

Structured Abstract
Objective
Lipid phosphate phosphatase 3 (LPP3), encoded by the PPAP2B gene, is an integral membrane enzyme that dephosphorylates, and thereby terminates, the G-protein-coupled receptor-mediated signaling actions of lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P). LPP3 is essential for normal vascular development in mice, and a common PPAP2B polymorphism is associated with increased risk of coronary artery disease in humans. Herein, we investigate the function of endothelial LPP3 to understand its role in development and human disease.
Approach and results
We developed mouse models with selective LPP3 deficiency in endothelial and hematopoietic cells. Tyrosine kinase Tek (Tie2) promoter-mediated inactivation of Ppap2b resulted in embryonic lethality due to vascular defects. LPP3 deficiency in adult mice, achieved using a tamoxifen-inducible Cre transgene under the control of the Tie2 promoter, enhanced local and systemic inflammatory responses. Endothelial, but not hematopoietic, cell LPP3-deficiency led to significant increases in vascular permeability at baseline, and enhanced sensitivity to inflammation-induced vascular leak. Endothelial barrier function was restored by pharmacological or genetic inhibition of either LPA production by the circulating lysophospholipase D autotaxin or of G-protein-coupled receptor-dependent LPA signaling.
Conclusions
Our results identify a role for the autotaxin/LPA-signaling nexus as a mediator of endothelial permeability in inflammation and demonstrate that LPP3 limits these effects. These findings have implications for therapeutic targets to maintain vascular barrier function in inflammatory states.
Keywords: hematopoietic cells, endothelial cells, vascular inflammation, vascular permeability, lipid phosphate phosphatase 3, autotaxin, lysophosphatidic acid
Introduction
Lipid phosphate phosphatases (LPPs) are integral membrane glycoproteins, originally identified as phosphatidic acid (PA) phosphatases, but subsequently shown to dephosphorylate a broader range of lipid substrates, including lysophosphatidic acid (LPA), ceramide-1-phosphate (C1P), sphingosine-1-phosphate (S1P), and diacylglycerol pyrophosphate (DGPP). 1 In mammals, the three enzymes LPP1, LPP2, and LPP3 are encoded by the PPAP2A, PPAP2C, and PPAP2B genes, respectively. 2, 3 LPPs localize to both the plasma membrane and intracellular membrane organelles, with a predicted topology of 6 transmembrane domains and an active site on the extracellular, or luminal surface, of the membrane. Evidence from cultured cells and model organisms identifies an important role for LPPs in dephosphorylating, and thereby inactivating, the G-protein-coupled receptor-mediated extracellular signaling effects of the bioactive lipids LPA and S1P. 4–6 While the 3 mammalian LPPs display essentially identical enzymatic activities in vitro and overlapping expression patterns in adult tissues, they are not functionally redundant during development. Loss of Ppap2c 7 and gene-trap inactivation of Ppap2a 8 in mice does not result in phenotypic alteration, although circulating LPA levels may be lower in the later animals. In contrast, global deletion of Ppap2b in mice results in embryonic lethality, largely due to defects in extraembryonic vascular development. Whether the requirement for LPP3 in vascular development reflects roles for LPA and/or S1P, or non-enzymatic functions of LPP3 such as regulation of Wnt signaling 9–12 or integrin interactions, is not presently known.
Recent genetic evidence has focused attention on the possibility that LPP3 may not only be required for vascular development, but may also mediate human atherosclerotic disease. Two concurrently published genome-wide association studies, identified polymorphisms in the final intron of PPAP2B that associate with increased risk for human coronary artery disease (CAD). In a GWAS meta-analysis involving more than 86,000 individuals, the PPAP2B risk allele independently predicted CAD (OR 1.17; P = 3.81 × 10−19), and lacked association with traditional risk factors such as hypertension, cholesterol, diabetes, obesity or smoking. At present, it is not clear if the risk-associated polymorphism alters LPP3 expression. To provide mechanistic insight into potential roles for LPP3 in the vasculature, we recently reported that targeted deletion of Ppap2b in murine smooth muscle cells enhances vascular inflammation and promotes the development of intimal hyperplasia. In this study, we investigate the consequences of LPP3 deficiency in endothelial and hematopoietic cells. Our findings indicate that LPP3 serves as an intrinsic negative regulator of vascular inflammation through mechanisms that are essential for maintenance of endothelial integrity and protection from inflammation-induced vascular leak. These findings may have broad implications for the development of atherosclerosis and also other disease processes that involved endothelial inflammatory responses.
Methods and Materials
Materials and Methods are available in the online-only Data Supplement.
Results
Tie2-Cre mediated deletion of Ppap2b results in embryonic lethality in mice
We previously reported that yolk sac vasculature fails to form in mouse embryos lacking LPP3.13 To explore cell-specific roles for LPP3 during mouse development, we bred Ppap2bfl/fl mice to transgenic mice expressing Cre recombinase under the control of the Tie2 promoter. Heterozygous Tie2-Ppap2bfl/+ mice were viable, fertile, and indistinguishable from their wild-type littermates. Mating of male Tie2-Ppap2bfl/+ with female Ppap2bfl/+ mice on either a mixed B6/129 or pure background failed to generate any Tie2-Ppap2bΔ animals postnatally, suggesting that the absence of LPP3 expression in a combination of endothelial and hematopoietic cells results in embryonic lethality. Analysis at E9.5 and E10.5 revealed a normal Mendelian distribution of genotypes, but no viable Tie2-Ppap2bΔ embryos were identified after E12.5. Tie2-Ppap2bΔ embryos share many features observed in embryos globally lacking Ppap2b expression, such as a delay in development compared to littermates, vascular defects represented by pale yolk sacs with accumulation of erythrocytes and signs of hemorrhage in the embryo proper. Defective chorio-allantoic fusion was observed in some embryos. In an embryo pair matched to 22 pair of somites (Figure 1), the Tie2-Ppap2bΔ embryo displayed hemorrhage in the tail region, smaller common atrial chamber, an abnormal aortic sac, outflow tract lumen collapsed, an abnormal swelling of the 3rd branchial-arch artery, open atrioventricular canal and irregular intersomitic vasculature. Many of these characteristics are consistent with loss of hemodynamics and vascular permeability. These vascular abnormalities, along with the lack of survival at birth in Tie2-Ppap2Δ mice, are consistent with previous observations that in E10.5 chimeric embryos formed by injection of Ppap2b−/− embryonic stem cells into Rosa26 blastocysts, wild-type endothelial cells predominately formed umbilical cord vessels without significant contribution from Ppap2b−/− cells.14 Together, these observations indicate that endothelial LPP3 is essential for normal vascular development.
