

Abstract
P450-mediated bioactivation of azoxymethane (AOM), a colon carcinogen, leads to the formation of DNA adducts, of which O6-methylguanine (O6-mG) is the most mutagenic and contributes to colon tumorigenesis. To determine whether P450 enzymes of the liver and intestine both contribute to AOM bioactivation in vivo, we compared tissue levels of AOM-induced DNA adducts, microsomal AOM metabolic activities, and incidences of colonic aberrant crypt foci (ACF) among wild-type (WT), liver-specific P450 reductase (Cpr)-null (LCN), and intestinal epithelium-specific Cpr-null (IECN) mice. At 6 h following AOM treatment (at 14 mg/kg, s.c.), O6-mG and N7-mG levels were highest in the liver, followed by the colon, and then small intestine in WT mice. As expected, hepatic adduct levels were significantly lower (by >60%) in LCN mice but unchanged in IECN mice, whereas small-intestinal adduct levels were unchanged or increased in LCN mice but lower (by >50%) in IECN mice compared to that in WT mice. However, colonic adduct levels were unchanged in IECN mice compared to that in WT mice and increased in LCN mice (by 1.5–2.9-fold). The tissue-specific impact of the CPR loss in IECN and LCN mice on microsomal AOM metabolic activity was confirmed by rates of formation of formaldehyde and N7-mG in vitro. Furthermore, the incidence of ACF, a lesion preceding colon cancer, was similar in the three mouse strains. Thus, AOM-induced colonic DNA damage and ACF formation is not solely dependent on either hepatic or intestinal microsomal P450 enzymes. P450 enzymes in both the liver and intestine likely contribute to AOM-induced colon carcinogenesis.
Introduction
Azoxymethane (AOM) and its metabolic precursor, 1, 2-dimethylhydrazine (DMH), are commonly used carcinogens to study the molecular mechanisms of colon carcinogenesis in rodents.1−4 They are preferred model carcinogens because they induce tumors preferentially in the distal colon of rodents and because the tumors have pathological features known to be associated with human sporadic colorectal cancer.5 DMH and AOM are procarcinogens, which require metabolic activation by cytochrome P450 (P450) enzymes, primarily CYP2E1.6 DMH undergoes N-oxidation to form AOM, which, upon hydroxylation, yields methylazoxymethanol (MAM). MAM is unstable, with a half-life of ∼12 h. It subsequently decomposes to yield formaldehyde and a highly reactive methyldiazonium ion, which alkylates the DNA bases, resulting in the formation of DNA adducts, including O6-methylguanine (O6-mG) and N7-methylguanine (N7-mG).4 Persistence of O6-mG can lead to mutation in oncogenes and initiation of tumorigenesis.7
Previous in vitro studies have demonstrated that colon epithelial cells are capable of metabolizing DMH into carcinogenic metabolites, without the need for prior metabolism by other tissues or colonic bacteria.8−12 However, the prevailing hypothesis is that the liver plays a critical role in DMH/AOM bioactivation in vivo and that the reactive intermediates produced by the liver are transported to the colon via the blood or bile to induce carcinogenicity.13−15 This is a plausible hypothesis, given that the colon generally possesses much lower levels of P450 enzymes, relative to the liver. Nonetheless, the relative contributions of the liver and the intestine to DMH/AOM-induced DNA damage in the colon have not been directly determined, and the bioactivation in the target organ may also explain the organ-specific induction of tumors in the distal colon by AOM.
In the present study, we determined the respective roles of hepatic and intestinal P450 enzymes in AOM metabolic activation in vitro and in vivo by studying the liver-specific Cpr-null (LCN) mouse and the intestinal epithelium-specific Cpr-null (IECN) mouse.16,17 The cytochrome P450 reductase (CPR or POR) is required for the monooxygenase activity of all microsomal P450 enzymes. The LCN and IECN mouse models have been found valuable for differentiating between hepatic and extrahepatic18−22 or between intestinal and extra-gut17,23,24 P450 contributions to xenobiotic metabolism or toxicity. We chose to study AOM instead of its precursor DMH, owing to AOM’s higher potency and greater stability in dosing solutions. We compared tissue levels of O6-mG and N7-mG adducts in the liver, small intestine (SI), and colon among wild-type (WT), LCN, and IECN mice that were treated with AOM according to an established protocol for the induction of colon DNA damage.6,25,26 We further compared microsomal AOM metabolic activities in the various tissues among the three strains of mice. Finally, we assessed AOM-induced colonic aberrant cryptic foci (ACF) formation, which represents the precursor lesions of colon cancers.4,27,28 Our findings indicate that knockout of liver Cpr or IE Cpr alone was not sufficient to block or reduce colonic DNA adduction and ACF formation by AOM and that P450 enzymes in both the liver and intestine likely contribute to AOM-induced O6-mG formation and the eventual colon carcinogenesis in WT mice.
Materials and Methods
Chemicals and Reagents
AOM, O6-mG, and formaldehyde were purchased from Sigma-Aldrich (St. Louis, MO). N7-mG was obtained from Santa Cruz Biotechnology (Dallas, TX). The sources of O6-methyl-deoxyguanosine (O6-m-dG) and O6-trideuteriomethyl-deoxyguanosine (O6-CD3-dG) were the same as described previously.20 All solvents (acetonitrile, methanol, and water) were of high-performance liquid chromatography (HPLC) grade (ThermoFisher Scientific, Waltham, MA).
Mouse Breeding
All studies with mice were approved by the Wadsworth Center Institutional Animal Care and Use Committee. WT B6, Vil-Cre(+/−)/Cprlox/lox (IE-Cpr-null or IECN) mice (on B6 background), and Alb-Cre(+/−)/Cprlox/lox (liver-Cpr-null or LCN) mice (on B6 background)16 were obtained from breeding stocks maintained at the Wadsworth Center (Albany, NY) and used for CPR expression, AOM metabolism, and DNA adduct formation studies.
