

Abstract
Tropisetron was identified in a screen for candidates that increase the ratio of the trophic, neurite-extending peptide sAPPα to the anti-trophic, neurite-retractive peptide Aβ, thus reversing this imbalance in Alzheimer’s disease (AD). We describe a hierarchical screening approach to identify such drug candidates, moving from cell lines to hippocampal neuronal cultures to in vivo studies. By screening a clinical compound library in the primary assay using CHO-7W cells stably transfected with human APPwt, we identified tropisetron as a candidate that consistently increased sAPPα. Secondary assay testing in neuronal cultures from J20 (PDAPP, huAPPSwe/Ind) mice showed that tropisetron consistently increased the sAPPα/Aβ 1-42 ratio. In in vivo studies in J20 mice, tropisetron improved the sAPPα/Aβ ratio along with spatial and working memory in mice, and was effective both during the symptomatic, pre-plaque phase (5-6 months) and in the late plaque phase (14 months). This ameliorative effect occurred at a dose of 0.5 mg/kg/d (mkd), translating to a human-equivalent dose of 5 mg/day, the current dose for treatment of postoperative nausea and vomiting (PONV). Although tropisetron is a 5-HT3 antagonist and an α7nAChR partial agonist, we found that it also binds to the ectodomain of APP. Direct comparison of tropisetron to the current AD therapeutics memantine (Namenda) and donepezil (Aricept), using similar doses for each, revealed that tropisetron induced greater improvements in memory and sAPPα/Aβ1-42. The improvements observed with tropisetron in the J20 AD mouse model, and its known safety profile, suggest that it may be suitable for transition to human trials as a candidate therapeutic for mild cognitive impairment (MCI) and AD, and therefore it has been approved for testing in clinical trials to begin in 2014.
Keywords: Multi-functional drug, Improvement in sAPPalpha/Abeta42 biomarker ratio, Memory, Improvement, Orally effective, Alzheimer’s disease, Candidate therapeutic
1. Introduction
In the absence of development of new therapeutics, the number of cases of Alzheimer’s disease (AD) – estimated to be 5.4 million currently – will rapidly increase in the coming decades. Current treatments provide, at best, only modest and temporary symptomatic relief, without altering the underlying mechanisms that led to the onset, and mediate the progression of the disease. Alzheimer’s is characterized by two pathologies: amyloid-β (Aβ) plaques and tau neurofibrillary tangles. Aβ accumulation is the initiating factor that leads to AD, while tangles play a critical role in neuronal death and AD progression (Holtzman et al., 2011; Klunk et al., 2006). Early in disease pathogenesis, Aβ monomers aggregate into soluble Aβ oligomers and insoluble Aβ plaques, both of which can alter synaptic transmission and destroy neurons under certain conditions (Cheng et al., 2007). AD pathogenesis begins 10-15 years before the first memory and cognitive deficits of AD are apparent (Mintun et al., 2006; Morris and Price, 2001). By the time of diagnosis, substantial pathology is present and cellular damage has already occurred. Consequently, it is likely that anti-AD therapy will require initiation prior to symptom onset in order to prevent or delay the disease, which means it may have to be administered for a decade or more prior to onset. This chronic, pre-symptomatic treatment will make drug safety imperative. While it is possible that currently proposed anti-Aβ interventions, such as Aβ immunization or use of BACE or γ-secretase inhibitors, may ultimately be successful, in clinical trials thus far these approaches have shown substantial side effects and toxicities, and have not ameliorated disease symptoms (Holmes et al., 2008; Orgogozo et al., 2003; Serrano-Pozo et al., 2010). Disappointing clinical trial results with promising preclinical candidates such as Flurizan, Semagacestat, and Dimebon increase the critical need for new approaches to identify and develop AD therapeutics.
Here we utilized a straightforward approach to identify novel AD therapeutics based on their ability to increase the sAPPα to Aβ ratio in in vitro and in vivo screens. APP may be cleaved to produce four peptides—sAPPβ, Aβ, Jcasp, and C31—that mediate neurite retraction, synaptic loss, caspase activation, and ultimately programmed cell death; or, alternatively, to produce two peptides—sAPPα, and αCTF—that inhibit neurite retraction, caspase activation, and cell death, and mediate neurite extension (Bredesen, 2009) (Fig. 1). Thus the ratio of these peptide APP derivatives may be a physiological determinant of plasticity, and drugs that alter this ratio represent candidate AD therapeutics. In support of this notion, maintaining adequate levels of sAPPα has been shown to be vitally important to memory, supporting maintenance of synaptic connections (Fig. 1B) (Claasen et al., 2009). We therefore hypothesized that molecules that effect an increase in the sAPPα/Aβ1-42 ratio may be beneficial in ameliorating the AD phenotype.
Figure 1. Mediation of alternative plasticity states by APP.
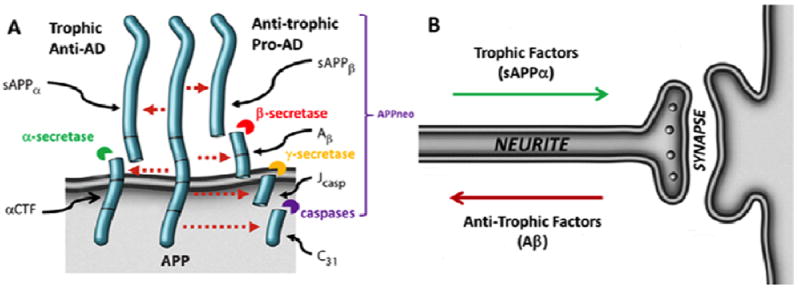
APP undergoes cleavage via two different pathways (A). The trophic pathway includes α-secretase cleavage, producing sAPPα and αCTF, which mediate neurite outgrowth, caspase inhibition, and synaptic (Syn) maintenance. Alternatively, cleavage by BACE, γ-secretase, and caspase produces sAPPβ, Aβ, Jcasp, and C31, mediating neurite (Nt) retraction, synaptic loss, caspase activation, and potentially cell death (B) (Descamps et al JAD 2011;25:51-57).
Such agents would not necessarily target secretases involved in the proteolytic processing of APP to Aβ or sAPPα directly, but could be pleiotropic in their mechanisms of action. To identify such compounds we screened a variety of small molecule libraries, including a clinical compound library comprised of molecules already FDA-approved for other indications. For our screen, we used a single readout to identify a ‘hit’: an increase in sAPPα over control. The ‘hits’ then went through secondary assay screening using primary hippocampal neurons from transgenic (Tg) J20 mice (APPSwe, Ind) mouse embryos. In the primary cultures, both sAPPα and Aβ1–42 were measured, in order to identify candidates that increased the sAPPα/Aβ ratio. Compounds that increased the ratio in primary cultures went into pharmacokinetic analysis for determination of brain uptake, and this was followed by pharmacodynamic analysis of selected candidates in mouse models of AD. Tropisetron ((1R, 5S)-8-methyl-8-azabicyclo [3.2.1] octan-3-yl 1methyl-indole-3-carboxylate) was a consistently successful candidate compound in this screen. It underwent initial pilot testing for effects on sAPPα and Aβ in the J20 mouse model. As J20 mice express human APP with two mutations - Swedish (K670N, M671L) and Indiana (V717F) - we also wished to determine whether similar biochemical alterations in sAPPα and Aβ could be seen upon treatment in a model expressing wildtype human APP (the I5 model), since the vast majority of human patients diagnosed with AD will have the wildtype sequence. (In this model tropisetron did not decrease the already very low Aβ levels, but did increase sAPPα.) Subsequent to this, further long-term testing in J20 mice was performed, using mice between 4.5-6 months of age, the time during which Aβ production is amplified and behavioral abnormalities are present, but prior to overt plaque formation. De novo Aβ production is more readily discernible from pre-existing plaque-bound Aβ at this stage. The Aβ pathology is in some aspects akin to the MCI stage of the disease in humans at this point, although it should be noted that many other AD-like symptoms (e.g., cognitive impairment) and pathological hallmarks (e.g., increased tau phosphorylation, reduction in calbindin, cFos, and synapse load) precede plaque formation in these mice (Hsia et al., 1999; Mucke et al., 2000; Palop et al., 2003). In these mice, short-term working memory and spatial memory improvements were determined using the Novel Object Recognition (NOR) and the Morris Water Maze (MWM) tests, respectively. The former became our testing paradigm of choice as it was found to be a more versatile, rapid, and useful method for pharmacological development, as has been noted previously (Alkam et al., 2011; Dere et al., 2007; Zhang et al., 2012).
To compare the efficacy of tropisetron to existing therapeutics for AD directly, additional testing was done in head-to-head comparisons with the NMDA receptor antagonist memantine (Namenda) and the acetylcholinesterase inhibitor donepezil (Aricept). Tropisetron, memantine, and donepezil all improved the sAPPα/Aβ1-42 ratio at similar doses, but only tropisetron improved working memory. These were subchronic studies using relatively low doses, and only served to show that under the conditions of these experiments, tropisetron exerted superior effects on memory.
Tropisetron is a dual-receptor binding molecule, and our studies herein (Fig. 2) showed binding to the 5-HT3 receptor (Ki ~ 3nM) and α7-nicotinic acetylcholine receptor (α7nAChR) (Ki ~ 470nM). Tropisetron increased the trophic APP peptide ratio upon treatment in our AD mouse model at 0.5 mpk per day. In functional studies, tropisetron has been shown to be a 5-HT3 antagonist and a partial α7-nicotinic receptor agonist, with low affinity for other nicotinic receptor subtypes (Macor et al., 2001). In addition to these interactions, we determined that tropisetron binds to the ectodomain of APP (Kd ~ 0.9uM, Fig. 3).
Figure 2. Molecular structure of tropisetron and receptor binding to 5-HT3 and α7nACh receptors.
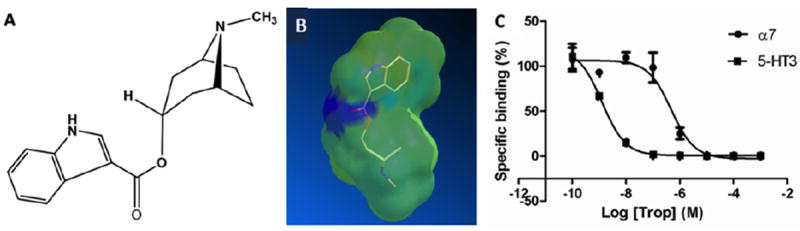
The molecular structure of tropisetron is shown in A and a molecular model is shown in B. The competitive binding curves for tropisetron at both the 5-HT3 and α7nACh receptors are shown in C.
Figure 3. Tropisetron binds directly to APP.
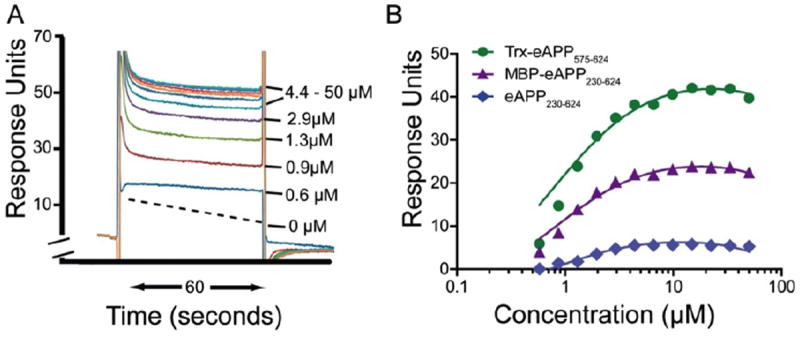
Surface plasmon resonance data was obtained as described in Methods. The sensograms obtained by flowing varying concentrations of tropisetron (0-50 μM) through the Trx-eAPP575-624 flow cell and before subtraction of the no-drug control is shown in A. The binding isotherms for tropisetron to various fragments of the ectodomain of APP695 are shown in B. The binding isotherms for tropisetron were fitted using a single binding site model. All three curves were constrained to share the same equilibrium constant with PRISM (GraphPad Inc). The resulting binding equilibrium constant was calculated to be 0.9μM ± 0.1μM with R2=0.99.
