

Abstract
Drugs and their metabolites often produce undesirable effects. These may be due to a number of mechanisms, including biotransformation by P450 enzymes which are not exclusively expressed by hepatocytes but also by endothelial cells in brain from epileptics. The possibility thus exists that the potency of systemically administered central nervous system therapeutics can be modulated by a metabolic blood–brain barrier (BBB). Surgical brain specimens and blood samples (ex vivo) were obtained from drug-resistant epileptic subjects receiving the antiepileptic drug carbamazepine prior to temporal lobectomies. An in vitro blood–brain barrier model was then established using primary cell culture derived from the same brain specimens. The pattern of carbamazepine (CBZ) metabolism was evaluated in vitro and ex vivo using high performance liquid chromatography–mass spectroscopy. Accelerated mass spectroscopy was used to identify 14C metabolites deriving from the parent 14C-carbamazepine.
Under our experimental conditions carbamazepine levels could not be detected in drug resistant epileptic brain ex situ; low levels of carbamazepine were detected in the brain side of the in vitro BBB established with endothelial cells derived from the same patients. Four carbamazepine-derived fractions were detected in brain samples in vitro and ex vivo. HPLC-accelerated mass spectroscopy confirmed that these signals derived from 14C-carbamazepine administered as parental drug. Carbamazepine 10, 11 epoxide (CBZ-EPO) and 10, 11-dihydro-10, 11-dihydrooxy-carbamazepine (DiOH-CBZ) were also detected in the fractions analyzed. 14C-enriched fractions were subsequently analyzed by mass spectrometry to reveal micromolar concentrations of quinolinic acid (QA). Remarkably, the disappearance of carbamazepine-epoxide (at a rate of 5% per hour) was comparable to the rate of quinolinic acid production (3% per hour). This suggested that quinolinic acid may be a result of carbamazepine metabolism. Quinolinic acid was not detected in the brain of patients who received antiepileptic drugs other than carbamazepine prior to surgery or in brain endothelial cultures obtained from a control patient.
Our data suggest that a drug resistant BBB not only impedes drug access to the brain but may also allow the formation of neurotoxic metabolites.
Keywords: Blood–brain barrier, Drug resistance, Drug metabolism, Drug delivery, Carbamazepine, Pharmacokinetics, In vitro models, Tissue engineering, Neurotoxicity, Pharmacogenomics
Introduction
Drug resistance in epilepsy represents an unsolved clinical problem. While a number of new anti-epileptic drugs (AED) have become available in the past years, seizures continue to be poorly controlled by these AEDs in approximately 25–30% of patients (Kwan et al., 2011). It is not clear whether AEDs fail to exert their effect due to pharmaco-kinetic or pharmacodynamic mechanisms, or a combination of the two. It has been suggested that multiple drug resistance may be a pathology distinct from epilepsy, one that requires a specific treatment (Granata et al., 2009). Recent evidence suggested the presence of P450 enzymes in the drug resistant epileptic (DRE) human brain (Ghosh et al., 2010, 2011a, 2011b). Specifically, enzymes responsible for the metabolism of AEDs (e.g., CYP3A4 metabolizing carbamazepine, CBZ) are over-expressed by drug resistant blood–brain barrier endothelial cells (DRE BBB) and by neurons. Interestingly, while other drug resistant molecules such as MDR1 are almost ubiquitously expressed in central nervous system (CNS) cells from epileptic resections, we failed to observe any glial expression of CYP3A4 (Ghosh et al., 2011a).
It still remains unclear whether brain-specific enzymatic machinery is responsible for the metabolism of AEDs and insufficient drug levels in the resistant brain. However, it is often assumed that most of the metabolic burden of drug metabolism is hepatic but increasing evidence points to the CNS as a drug metabolizing organ (Ghosh et al., 2010, 2011a, 2011b). The concomitant CNS expression of MDR1 and P450 enzymes in the drug resistant epileptic brain suggests that the bioavailability of AEDs is dictated by mechanisms similar to those used by the liver as the principal drug-metabolizing organ (Ghosh et al., 2010). The presence of a BBB machinery to manage drug fate is an important finding, since a local metabolic process may allow the formation and uncontrolled accumulation in the brain of molecules with unexpected functions or even toxicity. These may further confound brain pharmacology under pathological conditions associated with multiple drug resistance (Dombrowski et al., 2001; Loscher and Potschka, 2005; Marchi et al., 2010a).
While P450 enzymes are involved in virtually all CNS drug metabolisms, for this study we focused on the pattern of CBZ biotransformation. Similarly, while multiple drug resistance is a common clinical challenge (Granata et al., 2009; Loscher and Potschka, 2005), we focused on epilepsy due in part to the availability of tissue samples and detailed understanding of the molecular players involved. While details on intermediate metabolites of CBZ at the BBB are scant, hepatic CBZ metabolism has been extensively studied (Breton et al., 2005; Ju and Uetrecht, 1999). CBZ transformation by hepatocytes consists of an oxidation step by CYP3A4 and formation of carbamazepine-10,11-epoxide (CBZ-EPO). Excretion occurs after hydroxylation to CBZ-10,11-trans-dihydrodiol and Phase II conjugation. Since CYP3A4 is prominently expressed at the DRE BBB (Ghosh et al., 2010), we wished to test the hypothesis that a local CNS metabolism of CBZ occurs differently in drug resistant epileptic brain. This was achieved by use of a combined ex vivo (brain and blood samples)/in vitro (a model of the DRE BBB) experimental approach previously validated in a comparable experimental setting (Cucullo et al., 2007; Cucullo et al., 2008; Desai et al., 2002; Ghosh et al., 2010, 2011a; Grant et al., 1998). To detect and trace CBZ, we took advantage of a combination of analytical techniques; including high performance liquid chromatography (HPLC), mass spectrometry (MS) and accelerated mass spectrometry (AMS) (Tompkins et al., 2006) to profile the CBZ biotransformation by the BBB and in a drug resistant brain.
Materials and methods
Recruitment of subjects
The investigation conforms to the principles outlined in the Declaration of Helsinki. Patient consent was obtained as per the Institutional Review Board instructions. Epileptic patients were selected according to the antiepileptic drug administered before resection. Patients received carbamazepine or other AEDs (n=9) prior to surgery as part of their anti-epileptic drug schedule (Table 1). As a control we used a patient that was surgically operated due to cerebral aneurysm and administered AED prior to surgery. Brain and blood samples were collected as described in Fig. 1 and as reported in our previous studies (Ghosh et al., 2010, 2011a). Brain and blood samples were used for determination of CBZ metabolites using HPLC paired to accelerator mass spectroscopy (AMS) or conventional mass spectroscopy (MS). Primary brain endothelial cells were established from brain specimens and used for in vitro studies (Cucullo et al., 2008; Ghosh et al., 2010; Santaguida et al., 2006).
Table 1.