WT, IECN, and LCN mice used for studying AOM-induced colonic ACF formation were on the susceptible A/J background. IECN-A/J and LCN-A/J mice were generated by backcrossing IECN-B6 and LCN-B6 to a congenic (A/J-N11) strain of Cprlox/lox mice for 3–5 generations; the Cprlox/lox-A/J mice were produced by backcrossing the original Cprlox/lox-B629 to WT A/J mice (Jackson Laboratory, Bar Harbor, ME). WT-A/J mice used for experiments were produced from breeding pairs maintained at the Wadsworth Center.
All animals were 2- to 3-month old at the beginning of each study described below. Mice were genotyped using tail DNA for the Cre transgene and the Cpr allele, as described previously.16,29
Immunohistochemistry
The colon from male WT and IECN mice were obtained and cut into two equal halves, representing proximal and distal sections, and prepared as “Swiss rolls” for embedding and sectioning. Immunohistochemical analysis of CPR expression in paraffin sections of the colon was conducted as described previously for SI.17
Preparation of Microsomes from Intestine and Liver
SI mucosa from two mice or colonic mucosa from four mice were pooled for each microsomal sample, prepared as reported previously.30 Liver microsomes were prepared from individual mice as described31 but with use of protease inhibitors.30 Microsomes were stored at −80 °C until use. Microsomal protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce Chemical, Rockford, IL) with bovine serum albumin as standard.
Analysis of DNA Adducts in Tissue
For the determination of tissue levels of O6-mG and N7-mG, male WT, IECN, and LCN mice were treated with a single injection of AOM (at 14 mg/kg, s.c.)6 in saline. The liver, SI (duodenum, jejunum and ileum), and colon (proximal, distal) were obtained 6 h after AOM treatment. The SI and colon segments were slit open and rinsed with ice-cold saline before being stored at −80 °C until use. For DNA isolation, the middle lobe of the liver, the entire segments of duodenum, jejunum, ileum, proximal colon, and distal colon were homogenized in ∼4 mL of genomic DNA buffer (10 mM Tris-HCl, 100 mM NaCl, 2.5 mM EDTA, and 0.5% SDS). The homogenate corresponding to ∼100 mg of tissue was incubated with proteinase K (250 μg) at 55 °C for 2 h. DNA was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) (Invitrogen), and precipitated with ethanol. The resuspended DNA was incubated with RNase A (100 μg) and RNase T1 (0.5 μL) for 1 h at 37 °C to remove RNA contamination. The final DNA preparations were stored at −20 °C until used for adduct analysis.
Assay for DNA Adduct Formation in Microsomal Reactions in Vitro
The assay for AOM-induced in vitro DNA adduct formation was based on a published method.32 Briefly, microsomes (0.5–2.0 mg/mL) were incubated with calf thymus DNA (1 mg/mL) and AOM (200 μM) in a total volume of 1.0 mL. The assay buffer consisted of 0.1 M Tris–HCl (pH 7.4), 1 mM EDTA, 20 mM MgCl2, 0.3 M KCl, and 1.5 mM NADPH. Incubations were carried out at 37 °C for 60 min in a shaking water bath. An additional 30 nmol of NADPH was added after the first 30 min. The reaction was stopped by the addition of 0.5 mL of ice-cold 7.5 M ammonium acetate. DNA was then extracted as described above for tissue homogenates. Control incubations were performed without NADPH.
Detection of DNA Adducts by LC-MS/MS
Levels of O6-mG and N7-mG were determined, for both in vivo and in vitro DNA samples, essentially as described20 with minor modifications. Briefly, DNA samples (100–200 μg) were fortified with the internal standard O6-CD3-dG (6 pmol) and hydrolyzed in 0.1 N HCl at 80 °C for 90 min. The samples were allowed to cool, neutralized with NH4OH, and analyzed using LC-MS. Control genomic DNA from corresponding tissues were used for the preparation of calibration curves for the quantification of O6-mG and N7-mG, with O6-mG and N7-mG standards added in 40 to 1000 nM. Blank controls for the solvent and matrix were included in each set of calibration samples. The LC-MS method for the detection of O6-mG was according to ref (24). N7-mG was detected using the same method; the parent/product ion pairs were monitored at m/z 166/149 and m/z 166/124, using the MRM scan mode. The retention time was 7.2 min for N7-mG and 7.6 min for O6-mG and O6-CD3-dG. The detection limits for N7-mG and O6-mG were 0.02 pmol and 0.04 pmol (on column), respectively, with an injection of 6 μg of hydrolyzed DNA. The DNA adduct levels were normalized to guanine levels for all samples, determined using HPLC, as described previously.20
Assay for Methylazoxymethanol (MAM) Formation in Microsomal Reactions in Vitro
MAM formed from AOM in microsomal incubations was detected as formaldehyde using the chromotropic acid method.9 The microsomal incubations were performed at 37 °C, using 500 μM AOM and 0.2 mg/mL microsomal protein for 10 min. Control assays were performed in the absence of either AOM or microsomes to correct for any nonenzymatic conversion of AOM to formaldehyde.
AOM Induced Colonic ACF Formation
Male, 8–10 week old, WT-A/J, IECN-A/J, and LCN-A/J mice (8 per group) were treated with either saline or AOM (7.5 mg/kg BW, s.c.),33 once weekly for 3 weeks. Mice were sacrificed 6 weeks post-treatment for ACF detection, as described previously.33 The entire colon (from the cecum to anus) was excised (within 4 min from the time of euthanasia). A longitudinal incision was made along the entire length of the colon, which was further cut into two equal-length segments, representing proximal and distal portions of the colon. The segments were dipped in PBS to remove fecal pellets and then kept flat between filter papers in 10% buffered formalin for at least 24 h. Subsequently, the colons were immersed in freshly prepared 0.1% methylene blue for 10 min and rinsed briefly in deionized H2O to remove excess dye. The colon was mounted carefully on a microscope slide with the mucosal surface side up and viewed under a light microscope (Nikon TE2000) with 40× magnification. The ACF in the entire mucosal surface of the colon were counted blindly and independently by two investigators and recorded.