Both α7nAChRs and the 5-HT3 serotonin receptors belong to the Cys-loop ligand-gated ion channel receptor superfamily; other members of this receptor family include GABAA, GABAC, and glycine receptors. The α7nAChRs and 5-HT3 receptors are highly expressed in the frontal cortex and hippocampus, and are believed to play important roles in cognition and schizophrenia (Barnes and Sharp, 1999). Agonists of α7nAChRs have been shown to exert neuroprotective and neurotrophic actions, while Aβ binding has been reported to inhibit the activity of these receptors (Nagele et al., 2002). The 5-HT3 receptor is unique within the serotonin family of receptors, since all other 5-HT receptors are serpentine G-protein coupled receptors. Our in vivo screening revealed that a potent 5-HT3 antagonist, such as tropisetron, that is also an α7nAChR partial agonist, can reverse the AD phenotype in the mouse model, suggesting a therapeutic role for both of these receptors in the disease. Nicotinic activity is not general for this class of 5-HT3 receptor antagonists, as two other high-affinity 5-HT3 receptor antagonists - ondansetron and dolasetron - have no α7nAChR agonist activity (Toyohara and Hashimoto, 2010). Furthermore, as described below, we also discovered that tropisetron interacts directly with APP, with a sub-micromolar affinity. Thus, tropisetron interacts with three receptor targets that potentially modulate the AD phenotype, and this multi-functional mechanism makes it a unique candidate to develop as a potential therapeutic for AD and MCI (mild cognitive impairment).
Tropisetron is currently used primarily in the treatment of patients with chemotherapy-induced or post-operative nausea and vomiting (PONV) (Morrow et al., 1995). Treatment generally lasts for one week. Tropisetron has an excellent safety profile, is almost completely absorbed from the gastrointestinal tract after oral dosing, and is highly brain permeable. After the discovery of tropisetron in our in vitro screens, we hypothesized that treatment of our AD model mice with a human equivalent dose of tropisetron would be sufficient to induce both an improvement in memory and normalization of the aberrant APP processing resulting from the Swedish and Indiana mutations. The results of our studies presented here support our hypothesis, and suggest that tropisetron is an excellent candidate for clinical testing in AD and MCI. The data also suggest that other molecules with similar multifunctional properties (5-HT3R antagonist, α7nAChR agonist, and APP interactor) could be novel candidates as AD therapeutics.
2. Results
2.1 Tropisetron
Tropisetron is a tropinol ester: (1R, 5S)-8-methyl-8-azabicyclo [3.2.1] octan-3-yl 1methyl-indole-3-carboxylate hydrochloride (C17H20N2O2) with a molecular weight of 320.82 g/mol (as the HCl salt form). The chemical structure is shown in Fig. 2A and a molecular model in Fig. 2B.
2.2 Tropisetron receptor binding studies
Tropisetron displaced binding of the α7nAChR antagonist [125I]-α-bungarotoxin (α-bgt), with a Ki value of 470 nM (n = 3) in SH-SY5Y cells. Our studies also demonstrated selectivity for α7nAChRs, as it binds α4nACh receptors with a Ki ~ 20uM (n=3) and the muscle-type nicotinic (α1β1γδ nACh) receptor Ki ~ 15uM (n=3) similar to that reported previously (Macor et al., 2001). In a binding assay using human recombinant 5-HT3R expressed in CHO cells, tropisetron was shown to displace [3H]- BRL-43694, a 5-HT3R selective antagonist. In this study, tropisetron showed high affinity for 5-HT3Rs with a Ki value of 3 nM (n=3) (Fig. 2C).
2.3 Tropisetron binding to APP
Surface plasmon resonance (SPR) showed that tropisetron bound to APP. Fig. 3A shows example sensograms obtained with Trx-eAPP575-624 before subtraction of the 0μM sensogram. Fig. 3B compares the response for all three eAPP fragments. The different maximal responses reflect the varying amounts of protein cross-linked to the CM5 chip. TRX-eAPP575-624 was the densest at 11,700 RU, while eAPP230-624 was the least dense. Comparison of the curves obtained for the three fragments was performed with PRISM (GraphPad Inc., www.graphpad.com). The curves were fitted to a single-site saturation binding model in which the background and the non-significant binding contributions were constrained to be the same for all three proteins. The calculated KD was not significantly different between the proteins, suggesting that the binding site of ADDN-FO3 is most likely to be between residues 575-624 of the ectodomain of APP. Constraining the KD to be the same for all three proteins resulted in a value of 0.9μM ± 0.1 μM with R2=0.99.
2.4 Primary high-throughput screening
We used an AlphaLISA-based HTS formatable primary screening assay where we detected increases in sAPPα induced by a hit candidate in the cell media. The AlphaLISA-based primary assay works well with either CHO-7W cells or B103 APP cells stably transfected with wildtype human APP (APP 770), as well as SH-SY5Y neuroblastoma cells that express APP endogenously. For the assay that led to the identification of tropisetron, we used CHO-7W cells. As described in Methods, media from cells treated for 24 hours with compounds from an FDA-approved clinical compound library were assayed for sAPPα using a custom AlphaLISA assay and any compound that caused at least a 20% increase in sAPPα was considered a hit. All compounds were tested in triplicate with a reproducibility z-value ~ 0.6. The screen had a good signal over background (s/b) ratio of ~3.0. This screen yielded several initial ‘hits,’ with a hit-rate ~ 0.5%. Tropisetron increased sAPPα in media from 7W cells by ~ 30%, and was therefore tested in primary neuronal culture.
2.5 Secondary screening in primary hippocampal neuronal culture
As part of our iterative screening approach, hits identified in the primary assay were next evaluated in the secondary assay using primary neuronal assay. In the J20 model of AD, the hippocampus (Hip) and entorhinal cortex (ECx) show the most obvious pathological changes, and therefore hippocampal neurons were used for this secondary screen. As described in Methods, in neurons treated with tropisetron for 5 days, sAPPα was significantly increased (Fig. 4A) and Aβ1-42 (Fig. 4B) was significantly decreased.
Figure 4. Tropisetron increases sAPPα and decreases Aβ1-42 in primary hippocampal neuronal culture.
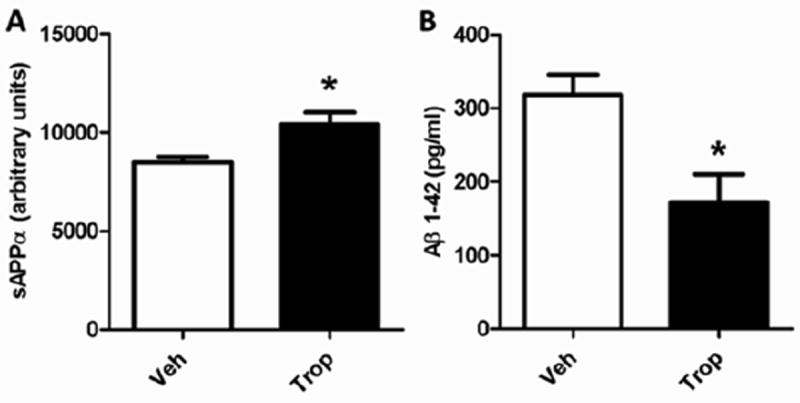
In primary hippocampal neuronal cultures, tropisetron at 1uM significantly (P = 0.0473) increased sAPPα (A) and significantly (P = 0.0486) decreased Aβ1-42 (B). Both t-test, two-tailed, unpaired statistical analyses.
2.6 Tropisetron increases sAPPα in the presence of ApoE ε4
Humans carrying one or more copies of the ApoE ε4 allele of apolipoprotein E are at higher risk for AD (Strittmatter and Roses, 1996). Co-transfection of huAPPwt and ApoE ε4 has been shown to significantly decrease sAPPα secretion, and reduce the sAPPα/sAPPβ or sAPPα/Aβ–1-40 ratio in several cell lines, and tropisetron – identified as “F03” - has been shown to reverse these reductions (Theendakara, 2013). Here we also show that tropisetron at 1μM significantly increased cell survival of both ApoE ε3- and ApoE ε4-transfected cells (Fig. 5).
Figure 5. Tropisetron increases cell survival after ApoE ε4 transfection.
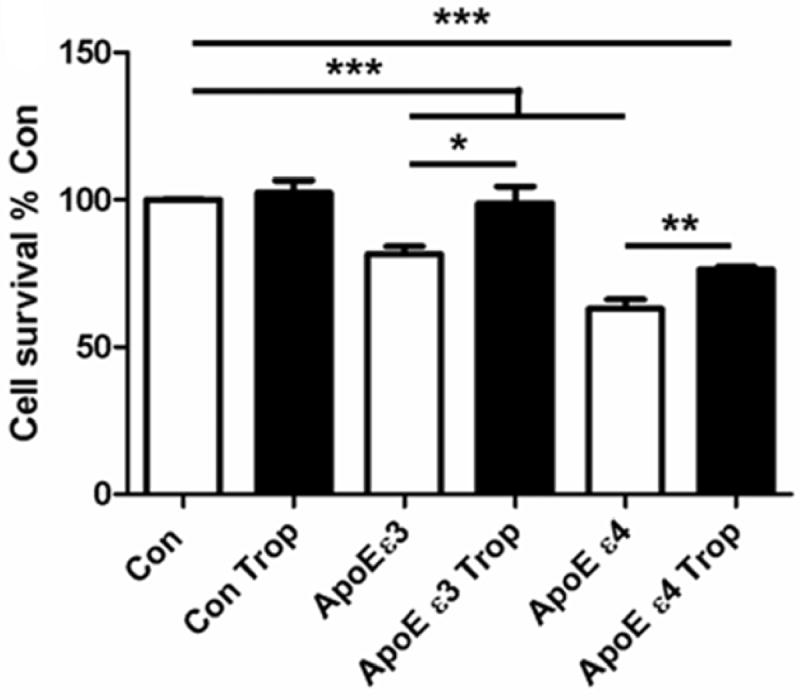
Transfection with either ApoE ε3 or ε4 decreased cell survival (P = 0.0001). Tropisetron treatment increased survival after transfection with either ApoE ε3 (P = 0.0207) or ε4 (P = 0.003); it did not change survival of untransfected cells. All t-test, two-tailed, unpaired statistical analyses.
2.7 Tropisetron exhibits excellent brain penetration
To determine the blood-brain barrier penetration of tropisetron, a pharmacokinetic (PK) study was performed as described in Methods. Tropisetron appeared in brain tissue at high levels at the first time point measured (1 hour), the drug concentration maximum (Cmax) was seen at 2 hours, dissipating by 6 hours post-injection. The brain/plasma ratio at Cmax was ~2.5. In contrast, MEM-3454 (M3454, RO5313534) a quinuclidinyl indazole that is currently in Phase II clinical trials that was also tested in our brain uptake studies, showed a brain/plasma ratio of only 0.1, which is consistent with previously reported brain permeablity values (Wallace et al., 2011) (Fig. 6).
Figure 6. Tropisetron exhibits superior brain penetration to MEM-3454.
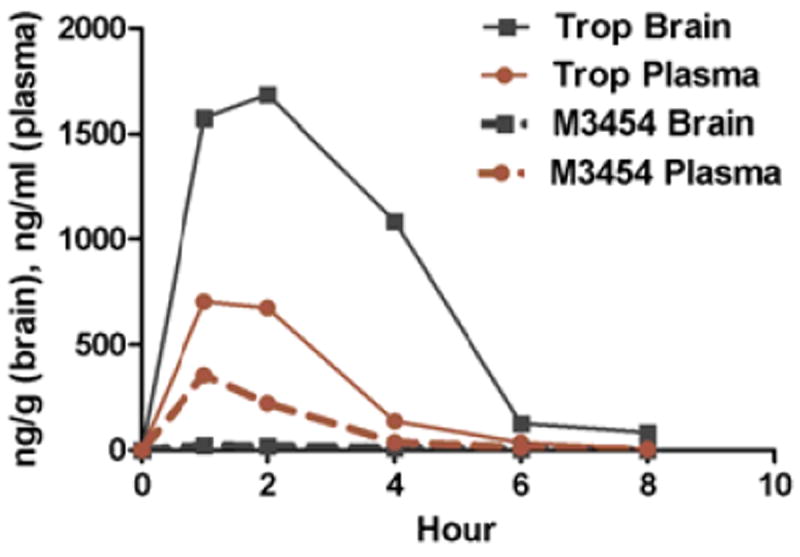
Tropisetron injected subcutaneously into adult mice at 10 mg/kg appeared in brain tissue at high levels at the first time point measured (1 hour), and dissipated by 6 hours post-injection. The brain/plasma level was 2.5 at the peak. A compound currently in clinical trials - MEM-3454 (M3454) - that also binds α7nAChR (but not APP) shows much lower brain penetration and a lower brain/plasma ratio when given at the same dose and route.