Demographic characteristics of the patients in this study.
| Patient id | Pathology | Age (yrs.) | Gender | Classification | Age of seizure onset | Seizure frequency | AED taken before surgery |
|---|---|---|---|---|---|---|---|
| Patient 1 | Drug resistant temporal lobe epilepsy (DRE-1) | 41 | F | Temporal lobe epilepsy | 18 months | Initially 4–8/month; with LEV 4/month | CBZ 12 h before surgery Other AEDs: LEV |
| Patient 2 | Drug resistant temporal lobe epilepsy (DRE-2) | 28 | F | Temporal lobe epilepsy | 14 yrs. old | 90/month | CBZ 2 h before surgery Other AEDs: LEV |
| Patient 3 | Drug resistant temporal lobe epilepsy (DRE-3) | 27 | M | Temporal lobe epilepsy | 20 yrs. old | 0.2/month | CBZ 14 h before surgery Other AEDs: LEV |
| Patient 4 | Drug resistant temporal lobe epilepsy (DRE-4) | 11 | M | Temporal lobe epilepsy | 4 yrs. old | 90/month | CBZ 17 h before surgery Other AEDs: LEV |
| Patient 5 | Drug resistant temporal lobe epilepsy (DRE-5) | 56 | F | Temporal lobe epilepsy | 46 yrs. old | 0.16 clusters/month | CBZ 18 h before surgery Other AEDs: LMT |
| Patient 6 | Drug resistant temporal lobe epilepsy (DRE-6) | 35 | F | Temporal lobe epilepsy | NA | NA | CBZ 8 h before surgery Other AEDs: TPM |
| Patient 7 | Cerebral aneurysm | 46 | F | Non-ruptured cerebral | NA | NA | TPM before surgery |
| Patient 8 | Drug resistant temporal lobe epilepsy (DRE-7) | 28 | F | aneurysmTemporal lobe epilepsy | 16 yrs. old | 6/month | LMT, GBP, PGB 2 h before surgery |
| Patient 9 | Drug resistant temporal lobe epilepsy (DRE-8) | 45 | F | Temporal lobe epilepsy | 40 yrs. old | 2–4 every few weeks in clusters | LMT 2 h before surgery |
AED Abbreviations: CBZ, carbamazepine; TPM, topiramate; LEV, levetiracetam; LMT, lamotrigine; GBP, gabapentin; PGB, pregabalin; NA, not available.
Fig. 1.
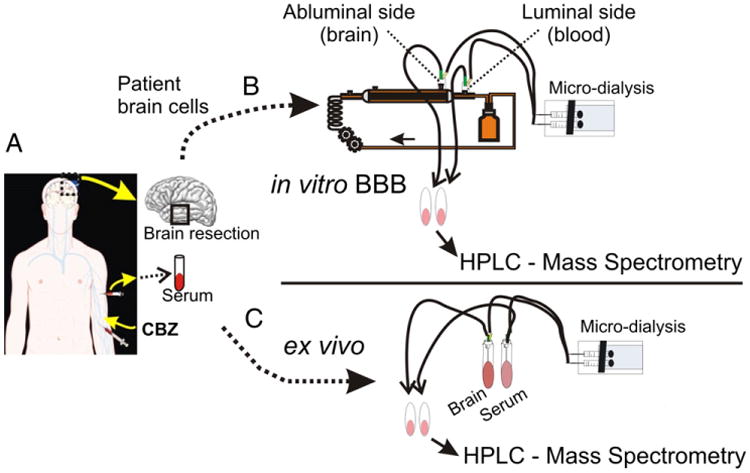
Experimental approach. (A) We selected drug resistant epileptic patients receiving CBZ prior to surgery. The exact time and dosage of drug administration were noted. Brain and blood samples were collected at time of resection. Each brain specimen was processed to give a sample for the establishment of primary brain endothelial cell culture and a comparable sample for ex vivo HPLC-UV analysis. (B) Primary brain endothelial cells were cultured using a flow-based in vitro system (DIV) allowing for the formation of a physiologic blood–brain barrier. CBZ and 14C-CBZ were added to the DIV-BBBs. (B–C) Levels of CBZ and its metabolites were measured ex vivo (brain and blood samples) and in vitro (luminal and extra-luminal sides of the DIV-BBB) using a combination of microdialysis, HPLC-UV, MS and AMS analysis.
Endothelial cells
Primary endothelial cells were isolated from secondary branches of middle cerebral arteries of brain specimens from patients undergoing temporal lobectomies to relieve medically intractable seizures or removed from aneurysm domes. Cell culture methods are as described in details elsewhere (Desai et al., 2002; Dombrowsk et al., 2001; Ghosh et al., 2010).
Dynamic in vitro model (DIV)
DIV modules were purchased from Spectrum (catalog. no 400–025, Spectrum Lab, CA, USA) or Flocel Inc. (Cleveland, OH). Each module consists of hollow polypropylene capillaries embedded in a clear plastic chamber. The capillaries are connected with a reservoir for media and a pulsatile pump apparatus. We used a flow rate of 4–5 ml/min corresponding to a shear stress of 3–10 dyn/cm2.
Drugs and sample preparations
Carbamazepine (CBZ, catalog number 4024), 14C-carbamazepine (14C CBZ, catalog number C-7737) and carbamazepine 10, 11 epoxide (CBZ-EPO, catalog number 4206) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Quinolinic acid (QA, catalog number P63204) was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Brain tissues were rinsed with PBS (0.01 M), and homogenized (10 ml/g) in methanol/water (60/40, v/v). Homogenates were vortexed and centrifuged at 2500 rpm for 15 min. Blood samples were centrifuged at 2500 rpm for 10 min to isolate serum. Microdialysis was used to collect free fractions of drug present in vitro and ex vivo samples. A gas-tight microdialysis syringe (5 ml) was filled with perfusion fluid (methanol/water 60/40, v/v) and DMEM (Dulbecco's Modified Eagle Medium for serum). The syringe is connected to the microdialysis probe (4 mm, catalog number MD-2204, Bioanalytical Systems, USA) using a plastic tube and operated with a pump (flow rate=1 μl/min, Bioanalytical Systems, Inc., IN, USA). Microdialysis probes have a 320 μm outer diameter, 4 mm membrane length, and a low molecular weight cut off membrane (<5000 Da).
HPLC analysis: CBZ, CBZ-EPO and QA measurement
Chromatographic conditions
Chromatographic separations (HPLC, Agilent 1200 series) were performed using a Zorbax Eclipse Plus C18 stainless steel column (4.6×150 mm, 3.5 μm), supplied by Agilent Technologies Inc, USA (Ghosh et al., 2010, 2011a). The mobile phase consisted of acetonitrile:methanol:water (18:19:63, v/v/v). Standard solutions: CBZ and CBZ-EPO were dissolved in methanol (1 mg/ml). Standards (0.5, 5, 10, 20, 40 and 60 μg/ml) were prepared by further dilution of stock solution. Quinolinic acid standard 5 mM stock was prepared in 1 N NaOH (following Sigma-Aldrich specification guidelines) and was further diluted to prepare calibration standards. Solutions were stored at −20 °C.