Other Methods
Statistical significance of differences among the three mouse strains in various parameters was examined using one-way analysis of variance (ANOVA), followed by Dunnett’s post-hoc test for pair wise comparisons, with the use of Graph Pad Prism 5 (Graph Pad, San Diego, CA). In all cases, p < 0.05 was considered statistically significant.
Results
Effects of Tissue-Specific Cpr Deletion on AOM-Induced DNA Adduct Formation in the Liver and Intestine
The respective roles of the liver and intestinal microsomal P450 enzymes in the bioactivation of AOM in vivo was determined by comparing levels of AOM-induced DNA adducts in WT, IECN, and LCN mice (Table 1). Levels of O6-mG and N7-mG, which are known to be produced by AOM treatment,6,34 were determined in the liver, duodenum, jejunum, ileum, proximal colon, and distal colon at 6 h after AOM treatment, at a dose (14 mg/kg) that was shown previously to be effective in inducing adduct formation in mice.6 The levels of N7-mG were (3–9 times) greater than O6-mG levels in the various tissues examined, which is consistent with previous reports.6,26,34 Regardless of the strain, the amounts of O6-mG and N7-mG produced by AOM were highest in the liver, followed by proximal and distal colons, which had similar levels, and then by duodenum, jejunum and ileum (Table 1). For O6-mG, the WT levels in the liver DNA was ∼5 times higher than that in colon DNA, ∼10 times higher than that in duodenal DNA, and ∼32 and 83 times higher than that in jejunum and ileal DNA.
Table 1. AOM-Induced DNA Adduct Formation in the Liver and Intestinea.
| pmol/μmol guanine |
|||
|---|---|---|---|
| tissue | strain | O6-mG | N7-mG |
| liver | WT | 413 ± 53 | 3062 ± 954 |
| IECN | 448 ± 68 | 2926 ± 564 | |
| LCN | 159 ± 24b | 1046 ± 311b | |
| duodenum | WT | 43 ± 10 | 168 ± 18 |
| IECN | 19 ± 4b | 63 ± 10b | |
| LCN | 71 ± 12b | 189 ± 32 | |
| jejunum | WT | 13 ± 2 | 41 ± 6 |
| IECN | 6 ± 1.2b | 17 ± 3b | |
| LCN | 19 ± 4 | 59 ± 16 | |
| ileum | WT | 5 ± 1.4 | 24 ± 7 |
| IECN | 2 ± 0.4b | 9 ± 2b | |
| LCN | 7 ± 1.3 | 28 ± 5 | |
| proximal colon | WT | 72 ± 15 | 343 ± 102 |
| IECN | 61 ± 12 | 375 ± 148 | |
| LCN | 108 ± 15b | 984 ± 294b | |
| distal colon | WT | 84 ± 17 | 480 ± 147 |
| IECN | 67 ± 11 | 384 ± 140 | |
| LCN | 128 ± 16b | 901 ± 232b | |
Male WT, IECN, and LCN mice were injected with AOM at a dose of 14 mg/kg (s.c.), and levels of O6-mG and N7-mG, as well as total guanine, were determined in the liver, SI (duodenum, jejunum, and ileum), and proximal and distal colon at 6 h after the injection. Data represent the means ± SD of eight determinations, each of a separate mouse.
P < 0.05, compared with the corresponding WT tissue; one way ANOVA with Dunnett’s post-hoc test.
The loss of P450 activity in the liver, in the LCN mice, caused a significant decrease in hepatic O6-mG and N7-mG levels to <40% of the WT level, confirming the role of microsomal P450 enzymes in AOM bioactivation in the mouse liver in vivo. In contrast, there was a significant increase in O6-mG levels (by ∼1.7-fold) in the duodenum and significant increases in both O6-mG (by ∼1.5-fold) and N7-mG (by ∼2.1–2.9-fold) levels in the proximal and distal colons of LCN mice compared to those in WT mice (Table 1). These data indicated that the AOM-induced DNA adduct formation in the SI and colon does not depend on bioactivation by hepatic P450 enzymes.
The loss of intestinal P450 activity, in IECN mice, did not change adduct levels in the liver, but it led to significant decreases in O6-mG and N7-mG levels in the duodenum, jejunum, and ileum to <50% of the WT level. Interestingly, in the proximal and distal colons, there was no significant difference in the abundance of DNA adducts formed between the IECN and WT mice (Table 1).
Previous studies have shown through immunoblot analysis that CPR protein expression was abolished in colonic microsomes from IECN mice.17 To be sure, we further examined CPR expression in the colon of WT and IECN mice by immunohistochemical analysis. As shown in Figure 1, the CPR protein was abundantly detected in the colon epithelium in both proximal and distal colons from WT mice, but it was absent in colons from IE-Cpr-null mice.
Figure 1.
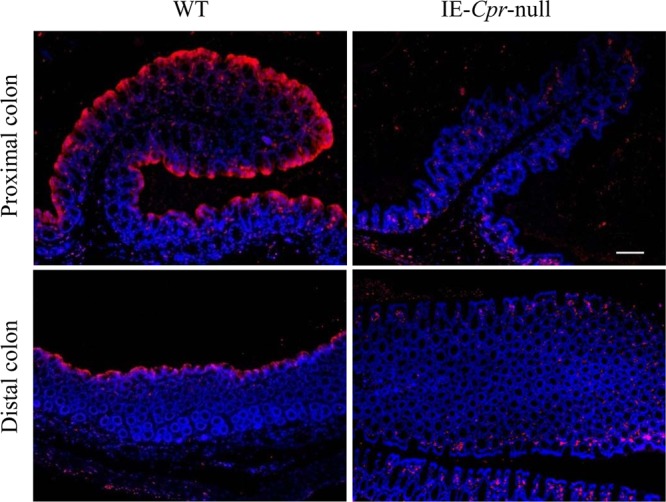
Immunohistochemical analysis of CPR expression in colon. Paraffin sections of the proximal and distal colon from 2-month-old male IE-Cpr-null mice and WT littermates were processed for immunohistochemistry. The tissue sections were incubated with a polyclonal rabbit anti-rat CPR antiserum. Antigenic sites were visualized with a peroxidase-conjugated goat anti-rabbit secondary antibody, with Alexa Fluor 594-conjugated tyramide as the peroxidase substrate. Sections were mounted with Prolong mounting medium with DAPI counter stain. Fluorescent signals were detected with a tetramethylrhodamine isothiocyanate filter (for Alexa 594, red) and a DAPI filter (for DAPI, blue); scale bar, 100 μm. No signal was detected in negative control slides (data not shown), which were incubated with a normal goat serum in place of the anti-CPR antibody. Results shown are typical of three mice per strain analyzed.
