experiments showed the introduction of cyclin D1 into previously drug sensitive cell lines to facilitate cell cycle entry even when BRAF was inhibited [54].
There is already good evidence from the breast cancer field that the expression and mutational status of the tumor suppressor PTEN is an important predictor of intrinsic resistance to targeted therapy agents such as trastuzamab and gefitinib [55]. In these instances, tumors that are PTEN negative, or those with high basal PI3K/AKT signaling showed a marked impairment of therapy-induced apoptosis and were associated with significantly worse therapeutic responses [55]. Our studies in melanoma support these ideas and identified loss of PTEN, observed in >10% of melanoma specimens, as being predictive for an attenuated apoptotic response following treatment with PLX4720 [10]. In the context of PTEN loss, BRAF inhibition led to an increase in AKT signaling that suppressed the pro-apoptotic protein BAD. The phosphorylation of BAD by AKT at Ser99 prevents the binding of BAD to Bax and relieves the antagonism of Bax on Bcl-2 and Bcl-XL [14]. In addition, the increase in AKT signaling observed following BRAF inhibition was also noted to suppress the expression of BIM through the phosphorylation and subsequent nuclear export of the transcription factor FOXO3a [10]. It was shown that intrinsic BRAF inhibitor resistance could be overcome by treating the BRAF V600E/PTEN null melanoma cell lines with the combination of a BRAF inhibitor and a PI3K inhibitor. This dual BRAF/PI3K inhibition restored the nuclear accumulation of FOXO3a, upregulated BIM expression and significantly enhanced the level of apoptosis [10]. In further support of a role for AKT activation in intrinsic BRAF inhibitor resistance, others have shown that the overexpression of myristolated (constitutively active) AKT3 prevents PLX4720-induced apoptosis through the downregulation of both BIM and BMF [15].
FOXO3a is a member of the Forkhead family of transcription factors that regulates cell survival and growth through the activation or suppression of a diverse array of oncogenesis-related genes such as BIM, Fas-Ligand, cyclin D1 and GADD45 [56]. Inactivation of FOXO3a occurs as a result of its phosphorylation by AKT/SGK (at Threonine-32, Serine-253 and Serine-315), ERK1/2 (at Serine-294, Serine-344 and Serine-425), CK1, IKKB, CDK2 and AMPK; which leads in turn to its nuclear exclusion and subsequent proteasomal degradation [56]. There is good evidence that inactivation of FOXO3a is a pre-requisite for the transformation of many cell types, and cytoplasmic FOXO3a accumulation is known to be a negative prognostic factor for breast cancer [56]. Studies in other tumor systems, including a limited number of melanoma cell lines, have also linked intrinsic MEK inhibitor resistance to the impaired activation of FOXO3a and a subsequent reduction in BIM promoter activity [57]. In this instance the combination of the MEK inhibitor AZD6244 with an inhibitor of AKT (API-2) was found to restore the nuclear localization of FOXO3a, upregulate BIM expression and enhance the levels of apoptosis [57].
Although the mechanisms underlying the BRAF inhibitor-induced increase in AKT signaling have not been fully elucidated, there is some suggestion that increased insulin like growth factor (IGF)-I signaling may be involved [10]. Similar findings implicating IGF-I signaling were also reported for melanoma cell lines showing intrinsic resistance to AZD6244 [58]. In this instance, intrinsic resistance could be overcome by treating the cells with AZD6244 in combination with an IGFR1, AKT or an mTORC1/2 inhibitor [58]. Other studies, performed in multiple myeloma, have also shown that increased IGF-I signaling suppressed BIM expression through post-translational mechanisms and the deregulation of FOXO3a [59].