Chromatography was performed at room temperature, flow rate of 1 ml/min and detection performed at 210 nm. Sampling and extraction procedure: Brain and serum microdialysis eluates (50 μl) were injected in the column CBZ and CBZ-EPO and QA standard chromatograms were matched with the profiles obtained from brain and serum samples. Fractions were collected for mass spectrometry analysis.
An alternative method was used for better resolution and separation of the QA-containing fraction and subsequent fractions. The mobile phase consisted of methanol:water:acetic acid (64:35.9:0.1, v/v/v, modified method of Husain et al., 1995). A Zorbax Eclipse Plus C18 stainless steel column (4.6×150 mm, 3.5 μm, Agilent Technologies, USA) was used. Samples were dissolved in the mobile phase. The analysis was performed under isocratic conditions at a flow rate of 0.2 ml/min at room temperature. Analytes were detected at 254 nm. Fractions were collected for mass spectrometry analysis. Both methods were validated based on a standard calibration curve and by determining the limit of quantification, precision and accuracy (see Table 2).
Table 2.
Precision of carbamazepine, carbamazepine-epoxide, quinolinic acid quantification.
| Drugs/metabolites | Concentration added (μg/ml) | Intraday | Correlation (r) | ||
|---|---|---|---|---|---|
|
| |||||
| Measured concentration (mean±SD) | CV (%)a | Accuracy (%)b | |||
| Carbamazepine | 1 | 1.043 ± 0.060 | 5.752 | 104.30 | 0.999 |
| 5 | 4.979 ± 0.157 | 3.153 | 99.58 | ||
| 10 | 10.201 ± 0.186 | 1.823 | 102.01 | ||
| 25 | 25.351 ± 0.312 | 1.230 | 101.40 | ||
| 50 | 49.761 ± 0.305 | 0.612 | 99.52 | ||
| Carbamazepine-epoxide | 1 | 0.994 ± 0.057 | 5.734 | 99.40 | 0.998 |
| 5 | 4.973 ± 0.183 | 3.679 | 99.46 | ||
| 10 | 9.952 ± 0.023 | 0.231 | 99.52 | ||
| 25 | 24.828 ± 0.192 | 0.773 | 99.31 | ||
| 50 | 49.939 ± 0.182 | 0.364 | 99.87 | ||
| QA method-1 | 0.84 | 0.838 ± 0.033 | 3.93 | 99.76 | 0.998 |
| 1.68 | 1.689 ± 0.016 | 0.945 | 100.53 | ||
| 4.20 | 4.198 ± 0.213 | 5.073 | 99.95 | ||
| 8.40 | 8.422 ± 0.089 | 1.056 | 100.26 | ||
| 16.80 | 16.762 ± 0.141 | 0.841 | 99.77 | ||
| QA method-2 | 0.84 | 0.849 ± 0.033 | 3.866 | 101.07 | 0.999 |
| 1.68 | 1.682 ± 0.018 | 1.070 | 100.11 | ||
| 4.20 | 4.221 ± 0.123 | 2.914 | 100.50 | ||
| 8.40 | 8.413 ± 0.312 | 3.708 | 100.15 | ||
| 16.80 | 16.81 ± 0.213 | 1.267 | 100.05 | ||
Mean values represent five different samples of each concentration.
Accuracy = 100 (observed concentration/theoretical concentration).
CV % or relative standard deviation % = 100 (standard deviation/mean).
Quantification and sensitivity
Quantifications of serum and brain concentrations of CBZ, CBZ-EPO and QA were achieved by relating peak-area ratios of the drug with the known concentrations on the calibration curve. The slope and correlation coefficients (r) were determined using the least-squares linear regression analysis method. Results from the linearity study showed 0.01–6.16% CV of standard curve based on absorbance values and a correlation of 0.998 between peak-area ratio and concentrations. The limit of quantification (LOQ) is the lowest amount of analyte that can be measured with the defined precision and accuracy. The limit of quantification was calculated by 10× (SD/S) where SD represents the standard deviation of the determination and S represents the slope of the calibration curve (Zhang et al., 2009). Under the experimental conditions used, the LOQ was approximately 0.04 (CBZ) and 0.05 (CBZ-EPO) and 0.01 μg/ml (QA). The coefficient of variation was found to be less than 7%.
Precision and accuracy
The intra-day precision was determined from a replicate analysis of pooled human serum/DMEM containing CBZ, CBZ-EPO and QA at five different concentrations covering low, medium and higher ranges of the calibration curve. Precision was expressed as the percent coefficient of variation (% CV), and accuracy as a percentage of the theoretical concentration (observed concentration×100/theoretical concentration). The intra-day precision ranged from 0.61 to 5.75% CV. Accuracy ranged from 99.40 to 104.30% (Table 2).
Mass-spectroscopy analysis
Samples were fractionated according to peak retention times. HPLC fractions were then collected and processed for MS analysis. Fractions were speed vacuumed, dried and reconstituted in 50 μl of 40% acetonitrile, 0.1% formic acid solutions. LTQ FT Ultra mass spectrometer (Thermo Fisher Scientific, CA) equipped with electrospray ionization (ESI) source was used for all ESI MS analysis. Each fraction was introduced into mass spectrometer by infusion. All MS spectra were acquired in the positive ion mode with the needle voltage of 4.5 kV. A full MS survey scan was recorded in the Fourier Transform (FT) analyzer at resolution R=100,000 for the m/z range of 50–300. The capillary temperature was set at 275 °C. Nitrogen was used as nebulizer and dry gas. The MS tandem analysis was used to confirm the parent drug by generating the fragment ions. For example, under CID conditions in the FTICR cell, the major product ion of m/z 194 was observed for CBZ precursor ion with m/z of 237 (Supplementary Fig. 1A). MS and tandem MS data were further analyzed by LC-MCD Trap 4.2 Quant Analysis Version 1.5 Software (Bruker Daltonnik GmbH, Bremen, Germany) and molecular ions identified by Scripps Center for Mass Spectrometry (http://metlin.scripps.edu/).
Accelerator mass spectroscopy, AMS (Tompkins et al., 2006)
This technique routinely prepares and analyzes samples containing extremely low concentrations of 14C generated in clinical trials. Instrument background levels were consistently in the low 10−16 range (14C/12C). Chemical background was approximately equivalent to 0.4% of natural background levels of 14C. When samples consisting of 14C levels 100 times above natural background were repeatedly processed, no increase in system background was observed, either during sample processing or during AMS measurement. Samples prepared and measured in duplicate had a correlation coefficient R2=0.9987. In AMS, carbon is required to make any measurement; therefore, it is not possible to have a blank sample without having carbon present. Unlabeled CBZ (final concentration in the DIV-BBB of 50 μg/ml and 14C-labeled CBZ (0.2 dpm/μl in DIV-BBB, specific activity = 22.6 mCi/mM)) was sufficient to provide detectable signal of the new peaks, possibly related to CBZ metabolism. Considering 1 mCi = 2.2×109 dpm, 0.2 dpm/μl in this study was equivalent to 100 pmol/μl.
Statistical analysis
We used Origin 7.0 (Origin Lab, Northampton, MA, USA) and Jump 7.0 software. For all parametric variables, differences between populations were analyzed by ANOVA In all figures, symbols with error bars indicate mean±SEM; *p<0.05 was considered statistically significant.