Effects of Tissue-Specific Cpr Deletion on AOM-Induced ACF Formation in the Colon
AOM is known to induce ACF in the colon as a downstream event to AOM-induced DNA adduct formation and a precursor to the eventual tumorigenesis.4,27 Thus, we further compared the extent of AOM-induced ACF formation in WT, IECN, and LCN mice after they were backcrossed 3–5 generations to the susceptible A/J background.
Irrespective of the mouse strain, no ACF was detected in the colons of saline-treated mice; in contrast, colonic ACF was detected in all three strains of AOM-treated mice (Figure 2). As shown in Figure 2A, the aberrant crypts in ACF, which are preneoplastic formations, are distinguished from normal crypts by their larger size, increased pericryptal area, irregular lumen with slit shaped appearance, greater staining intensity, and/or elevation above adjacent normal crypts.
Figure 2.

AOM-induced colonic ACF formation. (A) Morphology of AOM-induced ACF in colon. ACF was detected in methylene-blue-stained colon, as described in Materials and Methods. Representative images of ACF (arrows) with 2 (left) or 8 (right) aberrant crypts are shown. (B) Numbers of ACF detected in colon. Male WT-A/J, IECN-A/J, and LCN-A/J mice were treated with saline or AOM at a dose of 7.5 mg/kg (s/c), once weekly for three weeks, and sacrificed 6 weeks later for the detection ACF in the proximal and distal colon. Saline-treated mice did not develop any ACF (not shown). Data (means ± S.D., n = 4 −8) for proximal and distal colons, as well as the combined data for the entire colon (total), are presented. There was no difference between the two mouse strains (p > 0.05, compared to WT; one-way ANOVA with Dunnett’s test).
No significant difference in the total ACF counts was found among the strains for either the proximal or the distal region of the colon (Figure 2B). These results are largely consistent with the DNA adduct data, showing that colonic ACF formation is not critically dependent on AOM bioactivation by hepatic or intestinal microsomal P450 enzymes.
Effects of Tissue-Specific Cpr Deletion on Rates of Microsomal AOM Metabolism
To further confirm that the liver and intestinal Cpr deletion did reduce the capacity of the respective tissues to activate AOM to DNA-reactive electrophiles, we analyzed the alkylation of calf thymus DNA by reactive metabolites of AOM formed in incubations with the liver, SI, and colon microsomes from WT, IECN, and LCN mice (Table 2). Notably, although we performed analysis to detect both O6-mG and N7-mG, the levels of O6-mG were below the limit of detection. Therefore, only N7-mG levels are presented.
Table 2. Microsomal Activity toward AOM in Vitroa.
| rates
of microsomal metabolism |
||||||
|---|---|---|---|---|---|---|
| pmol N7-mG/μmol G/h/mg protein |
nmol formaldehyde/min/mg protein |
|||||
| strain | liver | SI | colon | liver | SI | colon |
| WT | 153 ± 57 | 10.3 ± 3 | 6.4 ± 1.8 | 34 ± 13 | 12.6 ± 7 | 6.4 ± 4.1 |
| IECN | 121 ± 15 | 1.3 ± 2.6b | <LOQc | 27 ± 11 | <LOQc | <LOQc |
| LCN | 6.4 ± 6.3b | 10.2 ± 1.1 | 4.8 ± 2.6 | <LOQc | 18.6 ± 7.3 | 7.7 ± 4 |
The rates of formation of formaldehyde or DNA adducts were measured for hepatic, SI, and colon microsomes of WT, IECN, and LCN mice on a B6 background. Each intestinal microsome preparation was obtained from pooled tissues from 2 to 4 adult male mice; three microsomal preparations were analyzed for each group. Hepatic microsomes were obtained from individual animals (n = 4). For DNA adduct formation, reaction mixtures contained 0.5–2.0 mg of microsomal protein, 200 μM AOM, 1.0 mg of calf thymus DNA, and other components as described in Materials and Methods in a total volume of 1.0 mL. Reactions were carried out at 37 °C for 60 min in the presence or absence of 1.5 mM NADPH. O6-mG formation was not detected under the conditions used (limit of detection was ∼2 pmol/μmol G/h/mg protein). For formaldehyde formation, reaction mixtures contained 0.2 mg of microsomal protein, 500 μM AOM, and other components as described in Materials and Methods in a total volume of 1.0 mL, and reactions were carried out for 10 min. Values represent the means ± SD (n = 3–4).
p < 0.01, compared with corresponding WT microsomes; one way ANOVA with Dunnett’s post-hoc test.
Below the limit of quantification (LOQ, ∼1).
On an equal protein basis, hepatic microsomes were much more active than SI and colon microsomes in NADPH-dependent AOM bioactivation and N7-mG adduct formation. Interestingly, colon microsomes were competent in this reaction, showing ∼60% of the activity seen in SI microsomes in WT mice (Table 2). Microsomes of the liver (but not those of the SI or colon) of LCN mice showed significantly lower activity in N7-mG formation compared to that of WT mice. Similarly, microsomes of the SI and colon from IECN mice showed minimal or no activity in the formation of N7-mG, whereas the liver microsomes from IECN mice had activity similar to that of WT in N7-mG formation (Table 2).