Figure 1. LPP3 is required for early vascular development.

Morphological analysis of Ppap2bfl/fl (top right panel) and Tie2-Ppap2bΔ (top left panel) embryos with 22 pairs of somites immunostained for PECAM1 and cleared with BB:BA. The Tie2-Ppap2bΔ embryo displays a smaller common atrial chamber (A) and open atrioventricular canal (AV). In addition, abnormalities of the aortic sac (as) and outflow tract (*), expansion of aorta (Ao) at the level of the 2nd branchial arch artery (baa), swelling of the 3rd bba and irregular intersomitic vasculature (ISV) were noted. Histologycal analysis (middle and bottom panels) in transverse sections of Ppap2bfl/fl (right panels) and Tie2-Ppap2bΔ (left panels) E9.5 embryos at the level of the AV canal. Note the reduction of mesenchymal (m) cells in endocardial cushions (EC) leading to a completely or partially open AV canal (arrows). LA, left component of atrial chamber; LV, left ventricle; RA, right component of atrial chamber; RV, right ventricle. Scale bar= 100 μm
Inducible inactivation of Ppap2b in adult mice
To generate a Ppap2b null allele in vascular endothelium of adult animals, Ppap2bfl/fl mice were crossed with transgenic mice expressing a recombinant estrogen receptor-Cre fusion protein under the control of the Tie2 promoter (ERT2-Cre) to generate ERT2-Ppap2bΔ mice. The activity of the promoter was regulated by administering the estrogen antagonist tamoxifen. Tamoxifen-treated Ppap2bfl/fl and ERT2-Ppap2bΔ mice were compared experimentally. Absence of LPP3 expression in ERT2-Ppap2bΔ mice was confirmed by immunostaining of endothelial cells in newly formed vessels in Matrigel implants supplemented with basic fibroblast growth factor (Figure 2A). Neovascularization was quantified with FITC-dextran administered intravenously. Vessel formation in the Matrigel plugs was scored by size (small [<10μm], medium [10–20μm], and large [>20μm]) (Figures 2B and 2C). In comparison to vessels that formed in Matrigel implanted in Ppap2bfl/fl mice, those in the ERT2-Ppap2bΔ mice were smaller and fewer vessels formed in the center of the plugs (P<0.001; Figure 2C), which is in keeping with previous reports that antibodies to LPP3 inhibited capillary morphogenesis in vitro.15
Figure 2. Tamoxifen inducible Tie2-Cre-mediated inactivation of Ppap2b results in loss of endothelial LPP3 and impairs angiogenesis.

(A) Matrigel plugs supplemented with bFGF were implanted in Ppap2bfl/fl (designated as fl/fl) and ERT2-Ppap2bΔ (designated as ERT2-Δ) mice treated with tamoxifen. Seven days later, animals were injected with FITC-dextran to visualize blood vessels. Sections of the explanted plugs were stained with antibodies to LPP3 (red) and PECAM (blue) to identify endothelial cells. (Mag 40X, Bar denotes 100μm). Matrigel sections from ERT2-Δ mice lacked endothelial LPP3 staining. (B) Representative whole mount images of FITC-dextran in explanted Matrigel plug from Ppap2bfl/fl (fl/fl) and ERT2-Ppap2bΔ (ERT2-Δ) mice taken at a lower magnification (10X) demonstrate that a more extensive vascular network forms in the fl/fl control mice. (C) Quantification of vessel size in the Matrigel implants from Ppap2bfl/fl (fl/fl) and ERT2-Ppap2bΔ (ERT2-Δ) mice demonstrates that smaller vessels form in the absence of LPP3. * P < 0.0001 by Fisher’s exact test.
Ppap2b mRNA levels were approximately 2-fold lower in lungs of ERT2-Ppap2bΔ mice (Figure 3A). No difference was observed in Ppap2a or Ppap2c mRNA levels, encoding LPP1 and LPP3 respectively. Immunoblot analysis of lung tissue indicated that LPP3 levels were reduced in ERT2-Ppap2bΔ lungs (Figure 3B), as was lipid phosphatase activity measured in LPP3 immunoprecipitates (Figure 3C). Immunohistochemical analysis of lung tissue indicated that LPP3 was not present in endothelial cells (Figure 3D). The remaining LPP3 expression and activity in lungs of ERT2-Ppap2bΔ mice is likely from epithelial, alveolar, or other cells. No difference was observed in complete blood counts (CBC) of the Ppap2bfl/fl and ERT2-Ppap2bΔ mice (Table 1). Likewise, heart rates were similar in Ppap2bfl/fl (645 ± 45 bpm; n = 16) and ERT2-Ppap2bΔ mice (630 ± 60 bpm; n = 16). Interestingly, systolic blood pressure was significantly lower in ERT2-Ppap2bΔ mice (92 ± 8 mmHg; n = 16) compared to Ppap2b fl/fl mice (100 ± 7 mmHg; n = 16; P<0.001).