Results
Patients received carbamazepine (CBZ) prior to surgery as part of their antiepileptic drug (AED) regimen. Brain and blood samples were collected during surgery as described in Fig. 1 and Table 1. These samples were used for brain and serum determination of CBZ metabolites by HPLC paired to accelerated (AMS) or conventional mass spectroscopy (MS). Brain endothelial cells from the same patients were used for in vitro studies (Fig. 1 and (Ghosh et al., 2010, 2011a; Santaguida et al., 2006). As expected (Potschka et al., 2001; Rambeck et al., 2006), CBZ had limited brain permeability (Figs. 2A1–A2). We found blood levels of free CBZ in the range of 1.38–1.50 μg/ml as described by others (Rambeck et al., 2006). While we could not detect CBZ in the brain by using ex-situ microdialysis, Rambeck et al. detected μg/g concentration of CBZ by using an intra-operative approach. The variability in the CBZ levels detected in the drug resistant epileptic brain may be due to the different techniques used. However, HPLC fractions in the 2 to 10 min interval contained known CBZ metabolic products. These were almost entirely segregated by brain-derived samples (fractions 1 and 4; note the statistically significant difference in A2).
Fig. 2.
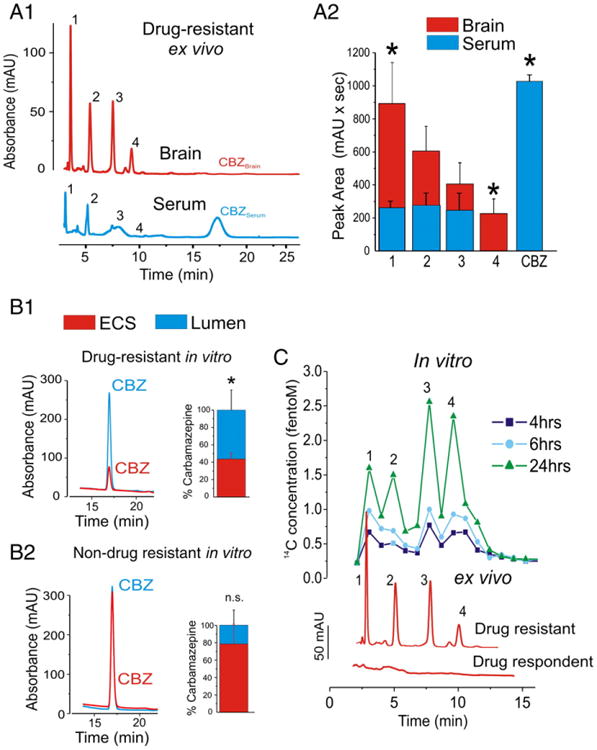
Brain endothelial cells metabolize CBZ. (A1–A2) HPLC quantification of serum and brain levels of CBZ. CBZ was absent in brain and detected in blood of drug resistant epileptic subjects (n=6). Note the appearance of signals (retention time: 2–10 min) possibly related to CBZ metabolic products. The latter were predominantly segregated in brain (P<0.05, fractions 1 and 4). (B1) Lack of CBZ penetration was detected across the in vitro drug resistant epileptic BBB. This is in contrast to what was seen with brain endothelial cells from a non-drug resistant patient (B2), where no barrier to CBZ passage was measured and where levels in the brain were comparable to serum. (C) Accelerated mass spectroscopy (AMS) analysis indicated that the fractions detected in the multiple drug resistant brains, in vitro and ex vivo, derived from the parent, radiolabelled 14C-CBZ. The figure shows 14C peaks measured in the brain side of the in vitro BBB between 2 and 10 min. Peak retention times matched in the two modalities. Note the time-dependent increase of radiolabelled CBZ metabolites. The bottom traces show a comparison of HPLC profiles obtained from a drug resistant epileptic and a control sample. Note the absence of signal in the adopted control brain. (* indicates p<0.05, data are presented as mean±SEM).
An obvious obstacle in these studies is the lack of “normal” comparison brain tissue. Thus, brain samples derived from drug resistant patients have no true “controls”, the closest being their “drug respondent” counterpart. However, these samples are very hard to access since epileptics responding to drug therapy almost never undergo brain resective surgery. Data from a patient (see Table 1) undergoing resection for a cerebrovascular event showed that reduced CBZ permeability across the blood–brain barrier appears to be an exclusive property of multiple drug resistant brain (Fig. 2B).
We demonstrated that in vitro human brain endothelial cells spare their in vivo properties of multiple drug resistance if grown under flow conditions (Desai et al., 2002; Ghosh et al., 2010, 2011a). P450 and MDR1 expression and physiological functions are thus preserved in endothelial cell cultured under dynamic conditions (Desai et al., 2002; Dombrowski et al., 2001; Ghosh et al., 2010). We took advantage of this system to evaluate the pattern of CBZ penetration and metabolism in the DRE and non-DRE in vitro BBB (Fig. 2B). Results were compared to data obtained ex vivo (Fig. 2C). Low amounts of CBZ were detected in the brain side of the DRE BBB model compared to the non-DRE (Figs. 2B1–B2). Thus, brain endothelial cells derived from drug respondent subjects do not appear to form a barrier to CBZ passage. HPLC-AMS analysis confirmed the presence of CBZ brain metabolites (fractions 1–4, Fig. 2C) which were not detected in non-DRE brain, but were produced by in vitro DRE BBB endothelial cells. Signals contained radioactive carbon and were therefore derived from exogenous 14C-CBZ (Fig. 1C). Signals were more pronounced in brain-side fractions ex vivo and in vitro. Taken together, these results support the notion that fractions 1–4 were derived from BBB metabolism of 14C-CBZ.
Fractions were subsequently analyzed using MS to determine the molecular mass of constituents. In fraction 1 we found a specific m/z of 167.96 and 168.0 in positive ion mode matching the molecular weight of quinolinic acid (QA, Fig. 3, see also Table 2). In the same samples, we measured the protonated form of kynurenine (a precursor of QA, m/z=209.03, Fig. 3, see also Table 3). In fraction 4 we found a molecular component with m/z correspondent to 10, 11-dihydro-10, 11-dihydrooxy-carbamazepine (DiOH-CBZ, m/z= 270.98 m/z, M+H+) (Fig. 3B). The molecular nature of known CBZ metabolites detected in brain samples was further confirmed using HPLC-UV. For example, we positively identified CBZ-epoxide in these samples (CBZ-EPO, see Supplementary Fig. 1 and Fig. 5). To confirm the presence of quinolinic acid (QA), fraction 1 was further analyzed by two ad hoc HPLC protocols (Fig. 4). Regardless of the protocol used, we found similar concentrations (8.09 μM and 7.56 μM) of QA in the drug resistant brain.
Fig. 3.