We also compared the liver, SI, and colon microsomes from WT, IECN, and LCN mice for their ability to metabolize AOM to formaldehyde via the formation of MAM (Table 2). In accordance with the in vitro N7-mG formation data, hepatic microsomes showed the highest activity in the hydroxylation of AOM, followed by SI and colon microsomes. With the loss of hepatic CPR, liver microsomes of LCN mice had no activity in formaldehyde formation, whereas the activities of the intestinal microsomes of LCN mice were comparable to those of WT mice. In IECN mice, neither SI nor colon microsomes had detectable activity in formaldehyde formation from AOM, while the liver activity was similar to that of WT (Table 2).
Discussion
Considering the high incidence of cancers in the colon and the fact that the gastrointestinal tract is a major portal of entry for myriad chemical carcinogens and toxicants, it is important to determine the metabolic mechanisms of chemical carcinogenesis in the colon, including the source of reactive intermediates, and the role of target tissue metabolic activation. Although the liver is the most abundant in P450 enzymes, some extrahepatic tissues, such as the lung and small intestine, have been shown to be highly efficient in target-tissue bioactivation of carcinogens and other toxicants, leading to tumorigenesis or tissue toxicity.20,23,24,35,36 The colon appears to possess a somewhat unique profile of P450 expression, both in terms of expression levels and isoforms present,12,37−39 and the role of colonic P450s in xenobiotic metabolism and toxicity is not well understood.
We studied AOM as a model colon carcinogen. Although human exposure to AOM, derived from a rare plant, is uncommon, our exposure to other, structurally related hydrazine derivatives that are found in mushrooms, tobacco, herbicides, rocket fuels, and drugs are frequent.40 Hence, the metabolic mechanisms of AOM may have broad implications. In that regard, while several studies had reported efficient in vitro metabolism of AOM by colon microsomes from rodents, hamsters, and humans,8,9,11,12 others failed to detect such activity in the colon.41 When non-P450-mediated bioactivation of MAM in colon was investigated in vitro, alcohol dehydrogenase (ADH) was implicated;42 however, a role for ADH was not confirmed in vivo.13,43
We determined the influence of tissue-specific suppression of P450 activity, via conditional Cpr deficiency, in the liver and intestine on AOM metabolism in vitro and AOM-induced DNA adduct formation in vivo. For in vivo studies, we chose subcutaneous AOM administration since it was reportedly the most effective route for producing colon tumorigenesis.44 We collected tissues for analysis at 6 h after AOM administration, a time point that was previously found to yield maximal alkylation of target tissue DNA.25,26 All bioactivation studies were conducted with mice on the B6 genetic background. In that connection, it should be noted that although mouse strain-related differential sensitivity was reported for AOM-induced carcinogenesis, with A/J being the most sensitive strain,28,33,45 a strain difference between B6 and A/J mice was not observed in AOM-induced DNA adduct formation (data not shown).
Our in vitro studies clearly demonstrated the predominant role of P450 enzymes in microsomal metabolism of AOM, in the liver, SI, and colon. It is interesting that SI and colon microsomes showed somewhat similar ability to bioactivate AOM, as indicated by their rates of in vitro formation of formaldehyde and N7-mG, even though the total P450 concentration in SI is reportedly several folds higher than that in the colon.46 This finding may be related to the reported presence of CYP2E1 in the colon,37,39,46 and the known activity of CYP2E1 toward AOM.6,47 The apparent absence of O6-mG adduct formation in vitro by microsomes from any of the mouse tissues analyzed was consistent with the fact that N7-mG was much more abundant than O6-mG in vivo, in AOM-treated mice (Table 1), which has been noted previously with adducts formed by another compound.48
The results of AOM-induced DNA adduct formation in vivo (Table 1) are more complex to explain. In AOM-treated LCN mice, compared to similarly treated WT mice, hepatic O6-mG and N7-mG levels were substantially decreased, consistent with a major role of hepatic P450 enzymes in AOM bioactivation in the liver. Note, however, that the contributions of hepatic microsomal P450s might have been underestimated by the data from the LCN mice, where the loss of hepatic microsomal P450-mediated AOM clearance would lead to increases in AOM concentrations in the liver and extrahepatic tissues, and consequently increased DNA adduct formation via alternative metabolic pathways and/or in extrahepatic tissues.
The DNA adduct levels were significantly increased, rather than decreased, in most parts of the intestine, including the colon, by the loss of hepatic microsomal P450 activity (Table 1). This result clearly indicated that hepatic microsomal P450-mediated metabolism is not required for AOM to induce DNA adduct formation in the colon. However, the data could not tell us whether liver-produced reactive AOM metabolites contributed any part to DNA adduct formation in the colon, given the likely increase in AOM bioavailability in the target organ (and thus a possible overestimation of local contribution to DNA adduct formation) in the LCN mice.