Figure 3. Tamoxifen inducible Tie2-Cre-mediated inactivation of Ppap2b lowers LPP3 expression in lung.

(A) Ppap2b mRNA expression was measured in lung tissue from Ppap2bfl/fl (fl/fl) and ERT2-Ppap2bΔ (ERT2-Δ) mice and reported relative to values in (fl/fl) lung (mean ± SD from n = 3 animals per genotype). (B) Immunoblot analysis of LPP3 expression in lung tissue with actin used as a loading control. Lipid phosphate phosphatase expression was normalized to actin staining (n = 3 animals) and presented as mean ± SD in arbitrary units in which the density of LPP3 in the Ppap2bfl/fl (fl/fl) samples was set to 1. (C) Lipid phosphate phosphatase activity determined in LPP3 immunoprecipiates from lung (n = 3 animals per genotype). Results are reported as pmol/min/μg protein (mean ± SD; n = 3 mice per genotype). (D) Representative images of immunostaining for PECAM (blue) and LPP3 (red) in lung vessels. Whereas endothelial PECAM and LPP3 expression overlap in Ppap2bfl/fl (fl/fl) tissue (magenta color), in ERT2-Ppap2bΔ (ERT2-Δ) mice, the residual LPP3 staining is not in endothelial cells. Scale bar= 100 μm. # P<0.05 by t-test versus control.
Table 1.
Blood counts in adult mice lacking LPP3 in hematopoietic and endothelial cells
| Genotype | N | WBC | NE | LY | MO | Hb | HCT | Plt | MPV |
|---|---|---|---|---|---|---|---|---|---|
| Ppap2bfl/fl | 15 | 6.94±1.95 | 1.91±0.81 | 4.49±1.22 | 0.35±0.13 | 13.11±2.27 | 39.15±6.33 | 884±288 | 4.78±0.35 |
| TieERT2-Ppap2bΔ | 18 | 6.44±1.76 | 1.73±0.89 | 4.79±1.29 | 0.37±0.18 | 12.11±2.15 | 40.38±4.32 | 809±273 | 4.86±0.69 |
Values mean ± SD
WBC = white blood cell count (x103/μl)
NE = neutrophil (103/μl)
LY = lymphocyte (103/μl)
MO = monocyte (103/μl)
Hb = hemoglobin (mg/dl)
HCT = hematocrit (%)
Plt = platelet count (106/μl)
MPV = mean platelet volume (nL)
Enhanced inflammatory responses in mice lacking LPP3 in hematopoietic and endothelial cells
Because the Tie2 promoter is also active in some hematopoietic cells, we investigated leukocyte LPP3 expression in the ERT2-Ppap2bΔ mice. We previously reported that neutrophils isolated from peripheral blood robustly express LPP3.16 At 4 hours following administration of thioglycolate i.p., significantly more leukocytes infiltrated the peritoneum of ERT2-Ppap2bΔ mice (Figure 4A); these infiltrating cells lacked LPP3 (Figure 4B). To determine if the ERT2-Ppap2bΔ mice display enhanced inflammatory responses to other challenges, endotoxin (LPS; 2 mg/kg i.p.) was administered to the animals. As expected, lipopolysaccharide (LPS) exposure elevated plasma inflammatory cytokines above vehicle treatment (Figure 4C and D). In ERT2-Ppap2bΔ mice, LPS-induced expression of IL-6 (Figure 4C) and KC (Figure 4D), were 3.3 ± 0.5-fold and 1.9 ± 0.6-fold higher, respectively, than in LPS-treated Ppap2bfl/fl mice. Analysis of plasma by cytokine antibody array confirmed the elevation in IL-6 and KC (Figure 4E), and indicated that MIP-2 and RANTES were also higher in ERT2-Ppap2bΔ plasma 4 hours after LPS administration, whereas G-CSF, sICAM-1, M-CSF, and CXCL9 appeared lower than in Ppap2bfl/fl mice. We investigate the role of monocytes in the LPS response by pretreating animals with liposomal-clondronate to deplete >90% of circulating monocytes. Plasma IL-6 levels in ERT2-Ppap2bΔ mice were not affected by monocyte deletion prior to LPS administration (8730 ± 1481 versus 8840 ± 433 in the absence or presence of clondronate).
Figure 4. Absence of LPP3 enhances inflammation.

(A) Leukocyte accumulation in the peritoneum 4 hours after i.p. thioglycolate challenge (n = 3 animals per genotype). Results are normalized to values obtained in Ppap2bfl/fl (fl/fl) mice. # P<0.05 by t-test. (B) Immunoblot analysis of LPP3 expression in neutrophils recovered from the peritoneum 4 hours after i.p. thioglycolate. LPP3 expression was normalized to CD18 staining (n = 3 animals) and plotted as mean ± SD in arbitrary units in which the density of LPP3 in the Ppap2bfl/fl (fl/fl) samples was set to 1. The results demonstrate reduced expression in leukocytes isolated from ERT2-Ppap2bΔ (ERT2-Δ) mice. # P<0.05 by t-test. (C) Plasma levels of IL6 and (D) KC at 4 hours after LPS treatment in Ppap2bfl/fl (fl/fl) and ERT2-Ppap2bΔ (ERT2-Δ) mice. # P<0.05 by ANOVA. (E) Cytokine expression by antibody blot array analysis of plasma from Ppap2bfl/fl (fl/fl) and ERT2-Ppap2bΔ (ERT2-Δ) mice (solid rectangle box marks IL6 reactivity and dashed rectangle box KC).