Mechanisms of acquired BRAF inhibitor resistance
Although very encouraging, the clinical responses seen so far to vemurafenib and GSK2118436 are relatively short-lived, with treatment failure and tumor progression occurring in nearly every case. These observations, where an initial period of response is followed by relapse and resistance has been seen for every targeted therapy evaluated so far, including imatinib in CML and GIST [60, 61], EGFR inhibitors in lung cancer and most recently hedgehog inhibitors in medulloblastoma [62, 63]. In nearly all of these examples, acquired drug resistance was associated with the acquisition of secondary mutations in the kinase being targeted. These mutations typically occurred at sites within the kinase ATP binding site that prevented the binding of drug to the hydrophobic pocket at so-called “gatekeeper” residues. Examples of clinically relevant gatekeeper mutations include T790M in the EGFR receptor and T315I in Bcr-ABL. A recent preclinical study identified the gatekeeper site in BRAF to be Threonine-259 (T259) [34]. Studies on COS7 cells showed that mutation of BRAF at T259 conferred resistance to SB590885 and PLX4720 whilst allowing oncogenic BRAF kinase activity to be maintained [34]. Intriguingly, similar BRAF gatekeeper mutations have not been observed in either BRAF inhibitor resistant melanoma cell lines or biopsies taken from melanoma patients failing vemurafenib therapy [34]. In the most detailed analysis performed so far, deep sequencing and ultra-deep sequencing of 14 biopsies from melanoma specimens from patients progressing on vemurafenib therapy showed no evidence of secondary BRAF mutations [64]. Further in depth sequencing of exon 13 of BRAF (where the T259 residue lies) also failed to show the presence of additional drug-induced BRAF mutations [64]. As a final confirmation that secondary BRAF mutations were not the mechanism of resistance in this patient cohort, the BRAF kinase was immunoprecipitated from vemurafenib resistant biopsy samples and found to retain drug sensitivity in an in vitro kinase assay [64].
The emerging data instead suggest that a diverse array of BRAF inhibitor resistance mechanisms exist [64–69]. In a recent report by Villanueva and colleagues, the acquisition of BRAF inhibitor resistance led to a recovery of MAPK signaling and was associated with an increase in CRAF protein expression [39, 66]. Intriguingly, shRNA knockdown of CRAF alone did not restore drug sensitivity and it was instead found that shRNA knockdown of both ARAF and CRAF was required to overcome resistance [66]. This flexible switching between RAF isoforms led to cross-resistance with other BRAF inhibitors but not MEK inhibitors and was not associated with acquired secondary mutations in BRAF, NRAS or PTEN. The nature of the upstream signal required for the ARAF/CRAF activation was not determined. Although inhibition of MEK was found to decrease the proliferation of the resistant cells and led to a G1 phase cell cycle arrest it did not induce apoptosis. As this suggested that other compensatory pathways could be involved, the authors performed phospho-receptor tyrosine kinase (RTK) arrays and identified the insulin like growth factor receptor (IGFR)-1 as being constitutively activated in the resistant cells [66]. Mechanistic studies showed IGFR1 signaling to mediate increased PI3K/AKT signaling in the resistant cells and that the resistance could be reversed by treating the cells with the combination of a PI3K and a MEK inhibitor or an IGF1R and a MEK inhibitor [66]. The translational relevance of this finding was confirmed by the observation that 1 out of 5 melanoma specimens from patients failing vemurafenib expressed increased levels of IGFR1, and that one other specimen expressed increased levels of IGFR1 in conjunction with PTEN loss [66].
A second recent paper showed acquired vemurafenib resistance to be associated with the upregulated expression of a number of RTKs [64]. Of the RTKs identified, increased expression and tyrosine phosphorylation of the platelet derived growth factor receptor (PDGFR)-β was demonstrated to be responsible for conferring BRAF inhibitor resistance [64]. The mechanism underlying the constitutive PDGFRβ signaling observed was not determined, but was not noted to be the result of an activating mutation or genomic amplification. The clinical relevance of this finding was demonstrated by increased level of PDGFRβ signaling in biopsies taken from 4 out of 12 patients failing vemurafenib therapy that was not observed in the pre-treatment biopsy samples [64]. The potential role of PDGFRβ signaling in resistance was further confirmed by overexpression studies, where introduction of PDGFRβ into treatment naïve cells decreased sensitivity to vemurafenib. Somewhat surprisingly it was also found that although siRNA knockdown of PDGFRβ reduced the growth and survival of the vemurafenib resistant cell lines, the resistant cell lines were not sensitive to the PDGFRβ inhibitor imatinib [64].