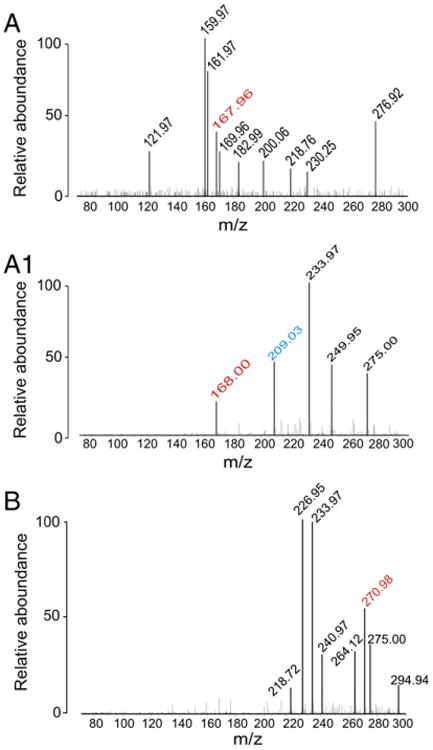
MS analysis reveals the presence of QA and DiOH-CBZ. (A–A1) Fraction 1 was collected by HPLC and analyzed by MS. We found m/z=167.96 and 168.0 in positive ion mode matching the molecular weight of quinolinic acid (QA, see also Table 2). A protonated form of kynurenine (a precursor of QA, m/z=209.03) was also detected in this fraction (see Table 2). (B) Fraction 4 contained 10, 11-dihydro-10, 11-dihydroxy-carbamazepine (DiOH-CBZ, m/z=270.98 m/z M+H+) and for molecular ion identification see Table 2. Known CBZ metabolites were identified in the remaining fractions (see Supplementary Fig. 1).
Table 3.
Molecular ions of each compound.
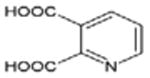
| Compounds | Mass | [M+H]+ | Formula | Structure |
|---|---|---|---|---|
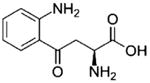
| Quinolinic acid | 167.02 | 168.02 | C7H5NO4 |
|
| Kynurenine | 208.08 | 209.08 | C10H5N2O3 |
|
| 10,11-dihydro-dihydroxy carbamazepine | 270.10 | 271.10 | C15H14N2O3 |
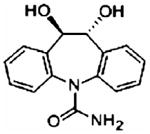
|
Fig. 5.
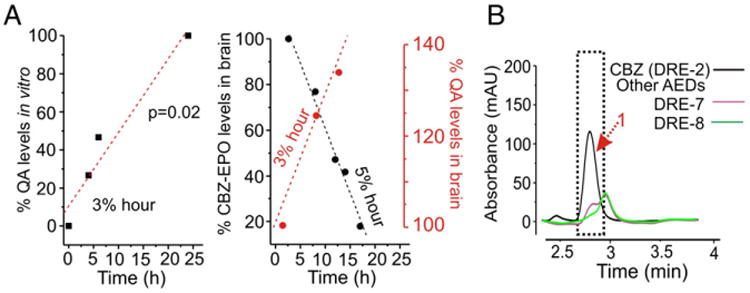
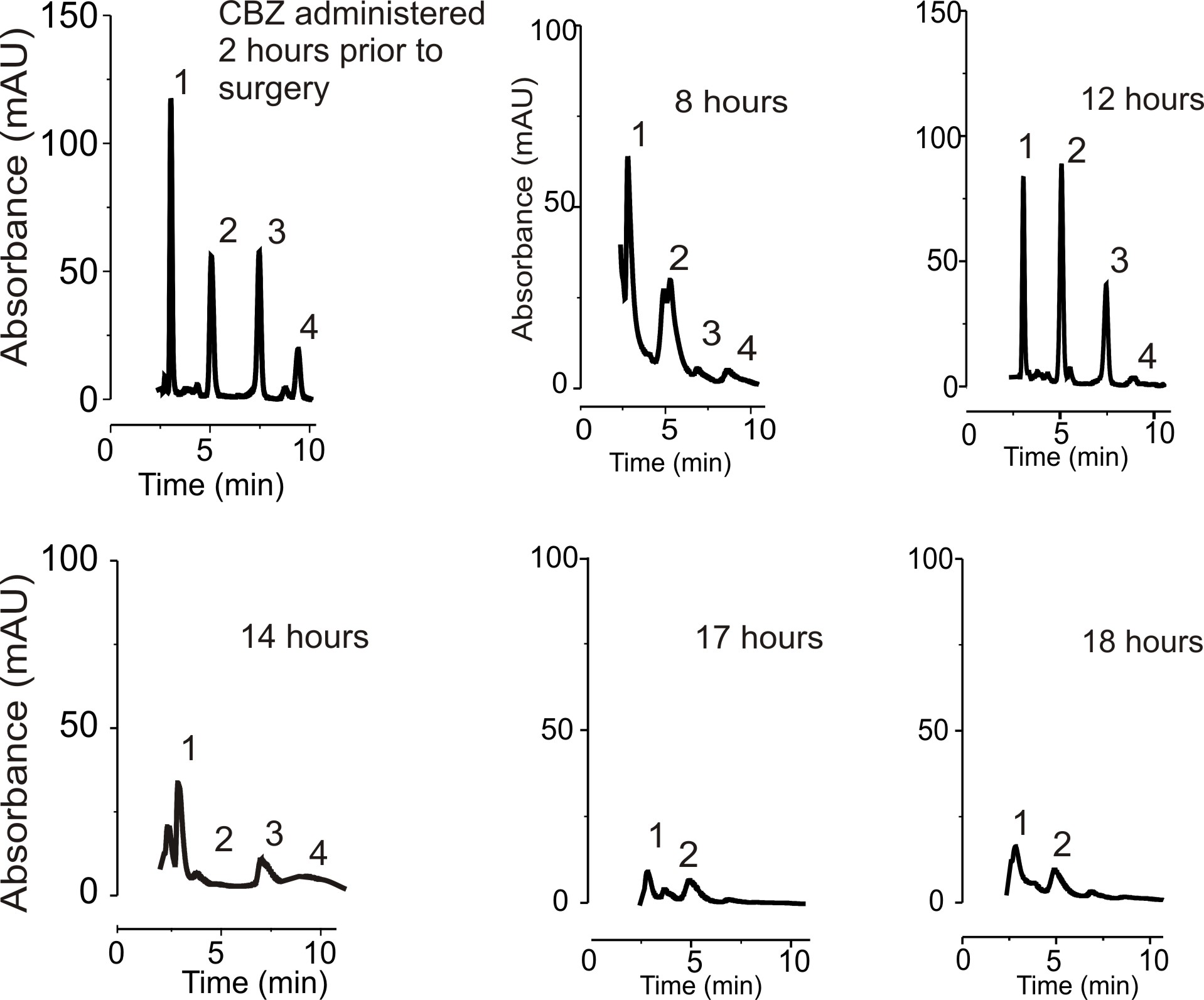
Time course of QA production. (A) Note that the axes in this panel refer to both CBZ-EPO (black ink) and QA (red ink). Ex vivo data: time-dependent disappearance of CBZ-epoxide (a known CBZ metabolite, black symbols) and appearance of QA (in red) occur at comparable rates. In vitro, brain EC produces QA at a rate of 3% per hour again similar to ex vivo; the figure depicts values obtained by MS-AMS analysis of 14C labeled CBZ and its products (see also Fig. 2C). (B) No QA was detected in samples from patients administered with AEDs other than CBZ.