Disruption in barrier function in mice lacking endothelial LPP3 is autotaxin and LPA-dependent
Inflammation is characteristically accompanied by an increase in vascular endothelial permeability. We therefore investigated extravasation of EBD into lung as a measure of the integrity of the endothelial barrier. The absence of LPP3 in ERT2-Ppap2bΔ mice resulted in a 2.2 ± 0.5-fold increase in basal vascular permeability, as measured by EBD accumulation in the lungs (Figure 5A). LPS increased vascular permeability in both Ppap2bfl/fl and ERT2-Ppap2bΔ mice, the later experiencing significantly more vessel leak (Figure 5A). To confirm that endothelial (but not bone marrow) cell-derived LPP3 was required for maintenance of the endothelial barrier, chimeric mice were created by bone marrow transplantation and then treated with tamoxifen. The vascular barrier defect followed the genotype of the recipient mice (lacking LPP3 in vessels) and not the genotype of the transplanted marrow (Figure 5B). These survival doses of LPS did not elicit significant inflammatory cell infiltration in lungs of either Ppap2bfl/fl or ERT2-Ppap2bΔ mice and no differences in macrophage accumulation in lung was observed between the two genotypes (Figure 5C).
Figure 5. Absence of endothelial LPP3 promotes vascular leak.
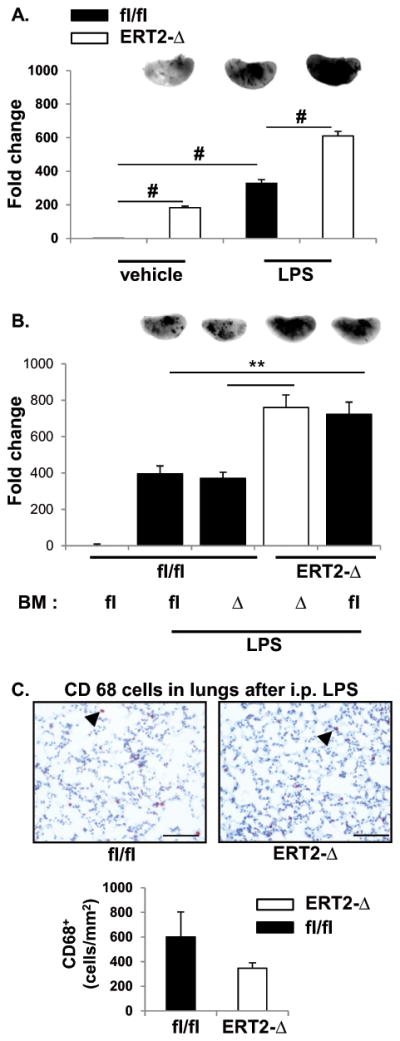
(A) LPS-induced lung endothelial permeability and accumulation of extravascular protein was measured by accumulation of EBD in lungs 2 hours after vehicle or LPS administration to Ppap2bfl/fl (fl/fl) or ERT2-Ppap2bΔ (ERT2-Δ) mice. The amount of EBD in left lung was measured by near-infrared imaging (LiCor) and is reported as mean ± SD. Representative images for each treatment and genotype are included at the top of the bar graphs. More EBD accumulates in the lung of ERT2-Δ mice at baseline (vehicle) and after LPS challenge. # P<0.05 by ANOVA. (B) Chimeric mice were created by bone marrow (BM) transplantation between fl/fl or ERT2-Δ animals, and the mice then treated with tamoxifen. Protein leak after vehicle or LPS treatment was measured as described above. The permeability defect segregated with absence of LPP3 in recipient mice (i.e., blood vessels) and not with the genotype of BM cells. # P<0.05 by ANOVA. (C) CD68-positive macrophage staining (arrowhead) in lung tissue 6 hours after LPS. The number of macrophages was scored per mm2 and graphed as mean ± SD. At the dose of LPS administered (2 mg/kg), the permeability defect in endothelial cells did not result in accumulation of macrophages in lung tissue as visualized by CD68 staining.
Since LPP3 may terminate vascular endothelial cell signaling responses to the bioactive lipids S1P and LPA, a lack of LPP3 could result in enhanced signaling actions of either bioactive mediator. Sphingosine-1-phosphate is known to maintain endothelial barrier function so we hypothesized that lower endothelial LPP3 expression might increase S1P signaling which, in turn, could downregulate S1P receptors, resulting in an increase in endothelial permeability, as has been reported with long-term administration of S1P agonists.17 To determine if ERT2-Ppap2bΔ mice demonstrated impairments in the response to S1P, the ability of S1P to promote endothelial barrier function was examined. In both Ppap2bfl/fl and ERT2-Ppap2bΔ mice, S1P enhanced barrier function following LPS administration, suggesting that the defect in the ERT2-Ppap2bΔ mice was not due to downregulation of S1P receptor signaling (Supplemental Figure I). In keeping with these observations, no difference in lymphocyte expression of S1PR1 between Ppap2bfl/fl and ERT2-Ppap2bΔ mice was observed by flow cytometry.
We next examined whether increases in LPA signaling could account for vascular leak in the ERT2-Ppap2bΔ mice, as LPA has been reported to promote endothelial permeability in culture models.18, 19 Two strategies were used to target LPA signaling. First, the potent autotaxin inhibitor PF8380 20 was administered to mice to reduce the generation of extracellular, bioactive LPA by autotaxin, which is a secreted lysophospholipase D responsible for production of bioactive LPA. In keeping with the ability to inhibit autotaxin, PF8380 decreased plasma LPA levels (control 248 ± 29, PF8380 173 ± 55 nM; p=0.0154 paired t-test). PF8380 also reduced vascular permeability in both Ppap2bfl/fl and ERT2-Ppap2bΔ in response to LPS (Figure 6A). Similarly, administration of the pan-LPA receptor antagonist, BrP-LPA, resulted in a 2-fold reduction in LPS-promoted permeability in ERT2-Ppap2bΔ mice at baseline and in response to LPS (Figure 6B).
Figure 6. Attenuating LPA production or signaling promotes endothelial barrier function.