A recent unbiased approach, in which 600 kinases and kinase-related open reading frames (ORFs) were expressed in melanoma cells identified nine candidate ORFs (including Axl, CRKL, ERBB2, FGR, MAP3K8, PAK3, CRAF, PKC-epsilon and PKC-eta) capable of mediating resistance to the BRAF inhibitor PLX4720 [68]. This study confirmed earlier work demonstrating a role for increased CRAF expression in mediating resistance to AZ628 and further identified MAP3K8 (COT) as a new candidate for the RAF-independent activation of ERK [68]. A number of cell lines were identified with genomic amplification of COT, all of which showed intrinsic BRAF and MEK inhibitor resistance. Further studies demonstrated that both COT shRNA knockdown and small molecule COT inhibitors reduced the MAPK signaling and survival of COT amplified cell lines. It was also shown that COT activated ERK through both MEK-dependent and independent mechanisms [68]. In melanoma cell lines where COT was overexpressed by lentiviral vector, drug resistance was overcome by treating the cells with a combination of a BRAF and MEK inhibitor. The clinical relevance of increased COT expression in the resistant phenotype was confirmed in a limited number of melanoma samples from patients failing BRAF and MEK inhibitor treatment.
As well as increased RTK activity, there is also evidence that genetic alterations in the Ras/Raf/MEK/ERK pathway also mediate acquired BRAF inhibitor resistance. A genetic analysis of biopsies from a patient failing vemurafenib therapy revealed the presence of an activating NRAS (Q61K) mutation that was lacking in the original tumor [64]. This apparent switch in mutational status was accompanied by the reactivation of the MAPK pathway upon vemurafenib treatment and appeared to be a relatively rare occurrence (only 1 patient identified so far). Of interest, it was noted that activating NRAS mutations were found in only 3 out of 6 sections from the same tumor specimen, suggesting the presence of different clonal populations. There is some evidence that melanomas consist of co-existent clones with different (e.g. both BRAF and non-BRAF) oncogenic mutations and at least two studies have now reported the existence of distinct BRAF mutant and NRAS mutant cells within the same melanoma specimen [70, 71]. The possible polyclonal nature of melanoma was further suggested by a single cell BRAF sequencing study, where both BRAF mutant and wild-type cells were derived from the same tumor [72]. If confirmed in a larger patient cohort, the issue of mutational polyclonality could have important clinical implications, particularly in light of the overwhelming pre-clinical evidence that BRAF inhibitors confer a growth advantage to NRAS-mutant melanoma cells [49, 51].
In addition to secondary Ras mutations, there is evidence from colon cancer that the genomic amplification of BRAF also mediates resistance to MAPK inhibition [73]. Analysis of drug naïve cell cultures using fluorescence in situ hybridization (FISH) identified limited numbers of cells with high BRAF copy number [73]. Treatment of these cultures with the MEK inhibitor AZD6244 led to an expansion of the BRAF amplified population, with resistance being reversed by the shRNA knockdown of BRAF [73]. The relevance of BRAF amplification to BRAF inhibitor resistance in melanoma is still currently under investigation.
Other studies have shown that mutations in MEK can also mediate BRAF inhibitor resistance. A massively parallel sequencing study of one biopsy sample from a melanoma patient failing vemurafenib therapy identified a novel codon 121 mutation in MEK1 (C121S) that conferred increased kinase activity in in vitro studies [65]. Overexpression of this novel MEK1 mutation in A375 melanoma cells was found to induce cross resistance to both MEK (AZD6244) and BRAF (PLX4720) inhibitors in vitro [65].
We still do not know whether therapeutic escape arises as the result of an evolutionary process within the melanoma or from the selection of pre-existing “resistant” clones that are already present prior to the initiation of therapy. Based upon the current data, both of these situations are likely to be true. A growing number of studies are now suggesting that the inherent phenotypic and genetic heterogeneity of cancer cell populations is a critical determinant of drug resistance. There is evidence that a transient drug-tolerant state can emerge through epigenetic means in individual cells, through activation of IGFR1 signaling and an altered chromatin state mediated through the histone demethylase RBP2/KDM5A/Jarid1A [74]. The drug tolerant cells were identified in cultures derived from a number of tumor types and seemed to be important in the escape response to both inhibitors of RTK signaling and cytotoxic chemotherapy drugs [74]. Interestingly, the drug tolerant population also emerged in cultures established from single cells, demonstrating the reversible, switchable nature of this phenotype. From a therapeutic standpoint, tolerance could be abrogated by the inhibition of IGFR1 signaling or through use of histone deacetylase (HDAC) inhibitors [74]. Of relevance to melanoma and BRAF inhibitor resistance, HDAC inhibition was found to induce at least some apoptosis in melanoma cells that were resistant to the BRAF inhibitor AZ628 [74].
