

Abstract
A convenient and efficient (one-step) oxidation is reported of commercially available tosylmethylisocyanide (TOSMIC) to form tosylmethylisocyanate, making this highly reactive bifunctional molecule a readily available synthetic reagent. Besides engaging in nucleophilic addition reactions with alcohols, amines and thiols, tosylmethylisocyanate also reacts with carboxylic acids to form tosylmethylamides, which undergo substitution reactions in the presence of organocopper and organomagnesium reagents.
Keywords: Isonitrile-based Multicomponent reactions, Tosylmethylisocyanide, p-Toluenesulfonylmethyl isocyanate, Tosylmethylisocyanate, Tosylmethylamide
Our laboratory recently reported a mild and efficient method for preparing isocyanates in high yield from the corresponding isonitriles by oxidation using dimethylsulfoxide (DMSO) in the presence of catalytic quantities of trifluoroacetic anhydride (TFAA).1 The mild, workup-free conditions seemed ideal for the synthesis of reactive or multifunctional isocyanates. One particularly interesting example that attracted our attention was p-toluenesulfonylmethyl isocyanate (TsCH2N=C=O), whose compact molecular framework combines a strongly acidic methylene group and a reactive tosyl leaving group together with a highly electrophilic isocyanate group.
The literature on TsCH2N=C=O consists of two published papers, although the compound is often confused with the much more well-known isonitrile TsCH2N=C: (tosylmethylisocyanide, or TOSMIC).2 Tosylalkylisocyanates have previously been synthesized by the Curtius rearrangement of alkanesulfonylacetyl azides,3 and by the chlorination of S-ethyl N-(1-tosylalkyl)thiocarbamates.4 The latter publication also described addition reactions of the isocyanates with methanol, ethanol, benzylamine, and dibenzylamine. Here we report an efficient one-step synthesis of TsCH2N=C=O from commercially available TOSMIC that makes the compound much more accessible. We also present more general findings on the reaction of TsCH2N=C=O with alcohols and amines, as well as new results with thiols and carboxylic acids. Besides being of general utility, our findings may be of particular interest to medicinal chemists seeking to introduce hydrophobic pharmacophores (such as tosyl) at the site of common nucleophilic functional groups.
Following our published procedure,1 treatment of TOSMIC with DMSO (1.1 equiv) and TFAA (0.05 equiv) at low temperature (CH2Cl2, −60 °C to rt, 10 min) resulted in a ~1:1 mixture of starting material and TsCH2N=C=O. Superior results were obtained using excess catalyst, leading to the optimal procedure depicted below (Eq. 1), which was routinely conducted on a 1–2 g scale in yields ranging from 50–60% after recrystallization. The clean oily crude product decomposed at rt, with a half-life of approximately 20 h, but crystallization from ether/hexanes afforded 2 as a white crystal (m.p. = 69–70 °C) that could be stored at −90 °C for several weeks.
 |
Eq 1 |
Consistent with earlier reports, isocyanate 2 formed the corresponding tosylmethylcarbamates in good yield when dissolved in neat primary and secondary alcohols and stirred at rt (Eq. 2).
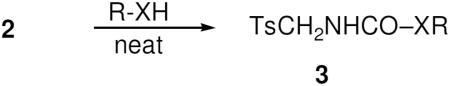 |
Eq 2 |
Reaction of 1.0 equiv of cyclohexanol or cholesterol with 3.0 equiv of 2 (rt, CHCl3 solution) also gave the expected carbamate, although only in ca. 30% yield. Reaction of 2 with butanethiol afforded the corresponding S-thiocarbamate in lower yield. Table 1 summarizes the synthesis of 3a–e.
Table 1.
Reaction of 2 with Alcohols and a Thiol
| RXH | Tosylmethylcarbamate 3 | % Yielda |
|---|---|---|
| EtOH (excess) | 3a RX= EtO | 71 |
| i-PrOH (excess) | 3b RX= iPrO | 60 |
| cyclohexanol (1 equiv) | 3c RX= cyclohexyl–O | 30 |
| cholesterol (1 equiv) | 3d RX= cholesteryl–O | 32 |
| n-BuSH (1 equiv) | 3e RX= n-BuS | 12 |
| n-BuSH (3 equiv) | 3e RX= n-BuS | 22 |
Conditions: stirring 16 h at rt, followed by concentration and purification by flash column chromatography.
Condensation of 2 with various primary and secondary amines (1 equiv) successfully afforded the corresponding N–tosylmethylureas 4 (Eq. 3), although the desired ureas could only be obtained by addition of a solution of 2 to a cold (−78 °C) stirred solution of the amine in THF. The inverse addition of amine to the isocyanate at any temperature led to the formation of insoluble polymeric material, possibly by deprotonation of the active methylene group in 2. Consistent with that hypothesis, the most basic amine (pyrrolidine) furnished carbamate in the lowest yield.
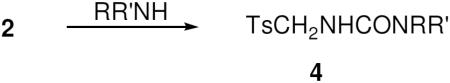 |
Eq 3 |
Isocyanates have long been known to undergo condensation reactions with carboxylic acids that proceed under certain conditions with loss of CO2 to afford carboxamides.5 Efforts to synthesize the corresponding tosylmethylamides 5 (Eq. 4) by condensing isocyanate 2 with benzoic acid were unsuccessful. However, by adapting the modification of Sasaki and Crich in which the carboxylic acid was first transformed into its salt using Hoenig's base,6 a series of tosylmethylcarboxamides 5a–e was prepared under mild conditions (Table 3).
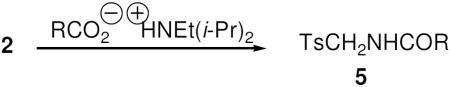 |
Eq 4 |
Table 3.