The autotaxin inhibitor PF8380 (1mg/kg) (A) or the LPA receptor pan-antagonist (5mg/kg) BrP-LPA (B) were administered 30 minutes prior to LPS, and protein leak in Ppap2bfl/fl (fl/fl) or ERT2-Ppap2bΔ (ERT2-Δ) lungs (n = 3/group/genotype) was measured with EBD as described. Both PF8380 and BrP-LPA reduced EBD accumulation in lung. # P<0.05 by ANOVA. (C) To verify that autotaxin was involved in LPS-mediated permeability, mice with reduced plasma autotaxin were generated using the Mx1-Cre transgene that is activated by administration of pI-pC. Plasma autotaxin (ATX) levels were lower in Mx1-Enpp2Δ mice (left) as was EBD accumulation in lungs after LPS (right). # P<0.05 by t-test. (D) The LPA receptor pan-antagonist BrP-LPA was administered to ventilated/perfused lungs prior to infusion of LPS via the pulmonary artery and protected from protein leak Ppap2bfl/fl (fl/fl) or ERT2-Ppap2bΔ (ERT2-Δ) lungs. # P<0.05 by ANOVA.
To complement these pharmacological studies of the role of LPA in regulation of endothelial permeability, we used mice with genetic inactivation of the LPA-generating enzyme autotaxin or G-protein coupled LPA receptors. Global deficiency of autotaxin (encoded by the Enpp2 gene) is embryonically lethal in mice. We established Enpp2fl/fl mice harboring the Cre recombinase under the control of the interferon-inducible Mx-1 promoter and stimulated expression of the Cre transgene with pI-pC (Figure 6C). The Mx1-Enpp2Δ mice displayed reduced plasma levels of autotaxin and were protected from endothelial permeability after LPS challenge (Figure 6C). Consistent with a role for autotaxin-generated LPA in LPS-induced vascular permeability, mice lacking LPA receptors 1 and 2 (LPA1, LPA2) or LPA receptor 4 (LPA4), demonstrated less EBD accumulation in lungs after LPS than did wild-type controls (Supplemental Figure II). Finally, we directly tested the ability of LPA to stimulate vascular permeability using the Miles assay. Intradermal administration of LPA dose-dependently, increased EBD accumulation in the skin of wild-type mice but not of LPA1−/−, LPA2−/−, or LPA4−/− mice, indicating that these receptors mediate LPA-induced vascular leak (Supplemental Figure II).
The observation that the barrier defect tracked with the absence of LPP3 in vascular (but not marrow) cells in chimeric mice suggested a critical role for endothelial LPP3. We used ventilated/perfused lungs to probe the role of endothelial LPP3 in inflammation-induced vascular permeability. In buffer-perfused lungs, LPS administered via the pulmonary artery increased EBD accumulation in the lungs of Ppap2bfl/fl mice and, to a greater extent, ERT2-Ppap2bΔ mice (Figure 6D). The pan-LPA receptor antagonist BrP-LPA attenuated endothelial permeability in both Ppap2bfl/fl and ERT2-Ppap2bΔ mice (Figure 6B).
The results described above establish a role for LPP3 in maintaining the integrity of the endothelial barrier that we hypothesize is due to the enzyme’s ability to terminate the barrier-enhancing actions of autotaxin-derived LPA that are mediated by G-protein-coupled LPA receptors. Therefore, we investigated whether plasma LPA levels were altered in the absence of LPP3, and observed no difference in circulating LPA levels in Ppap2bfl/fl and ERT2-Ppap2bΔ mice. We also measured the rate of elimination of exogenously-administered LPA and S1P. The lack of endothelial LPP3 in adult mice did not alter the rapid rate of elimination of single-bolus doses of either C17-LPA or C17-S1P from blood (Supplemental Figure III). Taken together, our findings suggest that endothelial LPP3 does not play a role in determining plasma LPA levels, or in the rapid elimination of LPA and S1P from plasma, but may function as a localized regulator of signaling through the autotaxin-LPA nexus.
Discussion
The endothelium forms a highly selective barrier between blood and tissue that is characteristically compromised during inflammation. We report that Tie2 promoter-driven deficiency of LPP3 impairs embryonic vascular development, resulting in embryonic lethality, and disrupts normal endothelial barrier function postnatally. While the Tie2 promoter strategy employed will result in deficiency of LPP3 in endothelial and some hematopoietic cells, the onset of the embryonic phenotype suggests a fundamental role for the enzyme in endothelial cells during early development. Endothelial and/or hematopoietic LPP3 deficiency also heightens inflammatory responses in adult animals. These findings imply that LPP3 expression is essential for normal prenatal vascular development and, in adult mice, LPP3 normally functions to maintain vascular integrity and to attenuate inflammation.
While the mechanistic basis of the requirement for LPP3 during vascular development remains to be established, our results suggest that in adult mice a major function of LPP3 is to attenuate LPA-mediated increases in permeability of the vascular endothelium. In support of this theory, we found that genetic and pharmacologic approaches to attenuate LPA-signaling or reduce LPA production by autotaxin, preserved endothelial barrier function in the setting of an inflammatory challenge. Moreover, LPA itself promoted protein extravasation from the vasculature in an LPA receptor-dependent manner. These observations are in keeping with reports in cell culture systems where LPA was found to increase endothelial permeability by stimulating Rho-mediated actomyosin contractility.21–23 Following bleomycin-induced lung injury, mice lacking LPA1 display reduced pulmonary fibrosis and less vascular leak,24 which is in agreement with our finding of a role for LPA1 and LPA4 receptors in mediating protein extravasation in lung after LPS challenge. Because genetic deficiency of LPP3 in adult mice was not associated with decreases in either circulating LPA levels or the rate of elimination of exogenously supplied LPA from the circulation, we consider it likely that LPP3 functions primarily to regulate vascular endothelial cell-localized LPA production and signaling pathways, leading to changes in vascular permeability. Taken together, these findings suggest that autotaxin-LPA signaling may elicit a loss of vascular integrity by preventing tight-junction formation, and that either autotaxin inhibitors or LPA-receptor antagonists might be useful in treatment of endothelial dysfunction and tissue injury in inflammation.