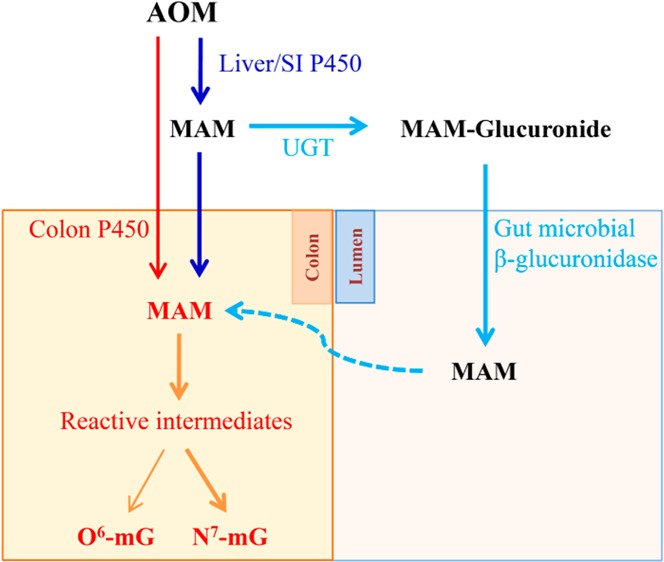
Given that AOM was administered subcutaneously and based on previous studies on pharmacokinetics of other compounds in the IECN mouse,17 we believe that the bioavailability of AOM was unchanged in IECN compared to that of WT mice. As expected, the lack of microsomal P450 activity in the intestine resulted in much reduced levels of AOM-induced DNA adducts in the SI but not in the liver. Surprisingly, DNA adduct levels in the colon were also unchanged, which contrasted with results of in vitro microsomal assays showing substantial decreases in AOM bioactivating activity in the IECN mice relative to that in WT mice (Table 2). The reasons for this apparent discrepancy between in vitro and in vivo results remain to be determined. However, it is worth noting that although the colon had the lowest in vitro activity among the three tissues analyzed (Table 2), the level of DNA adducts in the colon was the highest among all intestinal segments (Table 1). The latter fact was at least partly related to a possible colonic accumulation of the reactive metabolite derived from the proximal SI or to colonic bacterial β-glucuronidase activity, which can act on MAM-glucuronide (derived either from the liver or the SI) to release free MAM in the colon, as depicted in Figure 3. The regenerated MAM, once absorbed, can either undergo further bioactivation or spontaneously decompose, to reactive methyldiazonium ion in the colon, leading to adduct formation.49 The bacterial source of reactive AOM metabolites might have overshadowed the amounts of MAM generated directly from AOM by intestinal P450, thus obliterating any decreases in colonic DNA adducts resulting from the loss of intestinal P450 activity. At any rate, these data suggested that intestinal microsomal P450 enzymes are not essential to (though they can contribute to) AOM-induced DNA damage in the colon, under the conditions (dose and route) used in this study. Additional studies are warranted in order to determine whether intestinal (and hepatic) P450 plays a more essential role in colonic DNA adduct formation by orally ingested AOM.
Figure 3.
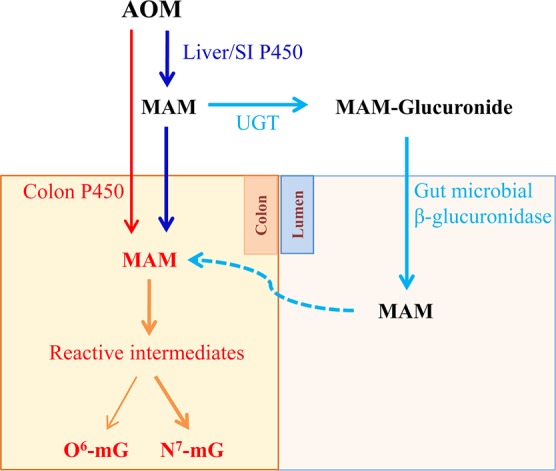
Schematic summary of sources of AOM reactive metabolites in the colon.
As expected, the results of the ACF studies were largely consistent with those of the DNA adduct studies, in that the loss of either hepatic or IE CPR did not lead to a noticeable decrease in colonic ACF formation. The multiplicity of colonic ACF was not different among the three mouse strains, even though colonic DNA adduct levels were ∼50% higher in the LCN mice than in WT mice. This latter discrepancy may be related to the different AOM dose and dosing regime used for the two studies. It should also be noted that the current study was focused on conventional ACF; further, more discriminating studies are required to elucidate the degrees of dysplasia in the ACF found in the different strains. Additionally, gross adduct levels may not accurately represent adduct levels in colonic stem cells, which may be more critical for ACF formation and carcinogenesis.
In summary, we show that liver, SI, and colon microsomal P450 enzymes are all capable of bioactivating AOM and that P450 enzymes in both the liver and intestine likely contribute to AOM-induced O6-mG formation and the eventual colon carcinogenesis in WT mice. Our results suggest that the overall (both hepatic and extrahepatic) bioactivation capacity is more useful than either hepatic or colonic activity alone for assessing the risks of chemical toxicity from AOM (and related compounds) in the colon.
Acknowledgments
We thank Dr. Robert Turesky for helpful discussions and Ms. Weizhu Yang for help with mouse breeding. We gratefully acknowledge the use of the Biochemistry, Histopathology, and Advanced Light Microscopy and Image Analysis Core Facilities of the Wadsworth Center.
Glossary
Abbreviations
- P450
cytochrome P450
- CPR
cytochrome P450 reductase
- NADPH
reduced β-nicotinamide adenine dinucleotide phosphate
- HPLC
high performance liquid chromatography
- LC-MS
liquid chromatography–mass spectrometry
- WT
wild-type
- B6
C57BL/6
- IECN
intestinal epithelial Cpr-null
- LCN
liver specific Cpr-null
- O6-mG
O6-methylguanine
- N7-mG
N7-methylguanine
- AOM
azoxymethane
- MAM
methylazoxymethanol
- DMH
1,2-dimethyl hydrazine
- ACF
aberrant cryptic foci
This work was supported in part by the National Institutes of Health [Grant CA092596 (to X.D.) and Grant GM082978 (to Q.Z.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Femia A. P.; Caderni G. (2008) Rodent models of colon carcinogenesis for the study of chemopreventive activity of natural products. Planta Med. 74, 1602–1607. [DOI] [PubMed] [Google Scholar]
- Magnuson B.; South E.; Exon J.; Dashwood R.; Xu M.; Hendrix K.; Hubele S. (2000) Increased susceptibility of adult rats to azoxymethane-induced aberrant crypt foci. Cancer Lett. 161, 185–193. [DOI] [PubMed] [Google Scholar]
- Perše M.; Cerar A. (2005) The dimethylhydrazine induced colorectal tumours in rat-experimental colorectal carcinogenesis. Radiol. Oncol. 39, 61–70. [Google Scholar]