Fig. 4.
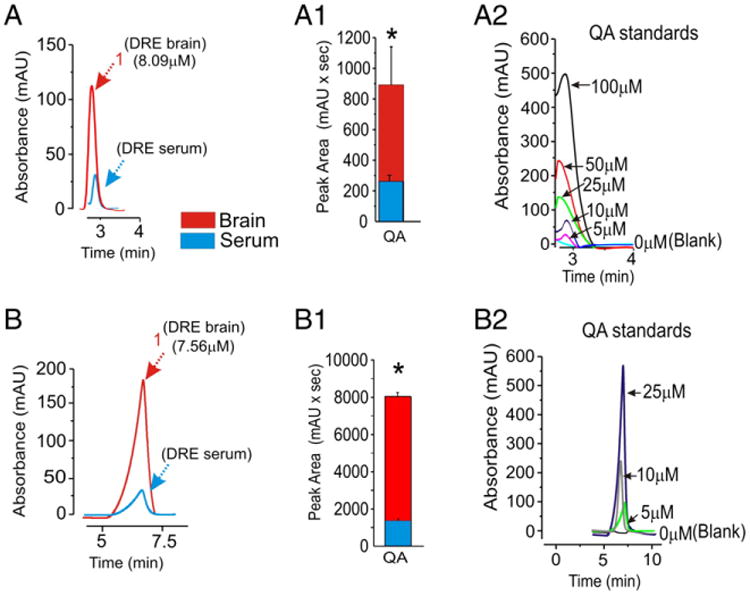
HPLC-UV determination of quinolinic acid (QA). (A–B) The presence of QA in fraction 1 was confirmed using two ad hoc HPLC-UV methods. (A1–B1) Levels of QA are significantly higher in the brain compared to serum. Brain levels detected using the two methods were identical (8.09 μM and 7.56 μM); note that the elution time of standards matched chromatograms of brain or serum samples (2.9 and 6.8 min in A and B respectively). (A2–B2) Dose response of QA was performed using two HPLC methods (* indicates p<0.05, data are presented as mean±SEM).
To determine the time course of QA appearance in brain samples, we compared its levels to the time interval from oral administration of CBZ. In the same samples, we determined the levels of CBZ and its metabolites. In the in vitro model of the BBB, radioactive fractions containing CBZ derivatives were isolated and analyzed with identical methods as ex vivo. A kinetic analysis of QA levels in brain showed that QA was formed at a comparable rate at which a surrogate of the parental drug was metabolized (CBZ-Epoxide; Fig. 5A and Supplementary Fig. 2). CBZ added to culture media in vitro was transformed in QA at a rate of 3% per hour, again comparable to what was observed in the brain (Fig. 5A). Finally, it was necessary to determine whether QA measured in the brain of epileptics was derived from endogenous metabolic processes, other therapeutics or derived especially from CBZ metabolism. The CBZ specificity of QA origin was demonstrated in two patients who took lamotrigine, pregabalin or gabapentin but never used CBZ (Fig. 5B). No QA was detected in the brain tissue from these patients, suggesting that CBZ and not the epileptic condition or associated AED may be the potential source of QA.
Discussion
Our results suggest that in human multiple drug resistant epileptic brain QA may be a result of P450-mediated CBZ metabolism. Our findings were obtained in a “translational” framework, since we explored drug metabolism directly in human subjects and in an in vitro model using the very same subjects' cells. These results also provide an insight into the unknowns and complexity of “personalized neurophar-macology”. Whether the brain itself or liver cells are responsible for this surprising finding remains undetermined, but our data suggest at least a partial metabolic role for CNS endothelial cells. The lack of measurable CBZ in the CNS may be due to the concomitant action of BBB metabolic machinery (Ghosh et al., 2010, 2011a, 2011b) along with efflux transporters (Loscher and Potschka, 2005; Marchi et al., 2010a; Rambeck et al., 2006).
While conclusive evidence linking QA to CBZ in human brain would require in vivo tracer experiments, we believe that the following considerations strongly support the presence of as yet unknown metabolic pathways responsible for CNS CBZ metabolism. The parental (CBZ) origin of QA was supported by the following: 1) 14C-CBZ metabolites were found in QA-containing fractions; 2) QA presence in fraction 1 was confirmed by MS and by two independent HPLC methods; 3) The rate of QA formation by brain EC in vitro was comparable to the rate of CBZ metabolism and QA production ex vivo, suggesting that a common pathway may be involved; and, 4) QA was not detected in brain of patients who took AEDs other than CBZ or in a control. The latter, in our opinion, suggests that the influence of CBZ on the traditional metabolic machinery for QA production is not necessary for CBZ conversion to QA.
Others have mapped QA to the epileptic brain, and its presence appears to be related to abnormal spiking activity typical of the seizure-prone brain (Fedi et al., 2003). Remarkably, all patients in this study were undergoing CBZ therapy, further supporting the exogenous origin of at least some of the QA present in the brain. However, CBZ enhances production of kynurenic acid (Kocki et al., 2004), a member of the tryptophan-QA pathway. Kynurenine (Kyn) was identified by us in fraction 1. Since the levels of quinolinic acid (seizure agonist) and kynurenic acid (seizure antagonist) are both elevated in certain pathologies (Stone and Darlington, 2002) the final outcome of their presence is dictated by their ratio. Thus, the true potentiality for an endogenous convulsant derived from CBZ ultimately rests on the availability of kynurenic acid (KA) as well as that of QA.
While the metabolic machinery involved in CBZ metabolism to QA is unknown, others have shown that CBZ is transformed into 9-hydroxymethyl-10-carbamoyl acridan (9-acridan) (Breton et al., 2005). In our experiments, CBZ was 14C-labeled in the carbonyl group which is preserved in 9-acridan and in QA as carboxylic acid (Fig. 6 and Supplementary Fig. 3). Thus, the initial steps of CBZ conversion into QA implicate a step consisting of 9-acridan formation carbamazepine-epoxide, CBZ-EPO followed by 10, 11-dihydro-10, 11-dihydrooxy-carbamazepine, DiOH-CBZ (Breton et al., 2005).
Fig. 6.