2.8 Meta analysis reveals significant increases in the sAPPα/Aβ1-42 ratio
Tropisetron effects were ascertained in ten in vivo studies, including several pilot studies (see Suppl. Methods). Meta analysis is a powerful way to determine the effect of sample size rather than an exact effect seen in smaller groups under a single set of conditions. Meta analysis of sAPPα and Aβ1-42 levels in hippocampi and entorhinal cortices of individual mice was performed by conversion of all data in individual studies to percentage of control for those studies. Analysis of all such data combined revealed tropisetron’s consistent ability to increase sAPPα and decrease Aβ1-42, even in short studies and at a variety of doses. These sAPPα increases (Fig. 7A) were highly significant (P = 0.0003), as were the Aβ1-42 decreases (0.0099; Fig. 7B); therefore the increase in the sAPPα/Aβ1-42 ratio was highly (p = 0.0001) significant (Fig. 7C). The J20 mice used in the studies contributing to meta analysis were all between 4.5 and 6 months of age, when brain Aβ concentration increases exponentially, resulting in plaque formation in almost all J20 mice by 7 months of age. The mouse-to-mouse variability in Aβ is high during this period, but tropisetron nonetheless effected a highly statistically significant reduction.
Figure 7. Meta-analysis of tropisetron effects on sAPPα and Aβ1–42 in hippocampus/entorhinal cortex.
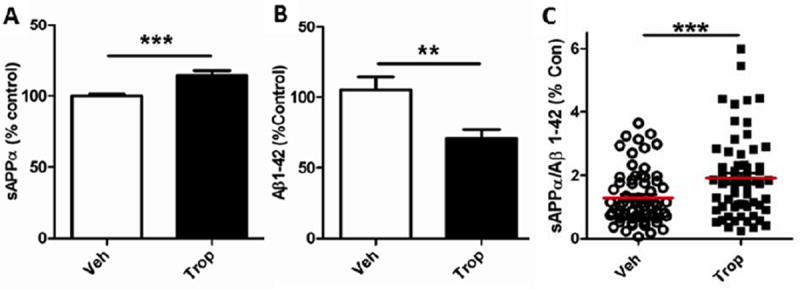
(A) When sAPPα values for individuals from eight separate in vivo experiments (vehicle n = 68, tropisetron n = 67) are converted to percentage of vehicle control for those experiments, the increase in sAPPα as a result of tropisetron treatment is very highly significant (P = 0.0001). (B) Decreases in Aβ1-42 are also highly significant (P = 0.0021). The sAPPα/Aβ1-42 is also highly significant (0.0008) (Fig. 7C). All t-test, two-tailed, unpaired statistical analyses.
2.9 Tropisetron improves performance in the Morris Water Maze (MWM)
Alzet pumps containing either tropisetron to deliver 0.5 mg/kg/day or vehicle only were implanted subcutaneously into 16-19 week-old J20 mice as described in Methods. During the last 2 weeks of the treatment period, mice performed the Morris Water Maze spatial memory task. Tropisetron-treated J20 MWM performance was intermediate between that of the NTg mice and vehicle-treated J20 mice for almost all parameters measured. In training, J20 mice treated with tropisetron found the hidden platform more readily than the vehicle-treated J20 mice (Fig. 8A), but not as quickly as NTg mice. In the hidden probe trials, tropisetron-treated J20s were intermediate again for time spent in the platform quadrant (Fig. 8B), crosses (Fig. 8C), and latency (Fig. 8D). J20 mice can manifest anxiety and helplessness in the MWM, as reflected by floating instead of active swimming. Tropisetron-treated J20s floated far less than vehicle-treated J20s (Fig. 8E). In the reversal, the platform is moved and the mice re-trained. Tropisetron-treated J20s were intermediate again for latency on the last day of training (Fig. 9A), time spent in platform quadrant in the probe (Fig. 9B), and crosses (Fig.9C), but notably, performed significantly better in the key parameter of reversal probe latency (Fig. 9D).
Figure 8. Tropisetron-treated mice show improvements in spatial memory.
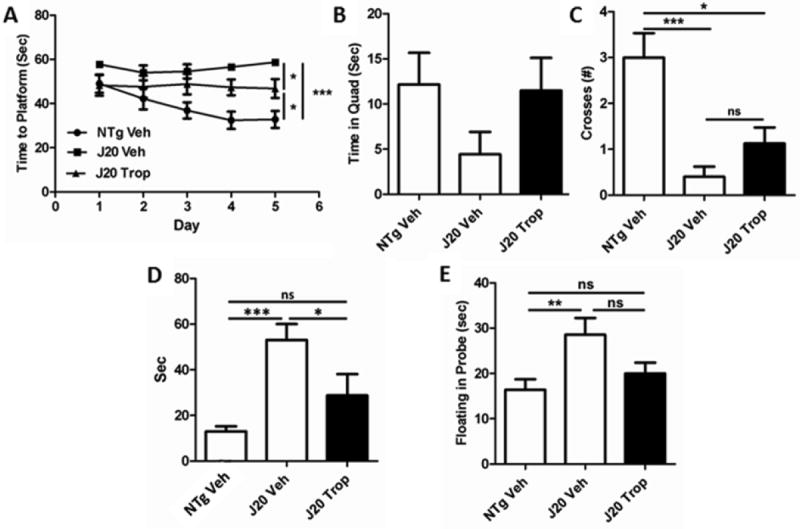
Mice were treated for 8 weeks as described in Methods. In the last 2 weeks of treatment, mice performed the Morris Water Maze (MWM) spatial memory task. (A) In training, J20 mice treated with tropisetron found the hidden platform significantly faster than vehicle treated J20s by Day 5 of training (P = 0.0113), but did not become significantly faster with days of training. In the hidden probe, where the platform was removed and attempts of the mice to find it were recorded, while tropisetron-treated J20s spent more time in the platform quadrant (B) than vehicle-treated J20s, and crossed the platform site (C) more frequently, these increases were not significant; and while tropisetron-treated J20s crossed the platform significantly fewer times than NTg (P = 0.0196), vehicle-treated mice crossed far less (P = 0.0005). (D) Tropisetron-treated J20s’ latency to the platform site was significantly improved over vehicle-treated J20 (P < 0.05) and not significantly different than NTg; vehicle-treated J20s were much slower (P = 0.0002) than NTgs (ANOVA, Bonferroni post-hoc). (E) All mice floated rather than initially swimming actively at first, with vehicle-treated J20s floating the most, significantly more than the NTgs (P = 0.0088); tropisetron-treated mice were intermediate.
Figure 9. Tropisetron-treated mice perform well in the MWM reversal.
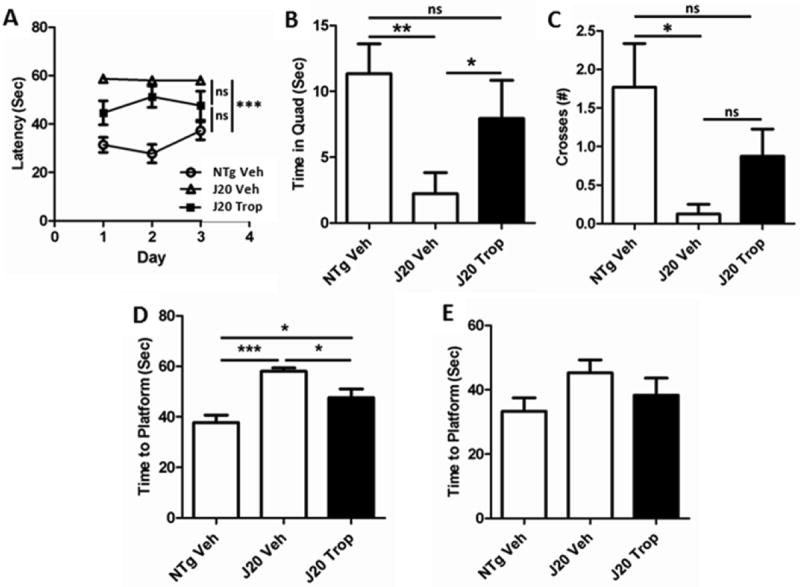
In the reversal, the platform is moved and the mice re-trained. (A) On the last day of training, vehicle-treated J20s took significantly longer to find the platform than NTg mice (P = 0.0004), tropisetron-treated J20s were neither significantly faster than vehicle-treated J20s nor significantly slower than NTgs. In the reversal probe, neither time spent in the platform quadrant (B) nor number of crosses of the platform site (C) by tropisetron-treated J20s was significantly less than that of NTg mice, but they spent significantly more time in the target quadrant (P < 0.05, ANOVA, Bonferroni post-hoc) and had a significantly shorter latency (D) than vehicle-treated J20s (P = 0.0061), although they were not as rapid as NTg mice (P = 0.034). (E) When the platform was visually cued, there was no significant difference in the time it took for mice to reach it, although vehicle-treated J20s floated more and therefore took slightly longer to reach the platform. All t-test, two-tailed, unpaired statistical analyses unless otherwise indicated.
2.10 sAPPα and Aβ also improved after 8-week tropisetron treatment
In hippocampi/entorhinal cortices, sAPPα was significantly higher in mice treated with tropisetron (Fig. 10A), and both Aβ1-40 and 1-42 were lower (Fig. 10B, C).
Figure 10. sAPPα and Abeta improved after eight-week tropisetron treatment.
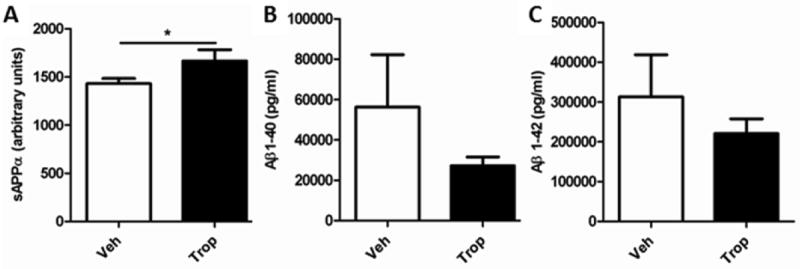
(A) sAPPα was significantly (P = 0.0425, t-test, one-tailed, unpaired) higher, and (B) Aβ1-40 and (C) 1-42 lower, but not significantly so in hippocampi/entorhinal cortices.
2.11 Tropisetron treatment improves Aβ similarly to memantine, but results in greater cognitive improvement
The starting human dose used for memantine in AD is 5 mgs per day with a maximum dose of 20 mgs per day, while the dose used for tropisetron in PONV is 5 mg per day (Garbe et al., 1994). In our pharmacokinetic studies, memantine brain levels (comparing Cmax values) were more than 5-fold higher (Fig. 11A) than tropisetron, and this was seen again after four-week treatment at the human equivalent doses of 0.4 mkd and 0.5 mkd for memantine and tropisetron, respectively (Fig. 11B). The Novel Object Recognition (NOR) task paradigm was used to quantify working object memory in this and further studies as it is a more facile method to discern differences as a result of pharmacological treatment of J20 mice, and furthermore allows determination of effects on the hyperactivity, which is part of the J20 phenotype. Neither compound significantly reduced hyperactivity in this study, although there was a trend toward lower activity in treated mice for both compounds (Fig. 12A). Only tropisetron improved working object short-term memory (Fig. 12B). sAPPα was unchanged with either compound (Fig. 13A), but both Aβ1-40 and 1-42 were lowered similarly (Fig. 13B, C). This indicates that, while both compounds have very similar effects on APP-generated cleavage products at equivalent doses, tropisetron confers an additional benefit of improvement in cognitive performance, possibly due to its impact on the cholinergic system. There may be a number of reasons for lack of improvement in cognition by memantine in our mouse model including less than optimal brain levels achieved at the dose we used. The final brain levels of memantine were ~ 0.16 uM at the 2h time point - which is below the range for optimal binding to the NMDA receptor and producing partial antagonism (Ki ~0.3 -1uM) (Parsons et al., 1999). In addition, memantine is known to antagonize α7nAChRs (Ki ~ 1uM; (Parsons et al., 1999), and may initially impair cognitive performance (Aracava et al., 2005). While further studies using a variety of dosages and duration of treatment could be run to further elucidate any differences between these compounds, this initial study indicates that, at least under the conditions used here, tropisetron may be more effective in improving cognition.
Figure 11. Memantine is more brain-penetrant than tropisetron.
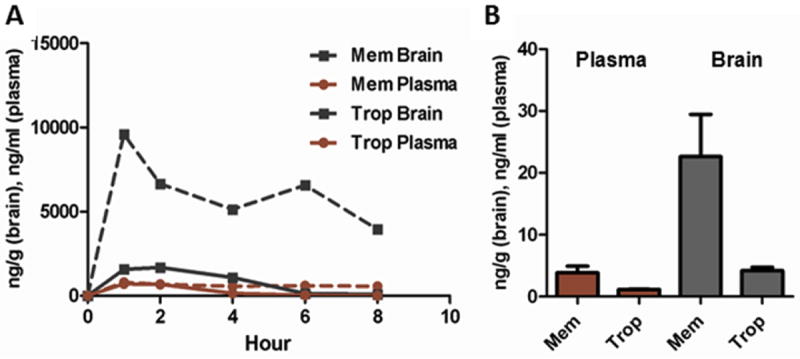
(A) After a single dose at 10mkd, memantine brain levels were more than 5-fold higher than those of tropisetron. (B) Plasma levels were higher for memantine in the comparative study and brain levels were again approximately 5-fold higher.