- Rosenberg D. W.; Giardina C.; Tanaka T. (2009) Mouse models for the study of colon carcinogenesis. Carcinogenesis 30, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet D. E.; Pierre F. (2005) How good are rodent models of carcinogenesis in predicting efficacy in humans? A systematic review and meta-analysis of colon chemoprevention in rats, mice and men. Eur. J. Cancer 41, 1911–1922. [DOI] [PubMed] [Google Scholar]
- Sohn O. S.; Fiala E. S.; Requeijo S. P.; Weisburger J. H.; Gonzalez F. J. (2001) Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 61, 8435–8440. [PubMed] [Google Scholar]
- Takahashi M.; Wakabayashi K. (2004) Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 95, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autrup H.; Harris C. C.; Schwartz R. D.; Trump B. F.; Smith L. (1980) Metabolism of 1, 2-dimethylhydrazine by cultured human colon. Carcinogenesis 1, 375–380. [DOI] [PubMed] [Google Scholar]
- Newaz S.; Fang W. F.; Strobel H. W. (1983) Metabolism of the carcinogen 1, 2-dimethylhydrazine by isolated human colon microsomes and human colon tumor cells in culture. Cancer 52, 794–798. [DOI] [PubMed] [Google Scholar]
- Oravec C. T.; Jones C. A.; Huberman E. (1986) Activation of the colon carcinogen 1, 2-dimethylhydrazine in a rat colon cell-mediated mutagenesis assay. Cancer Res. 46, 5068–5071. [PubMed] [Google Scholar]
- Sheth-Desai N.; Lamba-Kanwal V.; Eichholz A. (1987) Organ-specific effects of 1, 2-dimethylhydrazine in hamster. Jap. J. Cancer Res.: Gann 78, 117. [PubMed] [Google Scholar]
- Stralka D.; Strobel H. W. (1991) Characterization of cytochrome P450-dependent dimethylhydrazine metabolism in human colon microsomes. Cancer 68, 2363–2369. [DOI] [PubMed] [Google Scholar]
- Fiala E. S. (1977) Investigations into the metabolism and mode of action of the colon carcinogens 1, 2-dimethylhydrazine and azoxymethane. Cancer 40, 2436–2445. [DOI] [PubMed] [Google Scholar]
- Pozharisski K.; Kapustin Y. M.; Likhachev A. J.; Shaposhnikov J. (1975) The mechanism of carcinogenic action of 1, 2-dimethylhydrazine (SDMH) in rats. Int. J. Cancer 15, 673–683. [DOI] [PubMed] [Google Scholar]
- Zedeck M. S.; Grab D. J.; Sternberg S. S. (1977) Differences in the acute response of the various segments of rat intestine to treatment with the intestinal carcinogen, methylazoxymethanol acetate. Cancer Res. 37, 32–36. [PubMed] [Google Scholar]
- Gu J.; Weng Y.; Zhang Q.-Y.; Cui H.; Behr M.; Wu L.; Yang W.; Zhang L.; Ding X. (2003) Liver-specific deletion of the NADPH-cytochrome P450 reductase gene impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 278, 25895–25901. [DOI] [PubMed] [Google Scholar]
- Zhang Q.-Y.; Fang C.; Zhang J.; Dunbar D.; Kaminsky L.; Ding X. (2009) An intestinal epithelium-specific cytochrome P450 (P450) reductase-knockout mouse model: direct evidence for a role of intestinal P450s in first-pass clearance of oral nifedipine. Drug Metab. Dispos. 37, 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.; Behr M.; Xie F.; Lu S.; Doret M.; Luo H.; Yang W.; Aldous K.; Ding X.; Gu J. (2008) Mechanism of chloroform-induced renal toxicity: Non-involvement of hepatic cytochrome P450-dependent metabolism. Toxicol. Appl. Pharmacol. 227, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Cui H.; Behr M.; Zhang L.; Zhang Q.-Y.; Yang W.; Hinson J. A.; Ding X. (2005) In vivo mechanisms of tissue-selective drug toxicity: effects of liver-specific knockout of the NADPH-cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol. Pharmacol. 67, 623–630. [DOI] [PubMed] [Google Scholar]
- Weng Y.; Fang C.; Turesky R. J.; Behr M.; Kaminsky L. S.; Ding X. (2007) Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res. 67, 7825–7832. [DOI] [PubMed] [Google Scholar]
- Xie F.; Zhou X.; Behr M.; Fang C.; Horii Y.; Gu J.; Kannan K.; Ding X. (2010) Mechanisms of olfactory toxicity of the herbicide 2, 6-dichlorobenzonitrile: essential roles of CYP2A5 and target-tissue metabolic activation. Toxicol. Appl. Pharmacol. 249, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F.; Zhou X.; Genter M. B.; Behr M.; Gu J.; Ding X. (2011) The tissue-specific toxicity of methimazole in the mouse olfactory mucosa is partly mediated through target-tissue metabolic activation by CYP2A5. Drug Metab. Dispos. 39, 947–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Zhang Q.-Y. (2012) Role of intestinal cytochrome P450 enzymes in diclofenac-induced toxicity in the small intestine. J. Pharmacol. Exp. Ther. 343, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; D’Agostino J.; Xie F.; Ding X. (2012) Role of CYP2A5 in the bioactivation of the lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in mice. J. Pharmacol. Exp. Ther. 341, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H.; Buecheler J.; Kleihues P. (1978) DNA alkylation in mice with genetically different susceptibility to 1, 2-dimethylhydrazine-induced colon carcinogenesis. Cancer Res. 38, 3063–3065. [PubMed] [Google Scholar]
- Herron D. C.; Shank R. C. (1981) In vivo kinetics of O6-methylguanine and 7-methylguanine formation and persistence in DNA of rats treated with symmetrical dimethylhydrazine. Cancer Res. 41, 3967–3972. [PubMed] [Google Scholar]