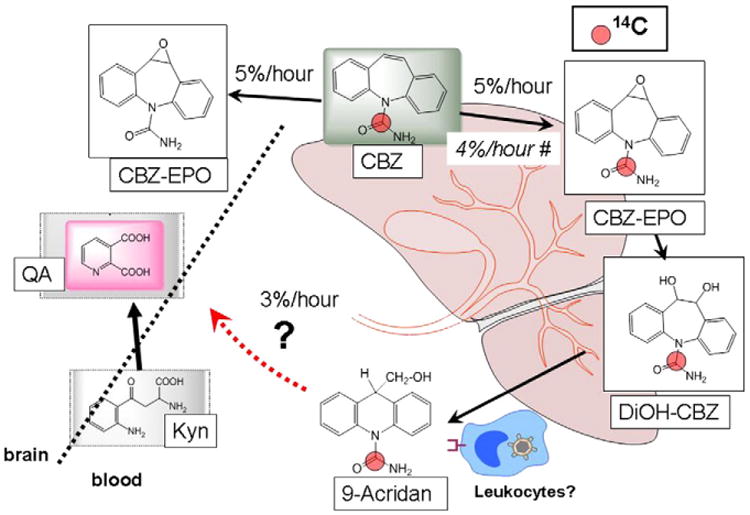
Proposed scheme of carbamazepine metabolism and quinolinic acid formation in the drug resistant epileptic brain. Carbamazepine metabolism by either liver (Fertig, 2008) or BBB EC (Ghosh et al., 2010, 2011a; Granata et al., 2009) results in the formation of known metabolites carbamazepine 10, 11, epoxide (CBZ-EPO) and 10, 11-dihydro-10, 11-dihydrooxy-carbamazepine (DiOH-CBZ). The published rate of CBZ metabolism ex vivo and in multiple drug resistant patients is approximately 4% (Fertig, 2008). BBB endothelial cell metabolism in vitro led to the formation of quinolinic acid (QA, Fig. 5) and perhaps of its precursor, kynurenine (Kyn). While the exact metabolic steps remain to be elucidated, we propose the following scenarios: 1) Liver enzymes (P450) produce CBZ-EPO and possibly QA, while brain cells may metabolize CBZ to QA; 2) In our experiments we could not estimate CBZ rate of metabolism by direct CNS determinations since no CBZ was found in the CNS (Fig. 2). We therefore used CBZ-EPO as a surrogate marker. The measured rate of CBZ metabolism to CBZ-EPO was similar to the measured appearance of brain QA; (∼3% per hour as shown in Fig. 5). The proposed link between known CBZ metabolites and QA also implicates 9-acridan, a product of DiOH-CBZ conversion by leukocytes (Breton et al., 2005). Again, whether this process in epileptics occurs in circulating or resident white blood cells remains unknown, but recent findings have shown limited presence of lymphocytes in the epileptic brain (Marchi et al., 2010b). Note that the radiolabelled carbon atom is shown as a red dot in parental and daughter drugs.
QA is an endogenous convulsive agent metabolized from tryptophan (Erhardt et al., 2009; Natsume et al., 2003) via the kynurenine pathway. Inflammatory conditions stimulate the breakdown of tryptophan (TRP) into L-kynurenine (Kyn), with the involvement of indoleamine 2,3-dioxygenase (IDO) thereby reducing the availability of TRP (Heyes et al., 1996; Raison et al., 2010). Subsequent reactions catalyzed by kynurenine 3-hydroxylase, kynureninase, and 3-hydroxyanthranilate-3,4-dioxygenase convert Kyn into a NMDA receptor agonist, QA, or may yield a NMDA receptor antagonist, KA (Heyes et al., 1996; Schwarcz et al., 1987). QA causes CNS damage by activating NMDA receptors (Stone and Perkins, 1981), partly by producing mitochondrial dysfunction (Bordelon et al., 1997) and partly by increasing free-radical generation (Behan et al., 1999). Additionally, a role for P450-mediated metabolic induction of KA and QA has been reported (DiNatale et al., 2010; Wu et al., 2011). P450 enzymes are found to be specifically upregulated in the BBB of multiple drug resistant epileptics (Ghosh et al., 2010). CBZ is transformed into 9-acridan by leukocytes (Breton et al., 2005). Leukocytes and macrophages also release QA (Heyes et al., 1996; Smith et al., 2001), and increasing evidence links activated leukocytes to epilepsy (Fabene et al., 2008; Marchi et al., 2009). Thus, while our results support an endothelial origin for QA in patients receiving CBZ, a contribution by leukocytes cannot be ruled out. In addition, whether or not CBZ metabolic conversion to QA implicates tryptophan or L-kynurenine is presently unknown (Fig. 6).
The concentrations measured in the human epileptic brain (∼10 μM) were in the range of neurotoxic and seizure promoting levels (During et al., 1989). Since QA levels in the brain are topographically overlapping with abnormal epileptiform activity, we propose a scenario where multiple drug resistance not only impedes CBZ access to the brain, but also leads to exacerbation of seizures by a powerful metabolic process. Our findings may thus reconcile two opposing views of pharmacoresistance, the “kinetic” and “dynamic” hypotheses, both mediated by the “drug resistant BBB phenotype”.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Accium BioSciences Inc. (Seattle, WA, USA) for generating AMS data. We are also indebted to Dr. Robert Schwarz (Professor of Psychiatry, University of Maryland, Baltimore, USA) who critically reviewed part of this work.
Funding: This work is supported by National Institute of Health — R01NS43284, R41MH093302 and R21HD057256 awarded to Dr. Damir Janigro and by Epilepsy Foundation Research Grant awarded to Dr. Nicola Marchi.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.nbd.2012.03.010.
Contributor Information
Nicola Marchi, Email: marchin@ccf.org.
Damir Janigro, Email: janigrd@ccf.org.