Conversion of 2 into Carboxamides 5
| Carboxylate salt (1.2 equiv) | Tosylmethylamide 5 | % Yielda |
|---|---|---|
| R=Ph | 5a | 56 |
| R= p-(OCH3)-C6H4 | 5b | 40 |
| R= o-(F)-C6H4 | 5c | 23 |
| R= 2-naphthyl | 5d | 53 |
| R= cinnamyl | 5e | 37 |
| R= p-(NO2)-C6H4 | 5f | 0 |
Conditions: DMF, −40 °C, 0.5 h; then stirring 2 h at rt, followed by partitioning between ethyl acetate and water, and purification by flash column chromatography.
Reactions of 2 with the salt of p-nitrobenzoic acid, as with salts of aliphatic carboxylic acids (hydrocinnamic acid, cyclohexanecarboxylic acid) afforded none of the corresponding tosylmethylamides, affording instead only decomposition products derived from 2. This unexpected lack of reactivity does not appear to trend with pKa values. However, a competition experiment in which equimolar quantities of benzoic and p-nitrobenzoic ammonium salts reacted with 2 afforded the benzamide 5a (39% yield), ruling out any interfering side reactions of 2 with the nitro group.
Besides presenting a potentially useful pharmacophore, tosylmethylamides 5 may undergo carbon-carbon bond-forming substitution reactions of the tosyl group using various organometallic reagents. To our surprise, an initial test coupling of amide 5a with di-n-butylcopperlithium (2.2 equiv) afforded not N-pentylbenzamide as expected, but furnished instead N-(n-butoxymethyl)benzamide 6 as the only detectable product in 72% yield (Eq. 5). Particularly characteristic was the sharp doublet for the O–CH2–N methylene group as well as the –CH2–O triplet in 1H NMR spectrum. The same product was obtained using n-BuMgCl (2.2 equiv, 52% yield) in place of the organocuprate. Compound 6 was also formed when the reaction was conducted using n-BuLi, but in much lower yield.
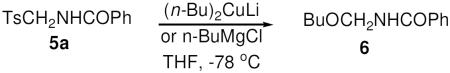 |
Eq 5 |
One plausible mechanism for this unusual transformation is depicted in Scheme 1 below.
Scheme 1.

Metallation of amide 5a followed by elimination of toluenesulfinate anion from 7 would afford intermediate imine 8. Back-addition of ArSO2− to form 9 followed by cyclization would lead to acyloxaziridine 10. Attack of the organometallic reagent at the oxaziridine oxygen in 10, which finds precedent in the work of Davis et al.,7 would furnish the N-(n-butoxymethyl)benzamide 6.
These preliminary findings indicate a convenient new route to alkoxymethylamides, a rare class of compounds that has proven challenging to prepare by other methods.7 The only alternative access to these compounds involves electrolysis of N-acylglycines (RCONHCH2COOH) in methanol, ethanol, or 2-propanol, as noted by Linstead et. al.8
In summary, we report an easy and efficient one-step synthesis of TsCH2N=C=O (2), making it a readily available synthetic reagent for nucleophilic addition reactions with various O, N and S nucleophiles. We also report that N-tosylmethylbenzamides such as 5a (formed by the reaction of 2 with benzoic acid) undergo an unusual substitution reaction in the presence of organocopper and organomagnesium reagents to afford N-(alkoxymethyl)benzamides like 6.
Supplementary Material
Table 2.
Reaction of 2 with Amines
| RR'NH | Tosylmethylurea 4 | % Yielda |
|---|---|---|
| n-BuNH2 | 4a R=H, R'= n-Bu | 61 |
| n-BuNH2 | 4b R=H, R'= t-Bu | 55 |
| pyrrolidine | 4c R,R'= (CH2)4 | 21 |
| aniline | 4d R=H, R'= Ph | 72 |
| o-phenylenediamine | 4e R=H, R'= (o-NH2Ph) | 54 |
Conditions: THF, −78 °C, 0.5 h; then stirring 1 h at rt, followed by concentration and purification by trituration or flash column chromatography.
Acknowledgments
Support of the Cornell NMR Facility has been provided by NSF (CHE 7904825; PGM 8018643) and NIH (RR02002).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data Supplementary data (representative experimental procedures and complete spectroscopic data for new reactants and products shown in Tables 1–3) associated with this article can be found, in the online version, at www.sciencedirect.com
References and Notes
- 1.Le HV, Ganem B. Org. Lett. 2011;13:2584–2585. doi: 10.1021/ol200695y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Online literature searches on TsCH2N=C=O returned many hits, but only two citations were relevant. The rest concerned TOSMIC, not TsCH2N=C=O.
- 3.Strating J, van Oven HO, van Leusen AM. Rec. Trav. Chim. Pays-Bas. 1966;85:631–633. [Google Scholar]
- 4.Olijnsma T, Engberts JBFN, Strating J. Rec. Trav. Chim. Pays-Bas. 1970;89:897–906. [Google Scholar]
- 5.Schuemacher AC, Hoffmann RW. Synthesis. 2001:243–246. [Google Scholar]
- 6.Sasaki K, Crich D. Org. Lett. 2011;13:2256–2259. doi: 10.1021/ol200531k. [DOI] [PubMed] [Google Scholar]
- 7.Davis FA, Mancinelli PA, Balasubramanian K, Nadir UK. J. Am. Chem. Soc. 1979;101:1044–1045. [Google Scholar]
- 8.Linstead RP, Shephard BR, Weedon BCL. J. Chem. Soc. 1951:2854–2858. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.