Although we did not establish the molecular mechanism underlying embryonic lethality in Tie2-Ppap2bΔ mice, emerging evidence supports a role for LPA in directing vascular development. Global deletion of autotaxin results in early embryonic lethality in mice, and knockdown of zebrafish autotaxin causes aberrant connections to form between segmental arteries that sprout from the dorsal aorta.25 These findings indicate that tight, spatially-restricted regulation of LPA metabolism and signaling is important for proper cell migration during embryonic development. Indeed, we recently reported that autotaxin localizes to the leading edge of migrating cells through integrin-mediated recruitment and directs the path of migration. 26 Based on these observations, we hypothesize that it may be possible that at least part of the role of LPP3 is to regulate localized LPA levels during development. In support, mammalian LPP3 can complement mutations in Drosophila LPPs encoded by the Wun and Wun2 genes, which guide migrating germ cells in the developing embryo through mechanisms that appear to involve localized inactivation of an attractive lipid signal. Experiments with ectopic expression of Wunens indicate that they regulate a diffusible lipid signal in a cell contact-independent manner, with an effective range of approximately 33 μm. 27 These observations are all consistent with ours and support the contention that LPA or another diffusible lipid signal could account for the requirement for endothelial LPP3 during development. In addition to LPA, S1P is a possible candidate mediator. Sphingosine-1-phosphate signaling through S1PR1 is also essential for blood vessel formation28; although the requirements for S1P and S1PR1 appear to be fundamentally distinct from those of LPP3. Additionally, LPP3 also has non-enzymatic functions that may be mediated by integrin binding or β-catenin signaling,12, 29, 30 which could be required during development.
In addition to the endothelial defects in ERT2-Ppap2bΔ mice, we observed heightened inflammatory responses in leukocyte accumulation in peritonitis and in cytokine responses to endotoxin, indicating that endothelial and/or hematopoietic LPP3 functions to limit systemic inflammation. These findings are similar to our observations that LPP3 attenuates inflammation following vascular injury on vascular smooth muscle cells. LPA regulates inflammation by stimulating the release of cytokines31–34 and inflammatory mediators that modulate movement of inflammatory cells. Thus, as with its role in regulation of endothelial permeability, the anti-inflammatory effects of LPP3 may stem from its ability to limit localized LPA signaling. Interestingly, although vascular permeability changes were observed with endotoxin, heightened leukocyte accumulation in lung was not demonstrated in ERT2-Ppap2bΔ mice. We do not have an explanation for why peritoneal but not pulmonary white cell accumulation was affected by LPP3, although the differences could be due to the nature of the inflammatory challenge or vascular bed specific mechanisms or both. The ability of LPP3 to modulate vascular inflammatory responses may provide a mechanistic link between the risk of CAD associated with a polymorphism in PPAP2B. Our observations indicate that genetic or environmental factors that downregulate LPP3 levels would predispose individuals to heightened vascular inflammation and permeability, and thereby might exacerbate atherosclerosis.
Research from our group and others indicates that circulating LPA levels are dynamically regulated with steady-state levels maintained by a balance between autotaxin-mediated LPA generation and less well-defined mechanisms, which result in rapid elimination of circulating LPA. 35, 36 Elimination of intravenously supplied LPA and S1P was unaffected in the ERT2-Ppap2bΔ mice, suggesting that either endothelial LPP3 is not responsible for the bulk of their clearance from plasma, or that different cells are involved. These observations are in keeping with our recent findings of elimination of LPA37. At the present time the mechanism(s) responsible for rapid removal of LPA and S1P from circulation are not known, although clearance by the liver is involved37.
In summary, our work substantiates and extends observations that the bioactive lipid LPA is an important regulator of systemic inflammation and vascular permeability and defines a previously unappreciated role for LPP3 as a negative regulator of these processes. Our observations have broad relevance for understanding the link between a common heritable genetic variant of the the PPAP2B gene that encodes LPP3 and human cardiovascular disease risk, and suggest that pharmacological approaches to limit LPA production and signaling would be of benefit for cardiovascular disease prophylaxis and also, perhaps, for the acute treatment of systemic inflammation and vascular permeability associated with sepsis.
Supplementary Material
Significance.
Lipid phosphate phosphatase 3 (LPP3), encoded by the PPAP2B gene, is essential for normal vascular development in mice, and a common polymorphism is associated with increased risk of coronary artery disease in humans. We identify a role for LPP3 and the autotaxin/LPA-signaling nexus in regulating endothelial permeability and inflammatory responses. These findings have broad implications for therapeutic targets to maintain vascular barrier function in inflammatory states and may help explain a role for LPP3 in the development of atherosclerosis and also other disease processes that involved endothelial inflammatory responses.
Acknowledgments
We thank University of Kentucky laboratory assistant Kelsey Johnson, for animal husbandry. M.P.: performed experiments, analyzed data, drafted manuscript; A.S.: performed experiments, analyzed data, drafted portions of the manuscript; S.M.: performed experiments, analyzed data, provided input on the manuscript; J.W.: performed experiments, analyzed data, provided input on the manuscript; P.P.: performed experiments, analyzed data, provided input on the manuscript; A.J.M.: Designed experiments, analyzed and interpreted data, provided reagents, wrote manuscript; D.E.A.: Designed and performed experiments, analyzed and interpreted data, wrote portions of manuscript; S.S.S.: Designed experiments, analyzed and interpreted data, provided reagents, wrote manuscript.