The characterization of the pre-existing sub-population of cells that escape BRAF inhibitor therapy is key to managing resistance. New insights into the nature of drug tolerant cells have come from a recent study identifying a minor subset of melanoma cells that were required for tumor maintenance and expressed high levels of the H3K4 histone demethylase Jarid1B [75]. These cells tended to be present at low levels within the melanoma population, proliferated very slowly and underwent a marked expansion when treated with either BRAF inhibitors or cytotoxic chemotherapeutic drugs [75]. Further study will be required to determine whether simultaneous treatment with inhibitors of BRAF and HDAC is sufficient to prevent the onset of resistance, and whether the expansion of Jarid1B expressing melanoma cells is a critical step in the emergence of drug resistance.
Future perspectives
One of the major challenges facing the melanoma field is how to develop strategies for overcoming intrinsic and acquired drug resistance to small molecule BRAF inhibitors. Virtually every small molecule kinase inhibitor evaluated in cancer so far has shown a similar pattern of response, relapse and resistance. Melanoma seems to be unique in terms of the sheer diversity of resistance mechanisms identified. Every report to date has identified a different resistance mechanism with further modes of therapeutic escape likely to be reported in the near future. Why this should be so is open to speculation, but may be linked to the vast number of genetic mutations found in a typical melanoma (up to 30,000 mutations reported in one melanoma cell line [76]). It is possible that this high degree of mutational diversity accounts for the heterogeneity of resistance mechanisms observed.
The management of BRAF inhibitor resistance is likely to be achieved through combination therapy approaches [77]. Although the resistance mechanisms identified so far are diverse, most seem to rely upon the reactivation of and dependence upon MEK/ERK signaling and increased signaling output through the PI3K/AKT/mTOR pathway. The finding that BRAF inhibitor resistance in melanoma is associated with re-activation of the MAPK pathway is not surprising. Melanomas harboring activating BRAF V600E mutations show a high degree of dependency upon MAPK and are exquisitely sensitive to pharmacological inhibitors of both BRAF and MEK. Indeed, a number of groups have already shown pre-clinically that dual BRAF and MEK inhibition may prevent or delay the onset of resistance [67, 68, 73] and that dual BRAF/MEK and PI3K/AKT/mTOR inhibition has synergistically pro-apoptotic effects [78, 79]. The combination of BRAF and MEK inhibitors is currently being evaluated clinically in a phase I/II clinical trial of the BRAF inhibitor GSK2118436 in combination with the MEK inhibitor GSK2110212 in BRAF V600E mutated melanoma patients who are treatment naïve (NCT01072175). Clinical trials combining PI3K and BRAF inhibitors and BRAF and AKT inhibitors are due to commence shortly.
It is likely that the existence of so many potential resistance mechanisms will require patient-specific approaches to the management of therapeutic escape and the further personalization of melanoma therapy. To achieve this will require greater insight into the genetic and cell signaling diversity of melanoma and improved knowledge on how this affects drug response. Progress in this area will require the continued efforts of basic scientists and clinicians and the ongoing support of the pharmaceutical companies. If these approaches are as successful as we anticipate, a future can be imagined where disseminated melanoma is no longer such a bleak prognosis and can instead be reduced to the level of a manageable, chronic disease.
Figure 1. Role of mutant BRAF in preventing apoptosis in melanoma cells.