Figure 12. Tropisetron, but not memantine, improved working memory.
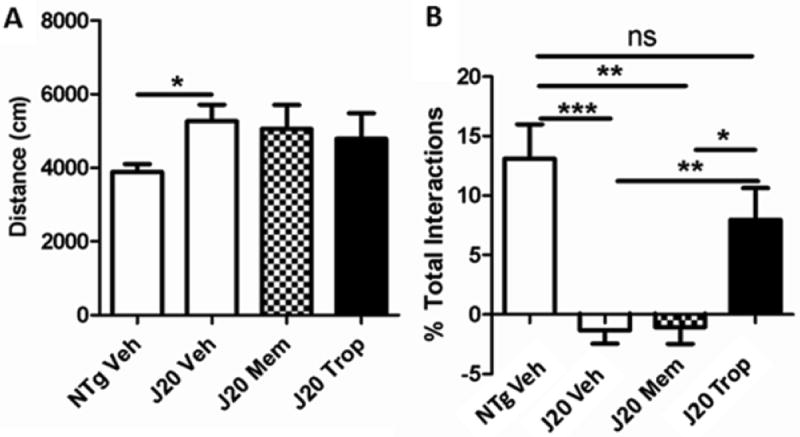
(A) No treatment significantly lowered hyperactivity in this study, but there was a trend for both memantine and tropisetron to do so. (B) Tropisetron treatment significantly improved novelty preference (NP) as compared to vehicle-treated J20 controls (P = 0.012) and memantine-treated J20s (P = 0.0297); and this NP was not significantly different from NTg controls. Memantine did not improve NP. All t-test, two-tailed, unpaired statistical analyses.
Figure 13. Tropisetron and memantine have similar effects on Aβ in hippocampus/entorhinal cortex.
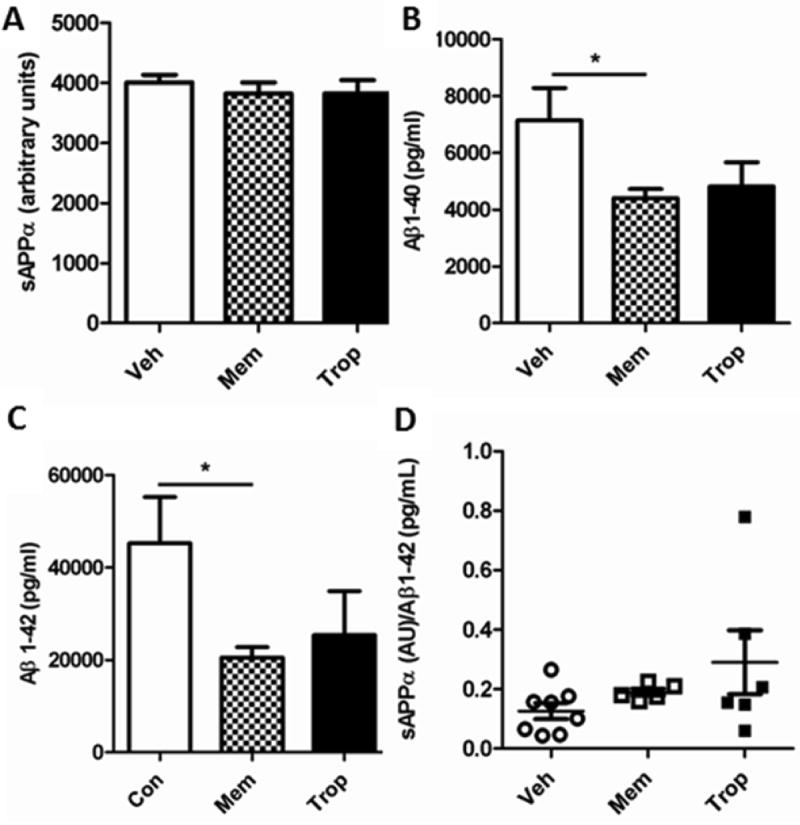
There was no increase in sAPPα as a result of treatment with either compound. Both Aβ1-40 (A) and Aβ1-42 (B) were lowered similarly by each compound, significantly so for memantine (P = 0.0475, P = 0.0415, respectively). All t-test, one-tailed, unpaired analyses. (C) Ratios for memantine and tropisetron. (D-E) Both plasma and brain levels of memantine were higher than tropisetron, as expected.
2.12 Tropisetron improves memory and lowers Aβ1-42 more effectively than donepezil at human equivalent doses
The human dose for donezepil in AD is 5-10 mg per day (Doody et al., 2008), while for tropisetron for PONV, it is 5 mg per day (Garbe et al., 1994). After 21 days of treatment at a human equivalent dose of 0.5 mkd, tropisetron improved short-term memory (Fig. 14B), decreased Aβ1-42 (Fig. 15C), and increased sAPPα (Fig. 15A), all significantly; therefore the sAPPα/Aβ1-42 ratio was markedly improved (Fig. 15D). At the human equivalent dose of 1 mkd for the same length of time, donepezil neither improved short-term object memory nor significantly lowered Aβ1-42, but did increase sAPPα significantly, and therefore the ratio. The brain levels of donepezil at the end of the study were ~70 ng/g or 0.2 uM and therefore in the range of the IC50 (G1 form= 340nM and G4 form = 200nM) for inhibition of AChE (Rakonczay, 2003). Brain levels of tropisetron were 0.34 uM (80 ng/g), well above the Ki of 5HT3, but slightly less than the Ki for α7nAChR agonism. Previous reports have shown cognitive improvement in other murine AD models with donezepil doses similar to that we used here (Dong et al., 2005; Van Dam et al., 2008; Zhang et al., 2012) after longer treatment. The efficacy reported in these studies with donezepil is seen despite high protein binding which has been reported to be ~96% (Tiseo et al., 1998), in comparison the protein binding for tropisetron is reported to be 71% (Simpson et al., 2000).
Figure 14. Tropisetron, but not donepezil, improved working memory at a human equivalent dose.
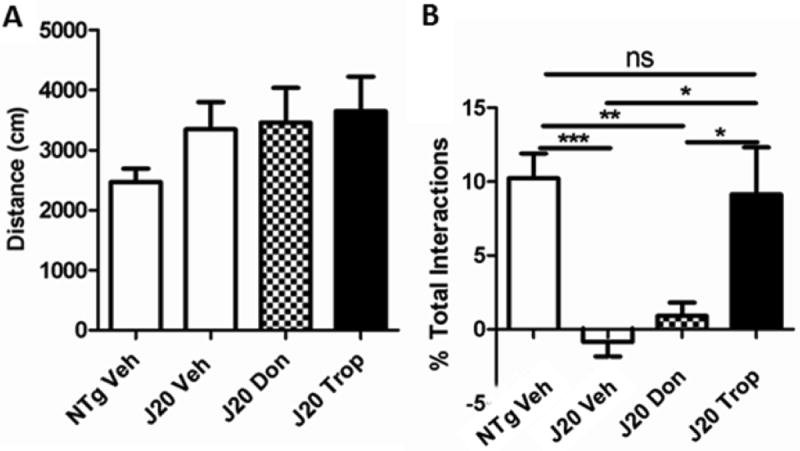
(A) There was no reduction in hyperactivity by any treatment in this study. (B) Only tropisetron significantly (P = 0.0258) improved novelty preference as compared to vehicle-treated J20s controls.
Figure 15. Tropisetron and donepezil effects on sAPPα and Aβ1-42.
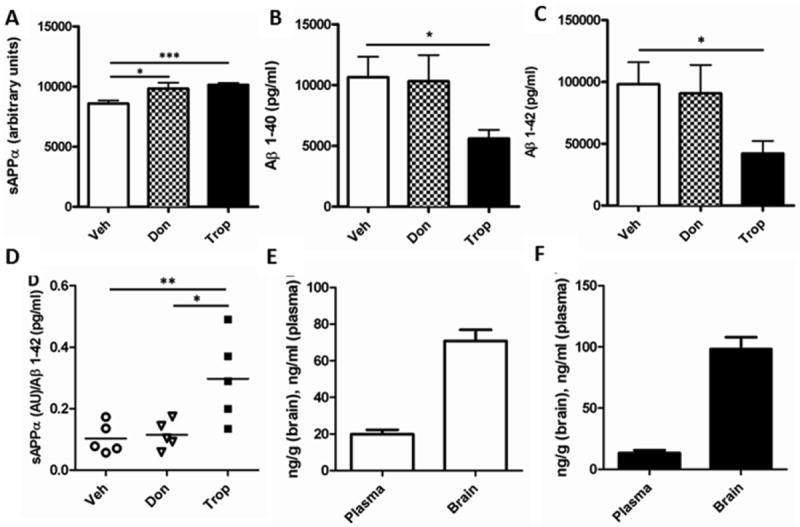
(A) Both tropisetron and donepezil increased sAPPα significantly (P = 0.0004, P = 0.0301, respectively). (B-C) Tropisetron-treated mice had significantly lower Aβ1-40 (P = 0.0353) and Aβ1-42 (P = 0.0446) than donepezil-treated J20s (t-test, one-tailed, unpaired). Hippocampi and entorhinal cortices from individual mice were used for analysis. There was no statistical difference between donepezil- and vehicle-treated mice. (D) The sAPPα/Aβ1-42 ratio was significantly higher for tropisetron-treated than both vehicle- (P = 0.0096) and donepezil-treated (P = 0.0122) mice. Plasma and brain levels of donepezil (E) and tropisetron (F) were similar. All t-test, one-tailed, unpaired statistical analyses.
While results of comparison of tropisetron to existing AD therapeutics under limited conditions and in a single mouse model are potentially informative, we recognize the limitation that these studies may not be a predictor of comparative efficacy in human patients.
2.13 Tropisetron is highly effective by oral delivery
We compared the effects of tropisetron on J20 mice when administered orally vs. subcutaneously. Both vehicle-treated J20s and J20s treated with tropisetron by pump had significantly higher activity than NTg mice (Fig. 16A); there was no significant difference in activity between NTg and J20 mice treated with tropisetron orally. In contrast, J20s treated with tropisetron orally had significantly reduced hyperactivity as compared to vehicle-treated J20s. Vehicle-treated J20s had significantly lower novelty preference as compare to NTg, but there was no significant difference between NTg and J20 mice treated with tropisetron either by pump or orally. Only J20 mice treated with tropisetron by pump had significantly greater novelty preference than vehicle-treated J20s, although there was a similar increase in novelty preference in mice treated orally with tropisetron, as well as greater individual variation (Fig. 16B).
Figure 16. Oral delivery of tropisetron is as effective as pump delivery for improving working memory.
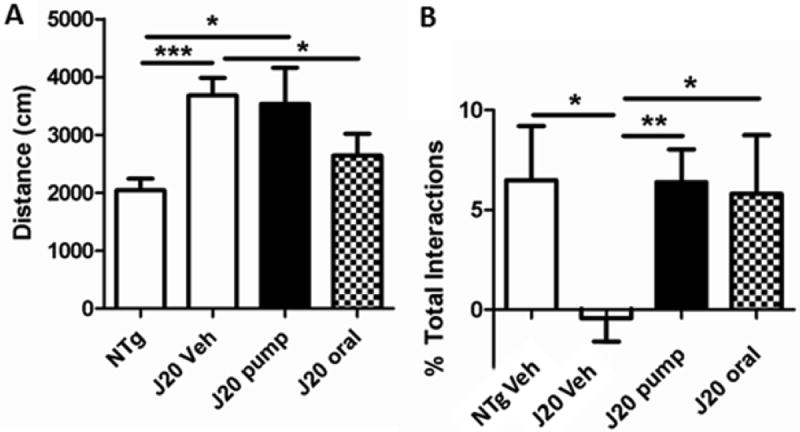
(A) J20 mice receiving either vehicle or tropisetron by pump were still hyperactive as compared to NTg mice (P = 0.0004 and P = 0.0303, respectively), but hyperactivity was reversed by tropisetron orally (P = 0.0461; not significantly different than control NTg). (B) Compared to NTg mice, vehicle-treated J20s had significantly (P = 0.0409) lower novelty preference (NP). There was no significant difference in NP between NTg and either tropisetron-treated group, but only mice treated with tropisetron by pump had significantly (P = 0.0075) greater NP than vehicle-treated J20s.