Source of Funding
This project was supported by a grant from the Heart Lung and Blood Institute (R01HL078663), from the General Medical Sciences Institute (GM050388), the National Center for Research Resources (P20RR021954), by an IDeA award from the National Institute of General Medical Sciences (P20GM103527) of the National Institutes of Health and CONACyT (165897); pilot project funding from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health through Grant UL1TR000117 and UL1RR033173; a predoctoral fellowship (to A.S), Beginning Grant-in-Aid (0950118G); and a Scientist Development Grant (10SDG4190036) from the American Heart Association. This material is also based on work supported in part by resources at the Lexington VA Medical Center. A portion of this work was presented in abstract form at the American Heart Association Scientific Sessions in 2009.
Abbreviations
- BB:BA
Benzyl benzoate:benzyl alcohol 2:1
- C1P
Ceramide-1-phosphate
- CAD
Coronary artery disease
- DGPP
Diacylglycerol pyrophosphate
- ER
Estrogen receptor
- EBD
Evans Blue Dye
- GWAS
Genome-wide association studies
- LPA
Lysophosphatidic acid
- LPP
Lipid phosphate phosphatase
- LPP3
Lipid phosphate phosphatase 3
- LPS
Lipopolysaccharide
- PA
Phosphatidic acid
- S1P
Sphingosine-1-phosphate
- Tie2
Tyrosine kinase Tek
Footnotes
Disclosures
All authors agree to the content contained in this manuscript and the use of their names as authors. Also, all authors declare there are no competing financial interests to disclose and that the manuscript meets the Journal’s requirements.
LITERATURE CITED
- 1.Sciorra VA, Morris AJ. Roles for lipid phosphate phosphatases in regulation of cellular signaling. Biochim Biophys Acta. 2002;1582:45–51. doi: 10.1016/s1388-1981(02)00136-1. [DOI] [PubMed] [Google Scholar]
- 2.Kai M, Wada I, Imai S, Sakane F, Kanoh H. Cloning and characterization of two human isozymes of mg2+-independent phosphatidic acid phosphatase. J Biol Chem. 1997;272:24572–24578. doi: 10.1074/jbc.272.39.24572. [DOI] [PubMed] [Google Scholar]
- 3.Roberts R, Sciorra VA, Morris AJ. Human type 2 phosphatidic acid phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface activity of the 2a isoform. J Biol Chem. 1998;273:22059–22067. doi: 10.1074/jbc.273.34.22059. [DOI] [PubMed] [Google Scholar]
- 4.Sigal YJ, McDermott MI, Morris AJ. Integral membrane lipid phosphatases/phosphotransferases: Common structure and diverse functions. Biochem J. 2005;387:281–293. doi: 10.1042/BJ20041771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pyne S, Long JS, Ktistakis NT, Pyne NJ. Lipid phosphate phosphatases and lipid phosphate signalling. Biochem Soc Trans. 2005;33:1370–1374. doi: 10.1042/BST0331370. [DOI] [PubMed] [Google Scholar]
- 6.Brindley DN, Pilquil C. Lipid phosphate phosphatases and signaling. J Lipid Res. 2009;50 (Suppl):S225–230. doi: 10.1194/jlr.R800055-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang N, Sundberg JP, Gridley T. Mice mutant for ppap2c, a homolog of the germ cell migration regulator wunen, are viable and fertile. Genesis. 2000;27:137–140. doi: 10.1002/1526-968x(200008)27:4<137::aid-gene10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN, Lynch KR. Lipid phosphate phosphohydrolase type 1 (lpp1) degrades extracellular lysophosphatidic acid in vivo. Biochem J. 2009;419:611–618. doi: 10.1042/BJ20081888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renault AD, Sigal YJ, Morris AJ, Lehmann R. Soma-germ line competition for lipid phosphate uptake regulates germ cell migration and survival. Science. 2004;305:1963–1966. doi: 10.1126/science.1102421. [DOI] [PubMed] [Google Scholar]
- 10.Sano H, Renault AD, Lehmann R. Control of lateral migration and germ cell elimination by the drosophila melanogaster lipid phosphate phosphatases wunen and wunen 2. The Journal of cell biology. 2005;171:675–683. doi: 10.1083/jcb.200506038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humtsoe JO, Feng S, Thakker GD, Yang J, Hong J, Wary KK. Regulation of cell-cell interactions by phosphatidic acid phosphatase 2b/vcip. The EMBO journal. 2003;22:1539–1554. doi: 10.1093/emboj/cdg165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humtsoe JO, Bowling RA, Jr, Feng S, Wary KK. Murine lipid phosphate phosphohydrolase-3 acts as a cell-associated integrin ligand. Biochemical and biophysical research communications. 2005;335:906–919. doi: 10.1016/j.bbrc.2005.07.157. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Juarez A, Morales-Lazaro S, Sanchez-Sanchez R, Sunkara M, Lomeli H, Velasco I, Morris AJ, Escalante-Alcalde D. Expression of lpp3 in bergmann glia is required for proper cerebellar sphingosine-1-phosphate metabolism/signaling and development. Glia. 2011;59:577–589. doi: 10.1002/glia.21126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escalante-Alcalde D, Hernandez L, Le Stunff H, Maeda R, Lee H-S, Gang-Cheng, Sciorra VA, Daar I, Spiegel S, Morris AJ, Stewart CL. The lipid phosphatase lpp3 regulates extra-embryonic vasculogenesis and axis patterning. Development. 2003;130:4623–4637. doi: 10.1242/dev.00635. [DOI] [PubMed] [Google Scholar]