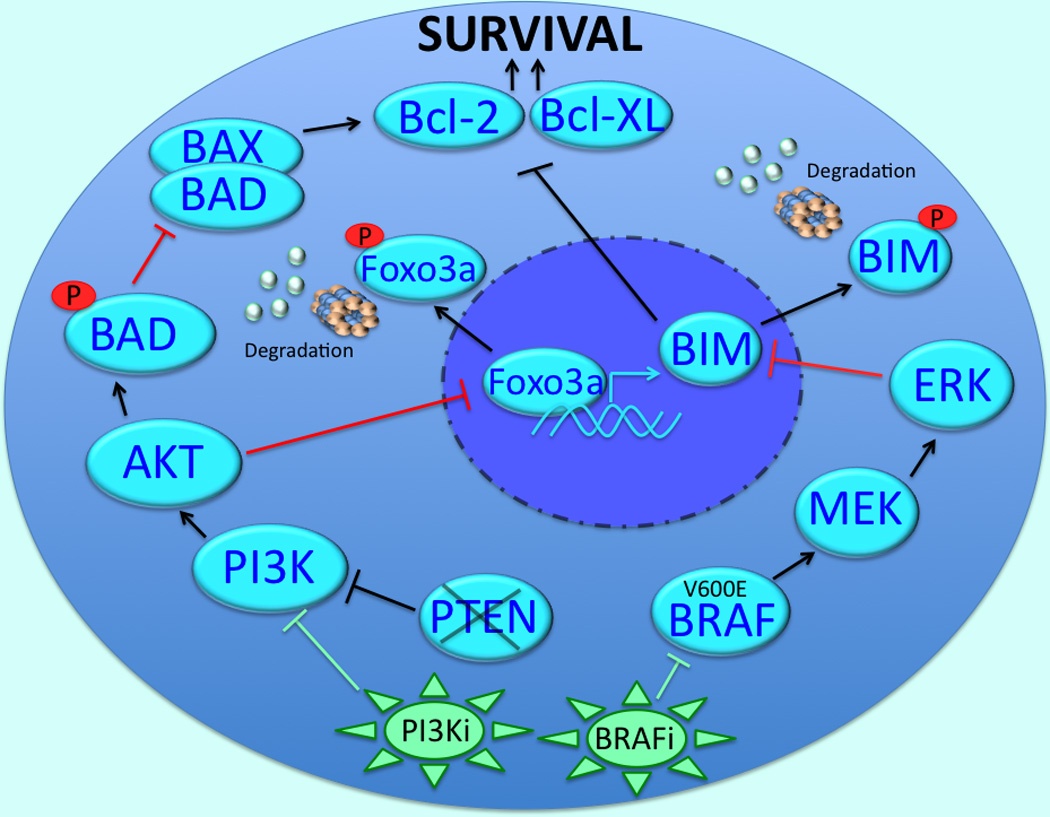
The inhibition of BRAF decreases the phosphorylation of BIM through the MEK/ERK pathway, preventing its proteasomal degradation. Once stabilized, BIM antagonizes the anti-apoptotic proteins Bcl-2 and Bcl-XL and leads to apoptosis induction. In PTEN-null melanoma cells, BRAF inhibition leads to the increased PI3K/AKT-mediated phosphorylation of FOXO3a resulting in reduced BIM transcription. Inhibition of BRAF in PTEN null melanoma cells also impairs apoptosis through the AKT-mediated phosphorylation and inactivation of BAD. The phosphorylation of BAD prevents its binding to Bax and relieves the antagonism of Bax on Bcl-2 and Bcl-XL. Intrinsic BRAF inhibitor resistance in the PTEN-null cells can be overcome through dual inhibition of BRAF and PI3K.
Figure 2. Mutated BRAF drives the invasion of melanoma cells.
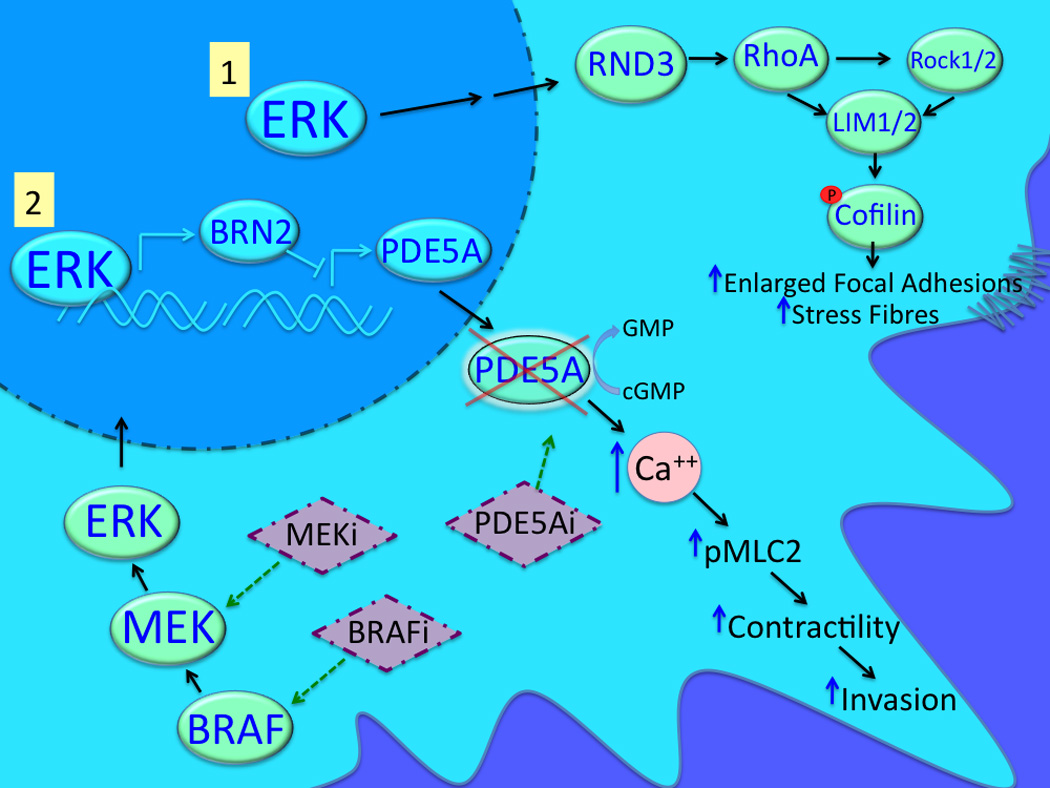