Only J20 mice treated orally with tropisetron had significantly greater sAPPα (Fig. 17A). J20 mice treated with tropisetron either by pump or orally had significantly lower sAPPβ than vehicle-treated mice (Fig. 17B). J20 mice treated with tropisetron either by pump or orally had lower Aβ1-40 and Aβ1-42 (Fig. 17C-D). The sAPPα/sAPPβ ratio was significantly increased for both pump and orally tropisetron-treated J20s, but the increase was greater for orally-treated mice (Fig. 17E). The sAPPα/Aβ1-42 ratio was higher for orally-treated mice (Fig. 17F), but not significantly so.
Figure 17. Tropisetron oral delivery effects on sAPPα.
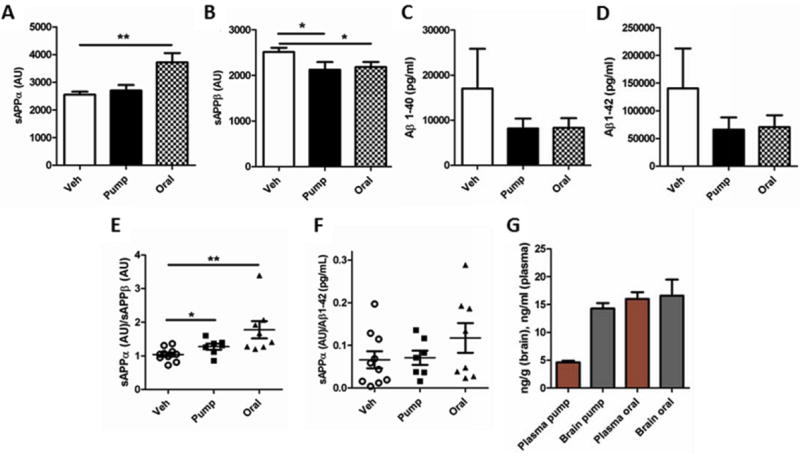
(A) Oral tropisetron treatment significantly (P = 0.0016) increased sAPPα. (B) sAPPβ was significantly lower with both pump and oral delivery (p = 0.0118, p = 0.012, respectively). (C, D) Aβ1-40 and Aβ1-42 were lower after either oral or pump delivery. (E) The sAPPα/sAPPβ ratio was significantly increased with either pump or oral delivery (p = 0.0435, p = 0.0072, respectively), but was greater for orally-treated mice. (F) The sAPPα/Aβ1-42 ratio was higher for orally-treated mice. All samples hippocampus/entorhinal cortex.
Brain levels were similar after pump and oral delivery (Fig. 17G). However, it is noteworthy that pump delivery is continuous, whereas the oral delivery was daily, therefore total exposure to compound would be less for oral delivery. Nonetheless it was more effective at increasing sAPPα.
2.14 NTg mice treated with tropisetron show improved memory
As a tropisetron-treated group of NTg mice was not originally included in the MWM studies, an additional study was run looking at performance in NOR. NTg mice receiving tropisetron showed reductions in activity (Fig. 18A) and increases in novelty preference (Fig. 18B). We speculate that tropisetron may improve behavior in the NOR in NTg mice either by altering processing of endogenous mouse APP and increasing sAPPα, by increasing acetylcholine as a result of 5-HT3 receptor antagonism, or both (see discussion). Endogenous murine sAPPα levels were below the level of detection in our assays; in ongoing studies, acetylcholine levels are being determined.
Figure 18. Tropisetron improves working memory in NTg mice.
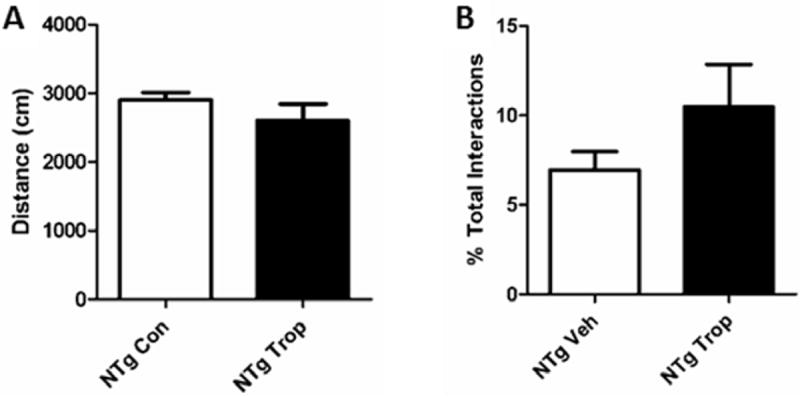
(A) Tropisetron reduced activity and (B) increased NP in NTg mice.
2.15 sAPPα increased in I5 mice
As it was important to determine if tropisetron’s sAPPα increasing and/or Aβ lowering effects could be seen in mice without APP mutations, and because sAPPα could not be determined by these methods in NTg mice, a single study of using I5 huAPPwt mice was performed using adult I5 mice. Hippocampi and entorhinal cortices were dissected separately rather than combined as we had determined Aβ was below the level of detection in entorhinal cortex from I5 mice in our ELISA. sAPPα, however, could be determined in entorhinal cortices in I5 mice, and so analysis of that region was included. Tropisetron-treated I5 mice showed increases sAPPα (Fig. 19A) in both hippocampi and entorhinal cortices; but the already very low Aβ1-42 in hippocampi was unchanged (Fig. 19B). This study did not include behavioral analysis as these mice did not show clear differences from NTg in NOR.
Figure 19. Tropisetron increased sAPPα in I5 mice.
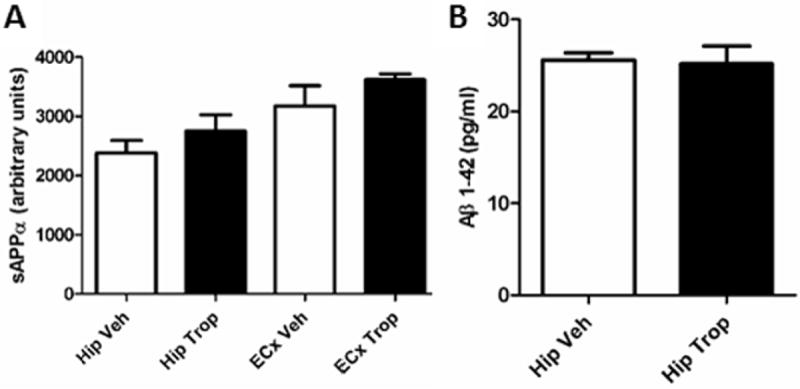
It increased sAPPα (C) but did not change Aβ1-42 (D) in hippocampi from I5 mice.
2.16 Improved cognition and sAPPα/Aβ ratio in plaque-bearing mice
In the majority of in vivo studies presented here, the J20 mice were in the symptomatic but pre-plaque stage. As we were interested in determining if tropisetron could reverse long-term cognitive deficits and lower Aβ even in the presence of plaques, a pilot study was performed using very old mice (12-15 m.o.); at this age, J20 mice have had plaques for approximately half of their lives. In these mice, after 14 days treatment by pump at 1 mkd, followed by an additional 21 days of oral treatment at 2 mkd, cognition was not only improved as compared to age-matched vehicle-only old J20 mice, it was not significantly different from that of younger (6 m.o.) NTg mice. Many old NTg mice showed cognitive impairment, and many old vehicle-only J20 mice were neophobic by the end of treatment (Fig. S3). Tropisetron-treated old J20 mice had higher sAPPα (Fig. S4A) and significantly lower sAPPβ (Fig. S4B), and therefore a significantly higher sAPPα to sAPPβ ratio (S4C). Measurement of sAPPβ was utilized in this study since sAPPβ is reflective of the amount of BACE cleavage; de novo Aβ generation is more difficult to determine by Aβ ELISA only in plaque-bearing mice. Both Aβ1-40 (Fig. S4D) and 1-42 (Fig. S4E) were lower, and the sAPPα to Aβ 1-42 ratio higher (Fig. S4F), but not significantly so.
3. Discussion
Using an iterative screening approach, we have identified a molecule—tropisetron—already approved for clinical use (for short-term use as an anti-emetic, outside the U.S.) that, at a human equivalent dose, reversed the phenotype in the PDAPP mouse model of AD, thus making it a promising therapeutic candidate for Alzheimer’s disease. Tropisetron increased the sAPPα/Aβ42 ratios in vitro in APP-expressing cells, in primary neuronal cultures, and in vivo in the J20 and I5 mouse models. This drug is a potent 5-HT3 receptor antagonist and partial α7nAChR agonist, and we have shown that it also interacts directly with APP. In our tests, tropisetron competitively displaced specific binding of a radioligand with a Ki of 3nM for the 5-HT3 receptor and of 470nM for the α7nAChR (Fig. 2C). Tropisetron selectively activates the α7nAChRs without affecting other nicotinic receptors, and has a greater-than 100-fold selectivity for α7 versus other nicotinic receptor subtypes. Our data suggest that both of these members of the Cys-loop family of receptors (Sine and Engel, 2006) play important roles in Alzheimer’s disease, and that molecules having such dual-receptor binding may be beneficial both as symptomatic and as disease modifying agents in the treatment of AD. Furthermore, as described above, we also discovered that tropisetron interacts directly with APP, with a sub-micromolar affinity. Thus tropisetron interacts with three receptors, and this multi-functional mechanism makes it a unique candidate to develop as a potential therapeutic for AD and MCI (mild cognitive impairment).
The two major subtypes of nicotinic receptors expressed in the brain are the α4β2 nicotinic receptor and the α7nAChR. The former provides high-affinity binding sites for nicotine in the brain (Flores et al., 1992). The α7nAChRs have been shown to bind to Aβ with high affinity, as well, resulting in increased tau phosphorylation (Wang et al., 2003), cholinergic neurotransmission defects (Lee and Wang, 2003), and neuronal cell death (Wang et al., 2000). While a prominent loss of cholinergic neurons in the basal forebrain and cerebral cortex is seen in AD, the involvement of α7nAChRs in this loss is not yet completely defined (Sugaya et al., 1990). A number of nicotinic receptor agonists or partial agonists such as A582941 have been shown to improve cognitive function (Tietje et al., 2008), provide neuroprotection (TC-1698) (Marrero et al., 2004), or lower Aβ (GTS-21) (Shimohama and Kihara, 2001). Some of these ligands are being tested in the clinic as possible AD therapeutics; these include RO5313534 (MEM-3454), reported to bind α7nAChRs with a Ki of 6nM. It is also a potent antagonist of the 5-HT3 receptor. However, as noted above, unlike tropisetron, MEM-3454 shows poor brain permeability and does not interact with APP. Nonetheless, this compound has reportedly shown beneficial effects in a Phase IIa study in mild to moderate AD patients (Sabbagh, 2009). Similarly, another potent α7nAChR partial agonist, EVP-6124, is reported to be in Phase II clinical testing for AD (Prickaerts et al., 2012).
Tropisetron is a potent antagonist of the 5-HT3 receptor with moderate α7nAChR binding and APP binding. As noted above, the 5-HT3 receptor is a member of the Cys-loop ligand-gated ion channel receptors, a family of receptors that also includes the nAChRs to which it is closely related by homology (Thompson and Lummis, 2007). 5-HT3 receptor antagonists have been used primarily as anti-emetics but have also been shown in preclinical testing to be of potential benefit for a number of CNS disorders including anxiety, cognitive dysfunction, and psychosis (Barnes and Sharp, 1999). Although it is not clear how 5-HT3 receptor antagonists alter cognition, antagonists such as ondansetron have been reported to improve water maze learning in aged rats (Pitsikas et al., 1993) and in rats with neurotoxic lesions of the basal forebrain (Hodges et al., 1996). Mechanistically, the 5-HT3 receptor antagonists have been implicated in enhancing the release of acetylcholine from cholinergic neurons, and 5-HT3 receptor antagonism by tropisetron has been shown to prevent the suppression of acetylcholine (ACh) release by 2-methyl-5-HT, a selective 5-HT3 agonist (Bianchi et al., 1990). 5-HT3 receptor antagonists also improve long-term potentiation (LTP) in the hippocampus through activation of GABAergic interneurons (Kawa, 1994; Morales et al., 1996; Morales and Bloom, 1997; Piguet and Galvan, 1994; Ropert and Guy, 1991; Tecott et al., 1993). In addition, 5-HT3 antagonists have been reported to reduce Aβ neurotoxicity (Ju Yeon and Yeon Hee, 2005).
One currently available AD therapeutic, memantine (Namenda), also interacts with the 5-HT3 receptor, as a non-competitive antagonist (Rammes et al., 2001), and has also been reported to be an antagonist at the α7nChR (Aracava et al., 2005). In our studies, at a dose similar to that found to be efficacious for tropisetron, memantine failed to improve memory. Similarly, donepezil, an acetylcholinesterase inhibitor, at human-equivalent dosage failed to improve memory in the PDAPP mice, at least under the conditions of the study performed here.