- Raju J. (2008) Azoxymethane-induced rat aberrant crypt foci: relevance in studying chemoprevention of colon cancer. World J. Gastroenterol. 14, 6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg D. W.; Liu Y. (1995) Induction of aberrant crypts in murine colon with varying sensitivity to colon carcinogenesis. Cancer Lett. 92, 209–214. [DOI] [PubMed] [Google Scholar]
- Wu L.; Gu J.; Weng Y.; Kluetzman K.; Swiatek P.; Behr M.; Zhang Q. Y.; Zhuo X.; Xie Q.; Ding X. (2003) Conditional knockout of the mouse NADPH-cytochrome P450 reductase gene. Genesis 36, 177–181. [DOI] [PubMed] [Google Scholar]
- Zhang Q.-Y.; Dunbar D.; Kaminsky L. S. (2003) Characterization of mouse small intestinal cytochrome P450 expression. Drug Metab. Dispos. 31, 1346–1351. [DOI] [PubMed] [Google Scholar]
- Fasco M. J.; Silkworth J.; Dunbar D. A.; Kaminsky L. S. (1993) Rat small intestinal cytochromes P450 probed by warfarin metabolism. Mol. Pharmacol. 43, 226–233. [PubMed] [Google Scholar]
- Guo Z.; Smith T. J.; Thomas P. E.; Yang C. S. (1991) Metabolic activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone as measured by DNA alkylation in vitro and its inhibition by isothiocyanates. Cancer Res. 51, 4798–4803. [PubMed] [Google Scholar]
- Papanikolaou A.; Wang Q.-S.; Papanikolaou D.; Whiteley H. E.; Rosenberg D. W. (2000) Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane-induced colon carcinogenesis. Carcinogenesis 21, 1567–1572. [PubMed] [Google Scholar]
- Papanikolaou A.; Shank R. C.; Delker D. A.; Povey A.; Cooper D. P.; Rosenberg D. W. (1998) Initial levels of azoxymethane-induced DNA methyl adducts are not predictive of tumor susceptibility in inbred mice. Toxicol. Appl. Pharmacol. 150, 196–203. [DOI] [PubMed] [Google Scholar]
- Ding X.; Kaminsky L. S. (2003) Human Extrahepatic Cytochromes P450: Function in Xenobiotic Metabolism and Tissue-Selective Chemical Toxicity in the Respiratory and Gastrointestinal Tracts. Annu. Rev. Pharmacol. Toxicol. 43, 149–173. [DOI] [PubMed] [Google Scholar]
- Megaraj V.; Zhou X.; Xie F.; Liu Z.; Yang W.; Ding X. (2014) Role of CYP2A13 in the bioactivation and lung tumorigenicity of the tobacco-specific lung procarcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK): in vivo studies using a CYP2A13-humanized mouse model. Carcinogenesis 35, 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkak R.; Korourian S.; Ronis M. J.; Ingelman-Sundberg M.; Badger T. M. (1996) Effects of diet and ethanol on the expression and localization of cytochromes P450 2E1 and P450 2C7 in the colon of male rats. Biochem. Pharmacol. 51, 61–69. [DOI] [PubMed] [Google Scholar]
- Kumarakulasingham M.; Rooney P. H.; Dundas S. R.; Telfer C.; Melvin W. T.; Curran S.; Murray G. I. (2005) Cytochrome P450 profile of colorectal cancer: identification of markers of prognosis. Clin. Cancer Res. 11, 3758–3765. [DOI] [PubMed] [Google Scholar]
- White T. B.; Hammond D. K.; Vásquez H.; Strobel H. W. (1991) Expression of two cytochromes P450 involved in carcinogen activation in a human colon cell line. Mol. Cell. Biochem. 102, 61–69. [DOI] [PubMed] [Google Scholar]
- Gamberini M.; Cidade M. R.; Valotta L. A.; Armelin M.; Leite L. (1998) Contribution of hydrazines-derived alkyl radicals to cytotoxicity and transformation induced in normal c-myc-overexpressing mouse fibroblasts. Carcinogenesis 19, 147–155. [DOI] [PubMed] [Google Scholar]
- Sandforth F.; Gutschmidt S. (1984) The intestinal monooxygenase enzyme system in DMH-induced rat colonic carcinogenesis. Z. Gastroenterol. 22, 586. [PubMed] [Google Scholar]
- Feinberg A.; Zedeck M. S. (1980) Production of a highly reactive alkylating agent from the organospecific carcinogen methylazoxymethanol by alcohol dehydrogenase. Cancer Res. 40, 4446–4450. [PubMed] [Google Scholar]
- Delker D. A.; McKnight S. J.; Rosenberg D. W. (1998) The role of alcohol dehydrogenase in the metabolism of the colon carcinogen methylazoxymethanol. Toxicol. Sci. 45, 66–71. [DOI] [PubMed] [Google Scholar]
- Izumi K.; Otsuka H.; Furuya K.; Akagi A. (1979) Carcinogenicity of 1, 2-dimethylhydrazine dihydrochloride in BALB/c mice. Virchows Arch. 384, 263–267. [DOI] [PubMed] [Google Scholar]
- Bolognesi C.; Mariani M. R.; Boffa L. C. (1988) Target tissue DNA damage in inbred mouse strains with different susceptibility to the colon carcinogen 1, 2-dimethylhydrazine. Carcinogenesis 9, 1347–1350. [DOI] [PubMed] [Google Scholar]
- Peters W.; Kock L.; Nagengast F.; Kremers P. (1991) Biotransformation enzymes in human intestine: critical low levels in the colon?. Gut 32, 408–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn O. S.; Ishizaki H.; Yang C. S.; Fiala E. S. (1991) Metabolism of azoxymethane, methylazoxymethanol and N-nitrosodimethylamine by cytochrome P450IIE1. Carcinogenesis 12, 127–131. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Bartels M. J.; Pottenger L. H.; Gollapudi B. B.; Schisler M. R. (2006) Simultaneous quantitation of 7-methyl-and O6-methylguanine adducts in DNA by liquid chromatography–positive electrospray tandem mass spectrometry. J. Chromatogr., B 833, 141–148. [DOI] [PubMed] [Google Scholar]
- Takada H.; Hirooka T.; Hiramatsu Y.; Yamamoto M. (1982) Effect of β-glucuronidase inhibitor on azoxymethane-induced colonic carcinogenesis in rats. Cancer Res. 42, 331–334. [PubMed] [Google Scholar]