Mutant BRAF regulates melanoma cell invasion through MEK/ERK-mediated signaling to RND3 and Rho/ROCK/LIM-mediated Cofilin phosphorylation. It is also known that ERK can upregulate BRN2 expression leading to the downregulation of PDE5A and cytoplasmic accumulation of cGMP and Ca2+. Increased intracellular calcium in turn leads to increased MLC2 phosphorylation, contractility, and invasion.
Figure 3. Mechanisms of acquired resistance to BRAF inhibitors.
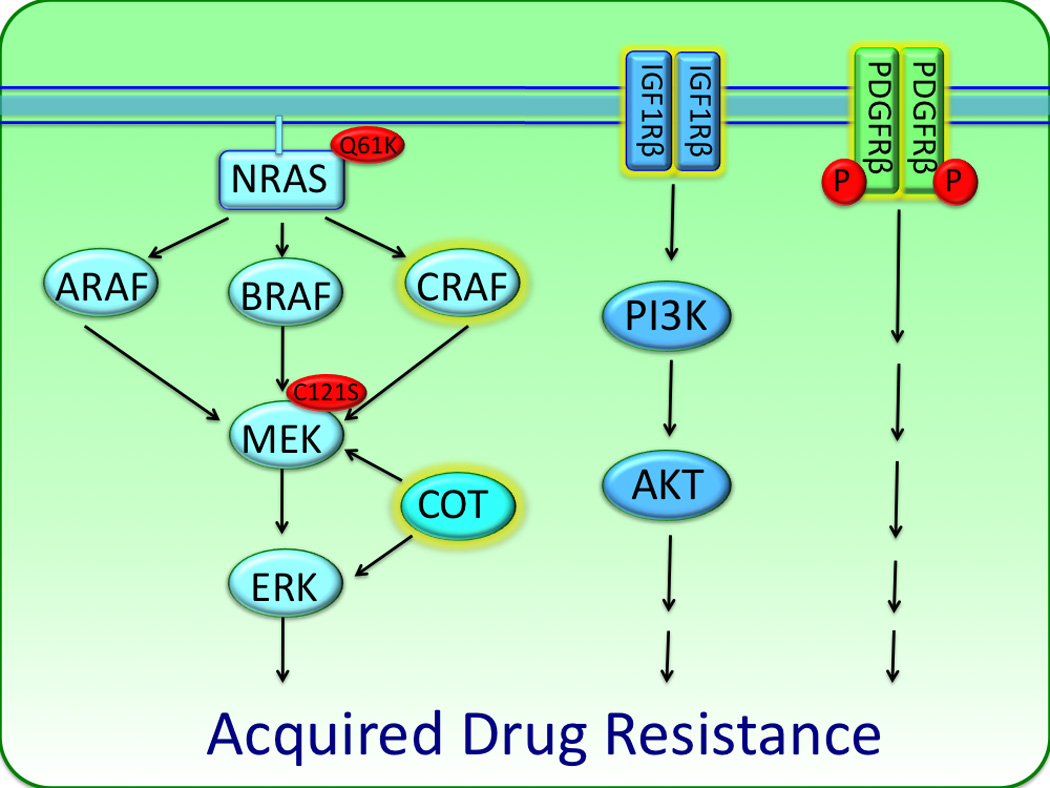
Multiple mechanisms of acquired BRAF inhibitor resistance have been identified. BRAF V600E melanoma cells chronically treated with BRAF inhibitors acquire drug resistance via switching between the three isoforms of RAF (ARAF, BRAF, CRAF) to activate the MAPK pathway. Increased IGFR1 and PDGFR signaling may also allow for resistance by activating PI3K/AKT signaling as well as other pathways. Resistance can also arise following the reactivation of the MAPK pathway, this can occur following the acquisition of activating mutations in NRAS (Q61K) and MEK (C121S) and the increased expression of the MAP3K8 (COT).