The lack of improvement in cognition with either donepezil or memantine in our studies could be due to a variety of factors, as noted above. In another study using the Tg2576 mouse AD model, donepezil did induced cognitive improvements when used at the dose used here, but the study was longer (6 weeks). Therefore, while it is possible that longer donepezil treatment may have resulted in cognitive benefits in J20 mice, our purpose here was to observe not only the comparative benefits, but also the speed at which they would be manifest. It is possible that the cognitive benefit from tropisetron was due to its multi-receptor binding activities, which offer the potential to enhance cholinergic function and APP non-amyloidogenic cleavage. Furthermore, the moderate α7nAChR activity of tropisetron may be useful in preventing desensitization of the receptor, a phenomenon seen with sustained exposure to a potent α7nAChR agonist (Revah et al., 1991; Seguela et al., 1993), while maintaining its pro-cognitive benefits. Overall, tropisetron proved superior to both donepezil and memantine in the studies performed here.
We chose sAPPα level as an initial readout for compound candidacy, rather than reduction of Aβ alone, as decreases in sAPPα are also associated with AD pathology. A recent publication has shown that in AD, there is an increased association of ADAM10—the α-secretase responsible for sAPPα production—and the protein AP2, resulting in increased removal of ADAM10 from synapses (Marcello et al., 2013). This removal of ADAM10 decreases sAPPα; therefore any compound that increases sAPPα may mitigate this aspect of the pathology underlying AD.
Reduction of Aβ in mice of 4.5 to 6 months of age reflects the ability of tropisetron to reduce net production of Aβ during a period of rapid amplification in this mouse model. Based on the studies presented here, tropisetron may have the effects in MCI; nonetheless, the reduction of Aβ production should also have benefits in AD. Pilot studies have already been performed using very old (12-15 m.o.) J20 mice, and not only were Aβ and sAPPβ reduced, but behavior in NOR was improved from pre-treatment baseline (Supplementary Figs. S3 and S4). Future studies include treatment for a longer duration to be able to quantify plaque reduction, as well as complete extensive pathological analysis including calbindin expression, synaptic load, and level of tau phosphorylation.
In summary, our studies show that tropisetron has excellent oral bioavailability, brain penetration, cognitive effects, and biomarker effects at currently used human equivalent doses, offering strong support for further evaluation of tropisetron in clinical trials in MCI and AD patients. Based on these results we have received approval to proceed with clinical testing of tropisetron in MCI patients. Tropisetron could potentially be useful as a single agent or in combination with complementary therapeutics for the treatment of Alzheimer’s disease and MCI.
Materials and Methods
Drugs
Tropisetron hydrochloride was obtained from Tocris Biosciences (cat# 2459) and stored with moisture absorbent drierite at 4°C. For the in vitro studies tropisetron was prepared as a 1mM stock solution in dimethylsulfoxide (DMSO, SigmaAldrich). For in vivo testing using subcutaneous injection, tropisetron hydrochloride was dissolved in 100% DMSO to generate a 5mg/ml stock solution. For Alzet pump studies, tropisetron was dissolved in 25% DMSO and 75% physiological saline (0.9%, pH 7.8) to give 5mg/ml. MEM3454 was obtained from Aquila Pharmatech (cat #: 411104). Memantine and donepezil were both purchased from Sigma-Aldrich (D6821 and M9292, respectively) and were prepared in the same way as tropisetron.
4.1 In Vitro testing
4.1.1 Receptor saturation and competition binding studies
Saturation binding analysis of the α7nAChR was carried out using endogenously expressed α7nAChRs in SH-SY5Y cells with increasing concentrations of the classical antagonist [125I]α-bungarotoxin (α-Bgt, 0.05nM). The competition assay was performed with 0.1nM-10uM tropisetron. The non-specific binding was defined by 1uM α-Bgt. Receptor binding for 5-HT3R was assessed using human recombinant 5-HT3 receptors expressed in CHO cells. Tropisetron was tested at 0.1nM-10uM to displace [3H]-BRL-43694 (Granisetron; 2 nM), a 5-HT3R selective antagonist. The non-specific binding was assessed with MDL-72222, a selective 5-HT3R antagonist and was defined as 10uM. Additional binding studies with α4nACh receptors and muscle-type nicotinic receptors ((α1β1γδ nACh) were also performed. All of the in vitro pharmacology studies with tropisetron were done at CEREP (Celle l’Eves cault, France).
4.1.2 Surface plasmon resonance (SPR)
SPR data were obtained with a Biacore T100 (GE Healthcare); the surfaces of all four flow cells (FC1, FC2, FC3, FC4) of a carboxymethylated-dextran (CM-5) chip were washed sequentially with 50mM NaOH, 1mM HCl, 0.05% H3PO4, and 20mM sodium phosphate pH 7.4, 125 mM sodium chloride in parallel using a flow rate of 30μl/min for 1 min. Three fusion proteins were immobilized via amine coupling using 20mM phosphate, 125mM sodium chloride pH 7.4. The three proteins were MBP-eAPP230-624 - a fusion protein containing maltose binding protein (MBP) and residues 230-624 of the ectodomain of APP (90-kDa) (FC4), eAPP230-624 – a protein that contains only residues 230-624 (45-kDa)(FC2), and TRX-eAPP575-624 – a fusion protein containing thioredoxin (TRX) and residues 575-624 of the ectodomain (20-kDa) (FC3). The proteins were produced as described in (Libeu et al., 2011). The proteins were concentrated to 2mg/ml in 20mM phosphate pH 6.5, 125mM sodium chloride, and then dissolved to a concentration of 50μg per ml in 20mM sodium acetate pH 5.0. FC1 served as a reference cell following a mock immobilization with buffer alone. For all cells, the flow rate was 10μl per min. The chip was blocked with 1M ethanolamine (pH 8.5). The final RU values were 11711 for TRX-eAPP575-624, 2156 for eAPP230-624 and 10275 for MBP-eAPP230-624.
Compounds were diluted from 10mM solutions in DMSO to 50μM in 1% DMSO, 20mM sodium phosphate pH 7.4, 125mM sodium chloride, 0.05% Tween, and then serially diluted by 1.5 for 10 steps. Binding traces were recorded for each dilution with a binding phase of 60 seconds and a dissociation phase of 240 seconds. Each cycle was performed at 20°C with a constant flow rate of 20μl/min. An additional 240 seconds of buffer flow at 60μl per min across the cells was applied as a regeneration phase to facilitate complete dissociation of the compound from the protein. The sensograms were obtained by subtraction of the reference and buffer signals using the Biacore T100 Evaluation software. The binding curves were modeled with the PRISM (Graphpad, Inc).
4.1.3 Primary screening assay
CHO-7W cells stable transfected with huAPPwt were used for these studies. Cells in 50μl of Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, were seeded at a density of 5 × 104 cells/well in 96 well plates overnight. The cells were then incubated for twenty-four hours with compounds from the clinical library prepared from a 10mM DMSO stock solution to achieve a final concentration in each well of 1μM. Cell media were then removed, Complete Protease Inhibitor (Roche cat# 1183617) added, and 2μl of the media obtained after a 500-fold dilution was made to the levels of sAPPα. This was done using a custom AlphaLISA assay (Perkin-Elmer), comprising acceptor beads that bind to the C-terminus of sAPPα (cat# AL231C Perkin-Elmer) and donor beads that bind to the N-terminus of biotinylated APP antibody (R&D cat# AF1168). After incubation in the dark for 1h, the AlphaLISA signal was measured using a Perkin-Elmer (PE) Enspire Multi-mode plate reader. Any compound that induced a 20% increase in sAPPα was considered a potential “hit”, and hits were re-tested to confirm initial findings. Tropisetron increased sAPPα levels consistently in the CHO-7W cells by ~ 30%.
4.1.4 Primary hippocampal neuronal cultures
Hippocampal neuronal cultures were generated from E18 embryos resulting from a J20 male, J20 female crosses. Such mating of heterozygous J20s typically results in 75% of the embryos, and therefore the cells in culture, carrying the transgene. Briefly, hippocampi were dissected from embryos using 12-22 embryos, and the dissected tissue combined in ice-cold Hank’s Balanced Salt Solution (HBSS, Invitrogen). The pooled hippocampi then underwent trypsinization and DNAase 1 (both Sigma-Aldrich, St. Louis MO) treatment to generate a single-cell suspension. Cells were plated onto poly-l-lysine (Sigma) coated 48-well plates at a density of 5 × 105 cells/well. The medium was Neurobasal with B27, glutamax and pen-strep (all Invitrogen, www.invitrogen.com). The cells were allowed to mature for four to five days before the addition of compound at 1μM final. Cells were treated once a day every day for four days, and both media and cells collected two hours after the last addition of compound on the 5th day. Partial media changes were timed so that the last day of treatment was the 3rd day after media change, when APP biochemical readouts were the highest. sAPPα was determined from media using the Perkin-Elmer AlphaLISA and Aβ1-42 was determined from media and cells using Invitrogen’s sensitive ELISA kit for Aβ1-42.
4.1.5 Apolipoprotein 4 (ApoE ε4) transfection
Human ApoE cDNA constructs were generously provided by Dr. Yadong Huang (Gladstone Institute of Neurological Disease, University of California, San Francisco, CA 94143). A172 human glioblastoma cells that endogenously express APP were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were transfected with human ApoE ε4 or ApoE ε3 using XtremeGENE HP (Roche, www.roche-applied-science.com) in OptiMEM I Reduced Serum Media (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours later, culture medium was replaced, and varying concentrations of the drug were added to the cells. Following an additional twenty-four hours, cell survival was determined by the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (M6494, www.invitrogen.com).
4.2 In vivo testing
4.2.1 Pharmacokinetic (PK) studies
Five adult (>3 months of age) NTg mice were used for pharmacokinetic analysis to determine brain and plasma levels of compounds. All were injected subcutaneously with 50μl of 5mg/ml tropisetron, MEM3454 (M3454), or memantine (Mem) in DMSO, and individual mice were anesthetized by ketamine/xylazine injection and blood collected via cardiac puncture followed by saline perfusion 1, 2, 4, 6, and 8 hours after injection. Brains were halved and frozen. Plasma was collected after centrifugation of whole blood at 5,000 rpm for 10 min. Plasma and hemi-brains were sent to IAS (Integrated Analytical Systems, Berkeley, CA) for compound level analysis with an LC-MS/MS approach.
4.2.2 Animals
The derivation and characterization of PDAPP huAPPSwe/Ind (J20) and huAPPwt (I5) mice have been described elsewhere (Hsia et al., 1999; Mucke et al., 2000). PDAPP mice were maintained by heterozygous crosses with C57BL/6J mice (Jackson Laboratories, jaxmice.jax.org). PDAPP mice were heterozygous with respect to the transgene. Non-transgenic littermates were used as controls. Mice were maintained in temperature controlled rooms with 12-hour light-dark cycle and all procedures were in compliance with the Buck Institute Animal Care and Use Committee and AAALAC guidelines.
4.2.3 I5 study
Adult I5 mice were injected subcutaneously with 50μl of a 5mg/ml stock solution of tropisetron in DMSO (10 mkd) every day for five days. Two hours after the last injection on Day 5, mice were anesthetized by ketamine/xylazine injection, blood collected, brain tissue saline perfused and hippocampi and entorhinal cortices dissected separately and snap frozen for biochemical and compound level analysis. Plasma collection and analysis were performed as in the PK studies. Experimental groups for I5 studies were n = 3 for both vehicle control and tropisetron; mice were males 4-5 months of age and were sibling-matched in each group. I5 mice do not develop plaques, but mice of the ages similar to those of J20 study mice were used for consistency. I5 mice do not show memory impairment in the testing paradigms used here, so this study was for analysis of sAPPα and Aβ 1-42 only.
4.2.4 Eight-week MWM study
Mice were implanted with subcutaneous Alzet pumps and the pumps were replaced at 28 days. All groups started with 12-13 mice; some mice died during the course of the study or were eliminated from analysis due to thigmotaxis (circling the perimeter of the pool) or excessive floating, therefore for MWM analysis non-transgenic (NTg) vehicle control n = 13, J20 vehicle control n = 8, and J20 tropisetron-treated n = 8. For biochemistry, J20 vehicle control was n = 7 (one mouse died after MWM was completed). All were 4-5 months old at the start of treatment and therefore 6-7 months old at the end, both males and females were used, and siblings sorted into each treatment group.