References
- Behan WMH, McDonald M, Darlington LG, Stone TW. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: protection by melatonin and deprenyl. Br J Pharmacol. 1999;128:1754–1760. doi: 10.1038/sj.bjp.0702940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordelon YM, Chesselet MF, Nelson D, Welsh F, Erecinska M. Energetic dysfunction in quinolinic acid-lesioned rat striatum. J Neurochem. 1997;69:1629–1639. doi: 10.1046/j.1471-4159.1997.69041629.x. [DOI] [PubMed] [Google Scholar]
- Breton H, Cociglio M, Bressolle F, Peyriere H, Blayac JP, Hillaire-Buys D. Liquid chromatography-electrospray mass spectrometry determination of carba-mazepine, oxcarbazepine and eight of their metabolites in human plasma. J Chro-matogr B. 2005;828:80–90. doi: 10.1016/j.jchromb.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Cucullo L, Hossain M, Rapp E, Manders T, Marchi N, Janigro D. Development of a humanized in vitro blood–brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia. 2007;48:505–516. doi: 10.1111/j.1528-1167.2006.00960.x. [DOI] [PubMed] [Google Scholar]
- Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, Janigro D. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. 2008;28:312–328. doi: 10.1038/sj.jcbfm.9600525. [DOI] [PubMed] [Google Scholar]
- Desai SY, Marroni M, Cucullo L, Krizanac-Bengez L, Mayberg MR, Hossain MT, Grant GG, Janigro D. Mechanisms of endothelial survival under shear stress. Endothelium (New York) 2002;9:89–102. doi: 10.1080/10623320212004. [DOI] [PubMed] [Google Scholar]
- DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ, Perdew GH. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski SM, Desai SY, Marroni M, Cucullo L, Goodrich K, Bingaman W, Mayberg MR, Bengez L, Janigro D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- During MJ, Freese A, Heyes MP, Swartz KJ, Markey SP, Roth RH, Martin JB. Neuroactive metabolites of L-tryptophan, serotonin and quinolinic acid, in striatal extracellular fluid — effect of tryptophan loading. FEBS Lett. 1989;247:438–444. doi: 10.1016/0014-5793(89)81387-0. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Olsson SK, Engberg G. Pharmacological manipulation of kynurenic acid potential in the treatment of psychiatric disorders. CNS Drugs. 2009;23:91–101. doi: 10.2165/00023210-200923020-00001. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Mora GN, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia LJ, Lowe JB, Mcever RP, Osculati F, Sbarbati A, Butcher EC, Constantin G. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedi M, Reutens DC, Andermann F, Okazawa H, Boling W, White C, Dubeau F, Nakai A, Gross DW, Andermann E, Diksic M. Alpha-[C-11]-methyl-L-tryptophan PET identifies the epileptogenic tuber and correlates with interictal spike frequency. Epilepsy Res. 2003;52:203–213. doi: 10.1016/s0920-1211(02)00216-4. [DOI] [PubMed] [Google Scholar]
- Fertig EJMRH. Carbamazepine. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Wolters-Kluwer; Philadelphia, PA: 2008. pp. 1543–1555. [Google Scholar]
- Ghosh C, Gonzalez-Martinez J, Hossain M, Cucullo L, Fazio V, Janigro D, Marchi N. Pattern of P450 expression at the human blood–brain barrier: roles of epileptic condition and laminar flow. Epilepsia. 2010;51:1408–1417. doi: 10.1111/j.1528-1167.2009.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Marchi N, Desai NK, Puvenna V, Hossain M, Gonzalez-Martinez J, Alexopoulos AV, Janigro D. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia. 2011a;52:562–571. doi: 10.1111/j.1528-1167.2010.02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh C, Puvenna V, Gonzalez-Martinez J, Janigro D, Marchi N. Blood–brain barrier P450 enzymes and multidrug transporters in drug resistance: a synergistic role in neurological diseases. Curr Drug Metab. 2011b;12:742–749. doi: 10.2174/138920011798357051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata T, Marchi N, Carlton E, Ghosh C, Gonzalez-Martinez J, Alexopoulos AV, Janigro D. Management of the patient with medically refractory epilepsy. Expert Rev Neurother. 2009;9:1791–1802. doi: 10.1586/ern.09.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GA, Abbott NJ, Janigro D. Understanding the physiology of the blood–brain barrier: in vitro models. News Physiol Sci. 1998;13:287–293. doi: 10.1152/physiologyonline.1998.13.6.287. [DOI] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain S, Khalid S, Nagaraju V, Rao RN. High-performance liquid-chromatographic separation and determination of small amounts of process impurities of ciprofloxacin in bulk drugs and formulations. J Chromatogr A. 1995;705:380–384. [Google Scholar]
- Ju C, Uetrecht JP. Detection of 2-hydroxyiminostilbene in the urine of patients taking carbamazepine and its oxidation to a reactive iminoquinone intermediate. J Pharmacol Exp Ther. 1999;288:51–56. [PubMed] [Google Scholar]
- Kocki T, Kocki J, Wielosz M, Turski WA, Urbanska EM. Carbamazepine enhances brain production of kynurenic acid in vitro. Eur J Pharmacol. 2004;498:325–326. doi: 10.1016/j.ejphar.2004.07.088. [DOI] [PubMed] [Google Scholar]
- Kwan P, Schachter SC, Brodie MJ. Current concepts drug-resistant epilepsy. N Engl J Med. 2011;365:919–926. doi: 10.1056/NEJMra1004418. [DOI] [PubMed] [Google Scholar]
- Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. 2005;6:591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- Marchi N, Fan QY, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Gonzalez-Martinez J, Nguyen MT, Granata T, Janigro D. Transporters in drug-refractory epilepsy: clinical significance. Clin Pharmacol Ther. 2010a;87:13–15. doi: 10.1038/clpt.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Teng QS, Ghosh C, Fan QY, Nguyen MT, Desai NK, Bawa H, Rasmussen P, Masaryk TK, Janigro D. Blood–brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010b;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume J, Kumakura Y, Bernasconi N, Soucy JP, Nakai A, Rosa P, Fedi M, Dubeau F, Andermann F, Lisbona R, Bernasconi A, Diksic M. Alpha-[C-11] methyl-L-tryptophan and glucose metabolism in patients with temporal lobe epilepsy. Neurology. 2003;60:756–761. doi: 10.1212/01.wnl.0000052682.99812.f5. [DOI] [PubMed] [Google Scholar]
- Potschka H, Fedrowitz M, Loscher W. P-glycoprotein and multidrug resistance-associated protein are involved in the regulation of extracellular levels of the major antiepileptic drug carbamazepine in the brain. Neuroreport. 2001;12:3557–3560. doi: 10.1097/00001756-200111160-00037. [DOI] [PubMed] [Google Scholar]
- Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, Spivey JR, Saito K, Miller AH. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15:393–403. doi: 10.1038/mp.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambeck B, Jurgens UH, May TW, Pannek HW, Behne F, Ebner A, Gorji A, Straub H, Speckmann EJ, Pohlmann-Eden B, Loscher W. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia. 2006;47:681–694. doi: 10.1111/j.1528-1167.2006.00504.x. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L. Side by side comparison between dynamic versus static models of blood–brain barrier in vitro: a permeability study. Brain Res. 2006;1109:1–13. doi: 10.1016/j.brainres.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Speciale C, French ED. Hippocampal kynurenines as etiological factors in seizure disorders. Pol J Pharmacol Pharm. 1987;39:485–494. [PubMed] [Google Scholar]
- Smith DG, Guillemin GJ, Pemberton L, Kerr S, Nath A, Smythe GA, Brew BJ. Quinolinic acid is produced by macrophages stimulated by platelet activating factor, Nef and Tat. J Neurovirol. 2001;7:56–60. doi: 10.1080/135502801300069692. [DOI] [PubMed] [Google Scholar]
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- Stone TW, Perkins MN. Quinolinic acid — a potent endogenous excitant at amino-acid receptors in CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- Tompkins EM, Farmer PB, Lamb JH, Jukes R, Dingley K, Ubick E, Turteltaub KW, Martin EA, Brown K. A novel C-14-postlabeling assay using accelerator mass spectrometry for the detection of O-6-methyldeoxy-guanosine adducts. Rapid Commun Mass Spectrom. 2006;20:883–891. doi: 10.1002/rcm.2370. [DOI] [PubMed] [Google Scholar]
- Wu Q, Zhang YH, Zhao X, Shi WL, Pu XP. Proteome studies on liver tissue in a phenobarbital-induced rat model. Eur J Pharmacol. 2011;670:333–340. doi: 10.1016/j.ejphar.2011.09.161. [DOI] [PubMed] [Google Scholar]
- Zhang XQ, He Y, Ding M. Simultaneous determination of tryptophan and kynurenine in plasma samples of children patients with Kawasaki disease by high-performance liquid chromatography with programmed wavelength ultraviolet detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1678–1682. doi: 10.1016/j.jchromb.2009.04.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.