Acknowledgements
We apologize to those authors whose important original works we could not cite due to space restraints. Research in the Smalley lab is supported by The Melanoma Research Foundation, The Harry Lloyd Trust, The Bankhead-Coley Research Program of the State of Florida (09BN-14), The American Cancer Society (#93-032-13) and the NIH/National Cancer Institute (U54 CA143970-01).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011 doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 4.Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, et al. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J Clin Oncol. 2011 doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 5.Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28:85–94. doi: 10.1038/onc.2008.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smalley KS. Understanding Melanoma Signaling Networks as the Basis for Molecular Targeted Therapy. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.177. [DOI] [PubMed] [Google Scholar]
- 7.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 8.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 9.Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2009;22:400–419. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen A, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009 doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartlidge RA, Thomas GR, Cagnol S, Jong KA, Molton SA, Finch AJ, et al. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 2008;21:534–544. doi: 10.1111/j.1755-148X.2008.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol Cancer Res. 2009;7:549–556. doi: 10.1158/1541-7786.MCR-08-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boisvert-Adamo K, Aplin AE. Mutant B-RAF mediates resistance to anoikis via Bad and Bim. Oncogene. 2008;27:3301–3312. doi: 10.1038/sj.onc.1211003. [DOI] [PubMed] [Google Scholar]
- 15.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu SY, Lin P, Hsueh AJ. BOD (Bcl-2-related ovarian death gene) is an ovarian BH3 domain-containing proapoptotic Bcl-2 protein capable of dimerization with diverse antiapoptotic Bcl-2 members. Molecular endocrinology (Baltimore, Md. 1998;12:1432–1440. doi: 10.1210/mend.12.9.0166. [DOI] [PubMed] [Google Scholar]
- 18.Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–1014. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- 19.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 20.Jiang CC, Lai F, Tay KH, Croft A, Rizos H, Becker TM, et al. Apoptosis of human melanoma cells induced by inhibition of B-RAF(V600E) involves preferential splicing of bim(S) Cell Death Dis. 2010;1:e69. doi: 10.1038/cddis.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smalley KSM. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? International Journal of Cancer. 2003;104:527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 22.Klein RM, Aplin AE. Rnd3 regulation of the actin cytoskeleton promotes melanoma migration and invasive outgrowth in three dimensions. Cancer Res. 2009;69:2224–2233. doi: 10.1158/0008-5472.CAN-08-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arozarena I, Sanchez-Laorden B, Packer L, Hidalgo-Carcedo C, Hayward R, Viros A, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45–57. doi: 10.1016/j.ccr.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 24.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res. 2006;4:779–792. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 26.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 27.Maldonado JL, Fridlyand J, Patel H, Jain AN, Busam K, Kageshita T, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–1890. doi: 10.1093/jnci/djg123. [DOI] [PubMed] [Google Scholar]
- 28.Edwards RH, Ward MR, Wu H, Medina CA, Brose MS, Volpe P, et al. Absence of BRAF mutations in UV-protected mucosal melanomas. J Med Genet. 2004;41:270–272. doi: 10.1136/jmg.2003.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landi MT, Bauer J, Pfeiffer RM, Elder DE, Hulley B, Minghetti P, et al. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006;313:521–522. doi: 10.1126/science.1127515. [DOI] [PubMed] [Google Scholar]
- 30.Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, et al. Improving melanoma classification by integrating genetic and morphologic features. PLoS medicine. 2008;5:e120. doi: 10.1371/journal.pmed.0050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 32.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 33.Hauschild A, Agarwala SS, Trefzer U, Hogg D, Robert C, Hersey P, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–2830. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 34.Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, et al. Gatekeeper Mutations Mediate Resistance to BRAF-Targeted Therapies. Science translational medicine. 2 doi: 10.1126/scitranslmed.3000758. 35ra41. [DOI] [PubMed] [Google Scholar]