4.2.5 Morris Water Maze (MWM)
The overall health of all mice was assessed on the first day of the MWM (Morris, 1984) study with special attention to vision and ability to swim. On the second day the mice were trained to swim to a 12 × 12 cm platform submerged 1.5 cm below the water surface in a rectangular enclosed pool (15 × 122 cm). The water maze itself consisted of a round pool (122 cm diameter) filled with water (21+/- 1°C) opacified with non-toxic white tempera paint powder. The pool was surrounded by distinct extra-maze cues (graphic patterns). The first 3 days of training comprised the “Visual” test. In this task, the platform was marked with a visible cue (15 cm tall, black and white striped pole placed on top of the platform). Mice were separated into 2 groups and each group underwent four trials per day, either in the morning or afternoon, with the order of the groups alternating daily. The platform location was changed each day, as were the four locations mice were lowered into the water (“drops”). Trials were aborted after 60 s. Hidden platform training was performed in the following week. For this training, the platform was again submerged 1.5 cm in the same location throughout hidden platform training, but the drop location varied semi-randomly between trials. Mice received one training session per day for 5 consecutive days comprised of 4 trials. Mice that did not find the platform within 60 s were guided to it and allowed to sit on it for 10 s. Hidden probe trials were run on Day 4 and Day 5. For the probe trials, the platform was removed, and mice were allowed to swim for 60 s before they were removed. The drop location was 180° from where the platform was placed during hidden platform training. The same drop location was used for all hidden probe trials. The following week, the mice underwent additional hidden platform training, but with the platform in a new location (“Reversal”). Reversal training and probe trials were just as described for the initial hidden training, but took place for only 3 consecutive days and with only one probe trial. Behavior was recorded with the Ethovision video tracking system (Noldus, www.noldus.com). Escape latencies, distance traveled, swim paths, swim speeds, percentage time spent in each quadrant, and platform crossings were recorded for subsequent analysis.
4.2.6 Biochemistry
In all in vivo studies with the exception of the I5 study, a single sample of combined hippocampus and entorhinal cortex from individual mice was used for biochemical analyses. In the I5 study, hippocampi and entorhinal cortices were dissected and analyzed separately. These brain regions were used because the pathological and biochemical alterations are the most pronounced in these areas in the J20. Aβ was only measurable in hippocampus in the I5 mouse. All tissues were weighed and sonicated in freshly prepared 5M Guanidine (Gdn)-HCl in 50mM Tris-NCl pH 8.0 at 20% weight/volume. Samples were rotated for 2-3 hours at room temperature after sonication and typically stored frozen before use. Invitrogen ELISA kits for Aβ1-40 and 1-42 were used according to the manufacturer’s instructions. For sAPPα level analysis, protein from 100μl of the Gdn sonicate was precipitated with 400μl of ice-cold methanol overnight at -20°C. The samples were centrifuged at 10,000 rpm for 15 minutes at 4°C to pellet the protein, and after all of the methanol was removed, the protein was resuspended in 100μl 1X AlphaLISA Hi-Block buffer (Perkin-Elmer) and sonicated on ice until the pellet was well-resuspended by microscopic inspection. The AlphaLISA for sAPPα described above was used according to the manufacturer’s instructions. In addition, Immuno-Biological Laboratories (IBL, www.ibl-america.com) ELISA for sAPPα was used to confirm AlphaLISA results in most studies, and for this assay, the Gdn sonicate was used.
4.2.7 Comparison to memantine
Mice received vehicle control, 0.5 mkd tropisetron or 0.4 mkd memantine in 28-day Alzet pumps implanted subcutaneously. The memantine dose was chosen to be similar to the tropisetron dose, which was a human-equivalent dose. Memantine is currently used at oral dose of 5-20 mgs per day for treatment of AD while tropisetron is used at oral dose of 5 mgs per day for PONV. Given memantine’s relatively high brain levels, the dose chosen was slightly lower than that for tropisetron. Each group started with 7-8 J20 4-5 m.o. mice. Mice underwent working object memory testing at 26 days using the NOR task at the end of the study, vehicle control n = 8, tropisetron n = 7, memantine n = 6. For biochemistry, vehicle control n = 8, tropisetron n = 6, memantine n = 5. Plasma and brain levels at the end of the study were determined by IAS using plasma collected post-centrifugation of whole blood and post-saline perfused brain tissue. Hippocampi and entorhinal cortices were combined from individuals for analysis.
4.2.8 Novel Object Recognition (NOR) Task
For most behavioral testing presented here, the NOR task, rather than MWM, was used to determine improvements in memory. A pilot study had shown that tropisetron treatment increased J20 performance in NOR, and the task had far fewer drawbacks than the MWM. Mice, and J20 mice in particular, are not good swimmers and often manifest anxiety in the form of floating or thigmotaxis (circling the perimeter of the pool) and many such mice have to be eliminated from data analysis. Great individual variation is seen in MWM and therefore large numbers of mice need to be tested to identify differences between groups. Others have also found that NOR may be a better testing paradigm for compound screening (Alkam et al., 2011; Dere et al., 2007; Zhang et al., 2012), particularly when the large animals numbers needed for MWM are not available, or shorter treatment periods are used. In addition, NOR allows for a determination of the activity level, and J20 mice are hyperactive compared to NTg. The NOR protocol used was based on that of Bevins and Besheer (Bevins and Besheer, 2006). Mice have a natural curiosity for the novel, and typically spend more time with a new object than one with which they have become familiar, provided that their memories for what is novel and what is familiar remain intact. Thus mice with impaired memories will often spend approximately equal time with the new and old objects. In addition, the J20 are hyperactive (Chen et al., 2000; Palop et al., 2003), and therefore often travel farther during the task. Individual mice were allowed to become acclimated to a 20 × 30cm black opaque box (“arena”) and the operator for 20 minutes per day for 2 days prior to testing. During testing, there was an “acquisition” phase wherein the mouse spent 10 minutes with two identical objects. Both the attention paid to each object (touching or coming within 2 cm of the object with the nose, designated as an interaction) and the distance the mouse traveled during the 10 minutes were recorded. After a 1-hour rest, the mouse was returned to the arena for short-term memory assessment; for this, one of the objects had been changed. Novelty preference (NP) was calculated as percentage of total interactions with both objects with 50% - or an equal number of interactions with each object – as zero preference. Mice were euthanized 24 hours after NOR testing.
4.2.9 Comparison to donepezil
Mice received vehicle, 0.5 mkd tropisetron, or 1 mkd donepezil in 14-day model 1002 Alzet pumps; at the end of the 14-day period, they received daily subcutaneous injections at those dosages for an additional 9 days. While the dose of donepezil was chosen to be similar to that of tropisetron, it is also equivalent to the human starting dose of 5 mg per day. The human equivalent dose was calculated through normalization of body-to-surface area using the formula:
where the mouse Km is 3 (and human Km is 37) (Reagan-Shaw et al., 2008) This dose also should be adequate to inhibit AChE (Rakonczay, 2003). Mice underwent short-term memory testing using the NOR task at 14 days (at which time no improvement was seen with donepezil, and so treatment was extended) and at 22 days. Injections were used for extended treatment as humans receive donepezil once a day rather than by continuous pump delivery, and we thought this may alter responses to treatment. Plasma and brain levels at the end of the study were determined as described above. Biochemical markers were assayed as described above from combined hippocampus and entorhinal cortex.
4.2.10 Comparison of oral and pump delivery
Mice received either 1mkd tropisetron by 28-day Alzet pump, or 4mkd tropisetron by a single oral dose. The oral dosing was performed by mixing 5μl of an appropriate stock solution of tropisetron with 5μl of liquid syrup to make the dose palatable. The mice were fed the 10μl total from the tip of a pipette, rather than gavage. As comparison of pharmacokinetics after oral and subcutaneous injection showed brain levels are 4-5 fold lower after oral dosing, the oral dose in this study was 4-fold higher. Mice underwent NOR testing at 21 days of treatment and were euthanized 24 hours later.
4.2.11 Meta-analysis of all in vivo studies
A total of ten in vivo experiments wherein J20 mice were treated with tropisetron were included in meta-analysis. For sAPPα, Aβ1-42, and the ratio, all values for individuals were converted to percentage of the average vehicle control value in each separate experiment, and then all of these converted values were analyzed together (N = 71 control, N = 72 tropisetron). All statistics used student’s t-test, unpaired, and two-tailed analyses unless otherwise specified.
4.2.12 Tropisetron treatment of NTg mice
NTg mice were treated with 0.5 mkd tropisetron for 28 days by Alzet pump as described above in the memantine study. At 27 days, mice performed the NOR task, and were euthanized 24 hours later.
4.3 Supplementary methods
4.3.1 Pilot studies
In addition to the in vivo studies presented in the body of the manuscript, five additional studies were run and included in meta-analysis. These included: 1) 4-day subcutaneous injection of 4-5 m.o. J20 males at 10 mkd, control n = 3, tropisetron n = 1 (all studies began with 3 or more mice per group, but some mice are not available for biochemical analysis); 2) 5-day BID subcutaneous injection at 10 mk (20 mkd) of 4-5 m.o. J20 females, control n = 4, tropisetron n =4; 3) 12-day subcutaneous injection at 10 mkd of 4-5 m.o. J20 males, control n = 4, tropisetron n = 4; 4) 5-day subcutaneous injection at 0.3 mkd of 4-5 m.o. J20 males, control n = 2, tropisetron n = 4; and 5) 28-day pump delivery of 0.5 mkd to 12-14 m.o. J20 males and females, control n = 2, tropisetron n = 2. All biochemical analysis was done using microdissected hippocampus and entorhinal cortex.
4.3.2 Pilot study in old, plaque-bearing mice
Old (12-15 m.o) J20 mice received 1 mkd tropisetron (n = 8) or saline vehicle (n= 9) by 14-day Alzet pump for 13 days, at which time no improvement in memory as determined by NOR testing was seen, therefore treatment was continued for an additional 21 days by oral dosing at 2 mkd. The oral dose was higher as oral availability is lower than subcutaneous. Aged-matched NTg mice were treated similarly with vehicle only (n = 5). All mice underwent baseline NOR testing 4 days before the commencement of treatment and, as many old NTg mice performed poorly in NOR, a relatively young (6 m.o.) NTg cohort was added and treated with vehicle as above. NOR testing was also performed at 13 days of treatment and the end of the study, a total of 35 days. Biochemical analysis of sAPPα, Aβ 1-40 and 1-42 from a single sample from each mouse comprised of combined hippocampus and entorhinal cortex was performed as described above, but here, analysis of sAPPβ was added. The Perkin-Elmer AlphaLISA for sAPPβ was used and the assay performed as described above for sAPPα. sAPPβ is the N-terminal fragment generated as a result of BACE cleavage of APP (Fig. 1) and as BACE cleavage is the first step in Aβ production it can be indicative of de novo Aβ production. This assay was used because in very old plaque-bearing mice there is an enormous amount of pre-existing, plaque-associated Aβ. To determine the effects of a compound such as tropisetron that reduces net Aβ production but does not necessarily clear existing plaques, an assay of sAPPβ in addition to Aβ itself was thought to provide additional information. In our hands, measurement of a soluble versus non-soluble fraction of Aβ in these very old mice did not prove to be useful, since very little soluble Aβ could be detected.
Supplementary Material
Highlights.
Dramatic improvement of memory and biomarker ratios in AD mouse model by a multi-functional drug candidate
Drug candidate is a potent 5HT-3 antagonist, a alpha7nicotinic receptor partial agonist and binds the ectodomain of APP.
Drug candidate has superior efficacy in the AD model in comparison to two current AD drugs
Suitable for human clinical trials as a candidate therapeutic for AD.
Acknowledgments
This research was supported by funding from the Douglas and Ellen Rosenberg Foundation, the Joseph Drown Foundation, and the NIH (AG034427 to D.E.B.). We thank Rowena Abulencia for her assistance in preparation of the manuscript. We thank Dr. Barbara Jagodzinska for the tropisetron model. We thank Dr. Edward Koo for providing the CHO cell line that overexpresses APP (7W) and Dr. Todd Golde for the neural cell line that overexpresses APP (H4APPwt).