- 15.Wary KK, Humtsoe JO. Anti-lipid phosphate phosphohydrolase-3 (lpp3) antibody inhibits bfgf- and vegf-induced capillary morphogenesis of endothelial cells. Cell communication and signaling : CCS. 2005;3:9. doi: 10.1186/1478-811X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panchatcharam M, Miriyala S, Salous A, Wheeler J, Dong A, Mueller P, Sunkara M, Escalante-Alcalde D, Morris AJ, Smyth SS. Lipid phosphate phosphatase 3 negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:52–59. doi: 10.1161/ATVBAHA.112.300527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1–phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. American Journal of Respiratory Cell and Molecular Biology. 2010;43:662–673. doi: 10.1165/rcmb.2009-0345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vermeer MA, van Hinsbergh VWM Amerongen GPvN. Role of rhoa and rho kinase in lysophosphatidic acid–induced endothelial barrier dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:e127–e133. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- 19.Bernas MJ, Cardoso FL, Daley SK, Weinand ME, Campos AR, Ferreira AJG, Hoying JB, Witte MH, Brites D, Persidsky Y, Ramirez SH, Brito MA. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protocols. 2010;5:1265–1272. doi: 10.1038/nprot.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gierse J, Thorarensen A, Beltey K, Bradshaw-Pierce E, Cortes-Burgos L, Hall T, Johnston A, Murphy M, Nemirovskiy O, Ogawa S, Pegg L, Pelc M, Prinsen M, Schnute M, Wendling J, Wene S, Weinberg R, Wittwer A, Zweifel B, Masferrer J. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J Pharmacol Exp Ther. 2010;334:310–317. doi: 10.1124/jpet.110.165845. [DOI] [PubMed] [Google Scholar]
- 21.Neidlinger NA, Larkin SK, Bhagat A, Victorino GP, Kuypers FA. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase a2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006;281:775–781. doi: 10.1074/jbc.M505790200. [DOI] [PubMed] [Google Scholar]
- 22.Sarker MH, Hu D-E, Fraser PA. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation. 2010;17:39–46. doi: 10.1111/j.1549-8719.2010.00001.x. [DOI] [PubMed] [Google Scholar]
- 23.Gan N, Yin F, Peng J, Wang WD. effect of lysophosphatidic acid increase the permeability of blood-brain barrier model. Zhonghua Yi Xue Za Zhi. 2008;88:416–418. [PubMed] [Google Scholar]
- 24.Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND, Hart WK, Pardo A, Blackwell TS, Xu Y, Chun J, Luster AD. The lysophosphatidic acid receptor lpa1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14:45–54. doi: 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- 25.Yukiura H, Hama K, Nakanaga K, Tanaka M, Asaoka Y, Okudaira S, Arima N, Inoue A, Hashimoto T, Arai H, Kawahara A, Nishina H, Aoki J. Autotaxin regulates vascular development via multiple lysophosphatidic acid (lpa) receptors in zebrafish. Journal of Biological Chemistry. 2011;286:43972–43983. doi: 10.1074/jbc.M111.301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, Vander Kooi C, Shah P, Charnigo R, Huang C, Smyth SS, Morris AJ. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2013 doi: 10.1096/fj.13-232868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee A, Neher RA, Renault AD. Quantifying the range of a lipid phosphate signal in vivo. J Cell Sci. 2013;126:5453–5464. doi: 10.1242/jcs.136176. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Wada R, Yamashita T, Mi Y, Deng C-X, Hobson JP, Rosenfeldt HM, Nava VE, Chae S-S, Lee M-J, Liu CH, Hla T, Spiegel S, Proia RL. Edg-1, the g protein–coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. The Journal of Clinical Investigation. 2000;106:951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chatterjee I, Humtsoe JO, Kohler EE, Sorio C, Wary KK. Lipid phosphate phosphatase-3 regulates tumor growth via beta-catenin and cyclin-d1 signaling. Mol Cancer. 2011;10:51. doi: 10.1186/1476-4598-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humtsoe JO, Liu M, Malik AB, Wary KK. Lipid phosphate phosphatase 3 stabilization of beta-catenin induces endothelial cell migration and formation of branching point structures. Mol Cell Biol. 2010;30:1593–1606. doi: 10.1128/MCB.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, He D, Zhao J, Wang L, Leff AR, Spannhake EW, Georas S, Natarajan V. Lysophosphatidic acid induces interleukin-13 (il-13) receptor alpha2 expression and inhibits il-13 signaling in primary human bronchial epithelial cells. J Biol Chem. 2007;282:10172–10179. doi: 10.1074/jbc.M611210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. Regulation of lysophosphatidic acid-induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase cdelta, lyn kinase, and matrix metalloproteinases. J Biol Chem. 2006;281:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Natarajan V. Lysophosphatidic acid signaling in airway epithelium: Role in airway inflammation and remodeling. Cell Signal. 2009;21:367–377. doi: 10.1016/j.cellsig.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saatian B, Zhao Y, He D, Georas SN, Watkins T, Spannhake EW, Natarajan V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 mapk and jnk in human bronchial epithelial cells. Biochem J. 2006;393:657–668. doi: 10.1042/BJ20050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hausmann J, Kamtekar S, Christodoulou E, Day JE, Wu T, Fulkerson Z, Albers HMHG, van Meeteren LA, Houben AJS, van Zeijl L, Jansen S, Andries M, Hall T, Pegg LE, Benson TE, Kasiem M, Harlos K, Kooi CWV, Smyth SS, Ovaa H, Bollen M, Morris AJ, Moolenaar WH, Perrakis A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat Struct Mol Biol. 2011;18:198–204. doi: 10.1038/nsmb.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. Journal of Biological Chemistry. 2011;286:34654–34663. doi: 10.1074/jbc.M111.276725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salous AK, Panchatcharam M, Sunkara M, Mueller P, Dong A, Wang Y, Graf GA, Smyth SS, Morris AJ. Mechanism of rapid elimination of lysophosphatidic acid and related lipids from the circulation of mice. J Lipid Res. 2013;54:2775–2784. doi: 10.1194/jlr.M039685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.