- 35.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010 doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kefford R, Arkenau H, Brown MP, Millward M, Infante JR, Long GV, et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. Journal of Clinical Oncology. 2010;28:8503. [Google Scholar]
- 38.King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–11105. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 39.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tap WD, Gong KW, Dering J, Tseng Y, Ginther C, Pauletti G, et al. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia. 2010;12:637–649. doi: 10.1593/neo.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 70:5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 44.Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–586. doi: 10.1038/sj.bjc.6603291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDermott DF, Sosman JA, Gonzalez R, Hodi FS, Linette GP, Richards J, et al. Double-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study Group. J Clin Oncol. 2008;26:2178–2185. doi: 10.1200/JCO.2007.14.8288. [DOI] [PubMed] [Google Scholar]
- 46.Flaherty KT, Puzanov I, Kim KB, Ribas A, MacArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hersey P, Smalley KS, Weeraratna A, Bosenberg M, Zhang XD, Haass NK, et al. Meeting report from the 7th International Melanoma Congress, Sydney, November, 2010. Pigment Cell Melanoma Res. 2011;24:e1–e15. doi: 10.1111/j.1755-148X.2010.00811.x. [DOI] [PubMed] [Google Scholar]
- 49.Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplan FM, Shao Y, Mayberry MM, Aplin AE. Hyperactivation of MEK-ERK1/2 signaling and resistance to apoptosis induced by the ongenic B-RAF inhibitor, PLX4720, in mutant N-Ras melanoma cell lines. Oncogene. 2010 doi: 10.1038/onc.2010.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 53.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smalley KS, Lioni M, Palma MD, Xiao M, Desai B, Egyhazi S, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 56.Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang JY, Chang CJ, Xia W, Wang Y, Wong KK, Engelman JA, et al. Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res. 2010;70:4709–4718. doi: 10.1158/0008-5472.CAN-09-4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70:8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Bruyne E, Bos TJ, Schuit F, Van Valckenborgh E, Menu E, Thorrez L, et al. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood. 2010;115:2430–2440. doi: 10.1182/blood-2009-07-232801. [DOI] [PubMed] [Google Scholar]
- 60.Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26:7560–7568. doi: 10.1038/sj.onc.1210558. [DOI] [PubMed] [Google Scholar]
- 61.Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 62.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science. 2009 doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010 doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J Clin Oncol. 2011 doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paraiso KH, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010 doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang CC, Lai F, Thorne RF, Yang F, Liu H, Hersey P, et al. MEK-Independent Survival of B-RAFV600E Melanoma Cells Selected for Resistance to Apoptosis Induced by the RAF Inhibitor PLX4720. Clin Cancer Res. 2010 doi: 10.1158/1078-0432.CCR-10-2225. [DOI] [PubMed] [Google Scholar]
- 70.Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene. 2006;25:3357–3364. doi: 10.1038/sj.onc.1209379. [DOI] [PubMed] [Google Scholar]
- 71.Jovanovic B, Egyhazi S, Eskandarpour M, Ghiorzo P, Palmer JM, Bianchi Scarra G, et al. Coexisting NRAS and BRAF mutations in primary familial melanomas with specific CDKN2A germline alterations. J Invest Dermatol. 2010;130:618–620. doi: 10.1038/jid.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin J, Goto Y, Murata H, Sakaizawa K, Uchiyama A, Saida T, et al. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Br J Cancer. 2011;104:464–468. doi: 10.1038/sj.bjc.6606072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF Gene Amplification Can Promote Acquired Resistance to MEK Inhibitors in Cancer Cells Harboring the BRAF V600E Mutation. Sci Signal. 2010;3 doi: 10.1126/scisignal.2001148. ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smalley KS, Flaherty KT. Integrating BRAF/MEK inhibitors into combination therapy for melanoma. Br J Cancer. 2009;100:431–435. doi: 10.1038/sj.bjc.6604891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 79.Lasithiotakis KG, Sinnberg TW, Schittek B, Flaherty KT, Kulms D, Maczey E, et al. Combined Inhibition of MAPK and mTOR Signaling Inhibits Growth, Induces Cell Death, and Abrogates Invasive Growth of Melanoma Cells. J Invest Dermatol. 2008 doi: 10.1038/jid.2008.44. [DOI] [PubMed] [Google Scholar]