Abbreviations
- AD
Alzheimer’s Disease
- APP
Amyloid precursor protein
- Aβ
amyloid beta
- sAPPα
soluble amyloid precursor protein alpha
- sAPPβ
soluble amyloid precursor protein beta
- CHO 7W
Chinese hamster ovary cells stably transfected with human APP wildtype
- α7nAChR
alpha 7 nicotinic acetylcholine receptor
- 5-HT3
5-hydroxytryptophan 3 (receptor)
- GABA
gamma-aminobutyic acid
- PONV
post-operative nausea and vomiting
- MCI
mild cognitive impairment
- BACE
beta-site APP cleaving enzyme
- Jcasp
juxtamembranar cytoplasmic domain
- C31
c-terminal fragment of caspase-cleaved APP
- αCTF
c-terminal fragment of ADAM10 (α-secretase) cleavage of APP
- βCTF
c-terminal fragment of BACE cleavage of APP
- NMDA
N-methyl-D-aspartate (receptor)
- NOR
Novel object recognition
- HTS
high-throughput screening
- MWM
Morris water maze
- AChE
Acetylcholine esterase
- ADAM10
a disintegrin and a metalloprotease 10
- PDAPP
platelet-derived growth factor promoter driven human APP gene
- HED
human equivalent dose
- BID
twice a day
- Mkd
mg/kg/day
Footnotes
Author Contributions: D.B. and V.J. conceptualized the study. O.D. performed HTS; D.B., V.J., and P.S. designed experiments. P.S., O.G., A.P., R.R., and K.P. performed experiments. P.S. analyzed data. P.S., V.J. and D.B. wrote the manuscript. Phone: +1 415-209-2000
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alkam T, Hiramatsu M, Mamiya T, Aoyama Y, Nitta A, Yamada K, Kim HC, Nabeshima T. Evaluation of object-based attention in mice. Behav Brain Res. 2011;220:185–93. doi: 10.1016/j.bbr.2011.01.039. [DOI] [PubMed] [Google Scholar]
- Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312:1195–205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc. 2006;1:1306–11. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Bianchi C, Siniscalchi A, Beani L. 5-HT1A agonists increase and 5-HT3 agonists decrease acetylcholine efflux from the cerebral cortex of freely-moving guinea-pigs. Br J Pharmacol. 1990;101:448–52. doi: 10.1111/j.1476-5381.1990.tb12728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE. Neurodegeneration in Alzheimer’s disease: caspases and synaptic element interdependence. Mol Neurodegener. 2009;4:27. doi: 10.1186/1750-1326-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 2000;408:975–9. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–28. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Claasen AM, Guevremont D, Mason-Parker SE, Bourne K, Tate WP, Abraham WC, Williams JM. Secreted amyloid precursor protein-alpha upregulates synaptic protein synthesis by a protein kinase G-dependent mechanism. Neurosci Lett. 2009;460:92–6. doi: 10.1016/j.neulet.2009.05.040. [DOI] [PubMed] [Google Scholar]
- Dere E, Huston JP, De Souza Silva MA. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci Biobehav Rev. 2007;31:673–704. doi: 10.1016/j.neubiorev.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Dong H, Csernansky CA, Martin MV, Bertchume A, Vallera D, Csernansky JG. Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer’s disease. Psychopharmacology (Berl) 2005;181:145–52. doi: 10.1007/s00213-005-2230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Corey-Bloom J, Zhang R, Li H, Ieni J, Schindler R. Safety and tolerability of donepezil at doses up to 20 mg/day: results from a pilot study in patients with Alzheimer’s disease. Drugs Aging. 2008;25:163–74. doi: 10.2165/00002512-200825020-00008. [DOI] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–7. [PubMed] [Google Scholar]
- Garbe C, Drechsler S, Fiedler H, Tilgen W, Landthaler M, Schroeder K, Kuehne KH, Faerber L. Dose comparison of tropisetron (Navoban) 5 mg and 10 mg orally in the prophylaxis of dacarbazine-induced nausea and emesis. Semin Oncol. 1994;21:12–6. [PubMed] [Google Scholar]
- Hodges H, Sowinski P, Turner JJ, Fletcher A. Comparison of the effects of the 5-HT3 receptor antagonists WAY-100579 and ondansetron on spatial learning in the water maze in rats with excitotoxic lesions of the forebrain cholinergic projection system. Psychopharmacology (Berl) 1996;125:146–61. doi: 10.1007/BF02249414. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–33. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Yeon B, Yeon Hee S. Blockade of 5-HT(3) receptor with MDL 72222 and Y 25130 reduces beta-amyloid protein (25--35)-induced neurotoxicity in cultured rat cortical neurons. Eur J Pharmacol. 2005;520:12–21. doi: 10.1016/j.ejphar.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Kawa K. Distribution and functional properties of 5-HT3 receptors in the rat hippocampal dentate gyrus: a patch-clamp study. J Neurophysiol. 1994;71:1935–47. doi: 10.1152/jn.1994.71.5.1935. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Mathis CA, Price JC, Lopresti BJ, DeKosky ST. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129:2805–7. doi: 10.1093/brain/awl281. [DOI] [PubMed] [Google Scholar]
- Lee DH, Wang HY. Differential physiologic responses of alpha7 nicotinic acetylcholine receptors to beta-amyloid1-40 and beta-amyloid1-42. J Neurobiol. 2003;55:25–30. doi: 10.1002/neu.10203. [DOI] [PubMed] [Google Scholar]
- Libeu CP, Poksay KS, John V, Bredesen DE. Structural and Functional Alterations in Amyloid-beta Precursor Protein Induced by Amyloid-beta Peptides. J Alzheimers Dis. 2011;25:547–66. doi: 10.3233/JAD-2011-101938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macor JE, Gurley D, Lanthorn T, Loch J, Mack RA, Mullen G, Tran O, Wright N, Gordon JC. The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist. Bioorg Med Chem Lett. 2001;11:319–21. doi: 10.1016/s0960-894x(00)00670-3. [DOI] [PubMed] [Google Scholar]
- Marcello E, Saraceno C, Musardo S, Vara H, de la Fuente AG, Pelucchi S, Di Marino D, Borroni B, Tramontano A, Perez-Otano I, Padovani A, Giustetto M, Gardoni F, Di Luca M. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. J Clin Invest. 2013;123:2523–38. doi: 10.1172/JCI65401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero MB, Papke RL, Bhatti BS, Shaw S, Bencherif M. The neuroprotective effect of 2-(3-pyridyl)-1-azabicyclo[3.2.2]nonane (TC-1698), a novel alpha7 ligand, is prevented through angiotensin II activation of a tyrosine phosphatase. J Pharmacol Exp Ther. 2004;309:16–27. doi: 10.1124/jpet.103.061655. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Morales M, Battenberg E, de Lecea L, Sanna PP, Bloom FE. Cellular and subcellular immunolocalization of the type 3 serotonin receptor in the rat central nervous system. Brain Res Mol Brain Res. 1996;36:251–60. doi: 10.1016/0169-328x(96)88406-3. [DOI] [PubMed] [Google Scholar]
- Morales M, Bloom FE. The 5-HT3 receptor is present in different subpopulations of GABAergic neurons in the rat telencephalon. J Neurosci. 1997;17:3157–67. doi: 10.1523/JNEUROSCI.17-09-03157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer’s disease. J Mol Neurosci. 2001;17:101–18. doi: 10.1385/jmn:17:2:101. [DOI] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Morrow GR, Hickok JT, Rosenthal SN. Progress in reducing nausea and emesis. Comparisons of ondansetron (Zofran), granisetron (Kytril), and tropisetron (Navoban) Cancer. 1995;76:343–57. doi: 10.1002/1097-0142(19950801)76:3<343::aid-cncr2820760302>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–7. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist--a review of preclinical data. Neuropharmacology. 1999;38:735–67. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Piguet P, Galvan M. Transient and long-lasting actions of 5-HT on rat dentate gyrus neurones in vitro. J Physiol. 1994;481(Pt 3):629–39. doi: 10.1113/jphysiol.1994.sp020469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsikas N, Brambilla A, Borsini F. DAU 6215, a novel 5-HT3 receptor antagonist, improves performance in the aged rat in the Morris water maze task. Neurobiol Aging. 1993;14:561–4. doi: 10.1016/0197-4580(93)90039-e. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, Reneerkens OA, Flood DG, Hilt D, Gawryl M, Bertrand S, Bertrand D, Konig G. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62:1099–110. doi: 10.1016/j.neuropharm.2011.10.024. [DOI] [PubMed] [Google Scholar]
- Rakonczay Z. Potencies and selectivities of inhibitors of acetylcholinesterase and its molecular forms in normal and Alzheimer’s disease brain. Acta Biol Hung. 2003;54:183–9. doi: 10.1556/ABiol.54.2003.2.7. [DOI] [PubMed] [Google Scholar]
- Rammes G, Rupprecht R, Ferrari U, Zieglgansberger W, Parsons CG. The N-methyl-D-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci Lett. 2001;306:81–4. doi: 10.1016/s0304-3940(01)01872-9. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–9. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Ropert N, Guy N. Serotonin facilitates GABAergic transmission in the CA1 region of rat hippocampus in vitro. J Physiol. 1991;441:121–36. doi: 10.1113/jphysiol.1991.sp018742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbagh MN. Drug development for Alzheimer’s disease: where are we now and where are we headed? Am J Geriatr Pharmacother. 2009;7:167–85. doi: 10.1016/j.amjopharm.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–27. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S, Kihara T. Nicotinic receptor-mediated protection against beta-amyloid neurotoxicity. Biol Psychiatry. 2001;49:233–9. doi: 10.1016/s0006-3223(00)01100-8. [DOI] [PubMed] [Google Scholar]
- Simpson K, Spencer CM, McClellan KJ. Tropisetron: an update of its use in the prevention of chemotherapy-induced nausea and vomiting. Drugs. 2000;59:1297–315. doi: 10.2165/00003495-200059060-00008. [DOI] [PubMed] [Google Scholar]
- Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. Nature. 2006;440:448–55. doi: 10.1038/nature04708. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- Sugaya K, Giacobini E, Chiappinelli VA. Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer’s disease. J Neurosci Res. 1990;27:349–59. doi: 10.1002/jnr.490270314. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Maricq AV, Julius D. Nervous system distribution of the serotonin 5-HT3 receptor mRNA. Proc Natl Acad Sci U S A. 1993;90:1430–4. doi: 10.1073/pnas.90.4.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theendakara V, Patent A, Peters-Libeu C, Philpot B, Flores S, Descamps O, Poksay K, Zhang Q, Cailing G, Hart M, John V, Rao R, Bredesen D. Neuroprotective sirtuin ratio reversed by ApoE4. PNAS. 2013 doi: 10.1073/pnas.1314145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC. The 5-HT3 receptor as a therapeutic target. Expert Opin Ther Targets. 2007;11:527–40. doi: 10.1517/14728222.11.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietje KR, Anderson DJ, Bitner RS, Blomme EA, Brackemeyer PJ, Briggs CA, Browman KE, Bury D, Curzon P, Drescher KU, Frost JM, Fryer RM, Fox GB, Gronlien JH, Hakerud M, Gubbins EJ, Halm S, Harris R, Helfrich RJ, Kohlhaas KL, Law D, Malysz J, Marsh KC, Martin RL, Meyer MD, Molesky AL, Nikkel AL, Otte S, Pan L, Puttfarcken PS, Radek RJ, Robb HM, Spies E, Thorin-Hagene K, Waring JF, Ween H, Xu H, Gopalakrishnan M, Bunnelle WH. Preclinical characterization of A-582941: a novel alpha7 neuronal nicotinic receptor agonist with broad spectrum cognition-enhancing properties. CNS Neurosci Ther. 2008;14:65–82. doi: 10.1111/j.1527-3458.2008.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiseo PJ, Foley K, Friedhoff LT. The effect of multiple doses of donepezil HCl on the pharmacokinetic and pharmacodynamic profile of warfarin. Br J Clin Pharmacol. 1998;46(Suppl 1):45–50. doi: 10.1046/j.1365-2125.1998.0460s1045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyohara J, Hashimoto K. alpha7 Nicotinic Receptor Agonists: Potential Therapeutic Drugs for Treatment of Cognitive Impairments in Schizophrenia and Alzheimer’s Disease. Open Med Chem J. 2010;4:37–56. doi: 10.2174/1874104501004010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, Coen K, De Deyn PP. Cognitive evaluation of disease-modifying efficacy of donepezil in the APP23 mouse model for Alzheimer’s disease. Psychopharmacology (Berl) 2008;197:37–43. doi: 10.1007/s00213-007-1010-x. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Callahan PM, Tehim A, Bertrand D, Tombaugh G, Wang S, Xie W, Rowe WB, Ong V, Graham E, Terry AV, Jr, Rodefer JS, Herbert B, Murray M, Porter R, Santarelli L, Lowe DA. RG3487, a novel nicotinic alpha7 receptor partial agonist, improves cognition and sensorimotor gating in rodents. J Pharmacol Exp Ther. 2011;336:242–53. doi: 10.1124/jpet.110.171892. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–32. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang HY, Li W, Benedetti NJ, Lee DH. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J Biol Chem. 2003;278:31547–53. doi: 10.1074/jbc.M212532200. [DOI] [PubMed] [Google Scholar]
- Zhang R, Xue G, Wang S, Zhang L, Shi C, Xie X. Novel object recognition as a facile behavior test for evaluating drug effects in AbetaPP/PS1 Alzheimer’s disease mouse model. J Alzheimers Dis. 2012;31:801–12. doi: 10.3233/JAD-2012-120151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.