

Background: Tropomodulins (Tmods) cap pointed ends of actin filaments in a tropomyosin (TM)-dependent manner.
Results: Tmod1 and Tmod3 similarly cap actin filaments with diverse TM and actin isoforms, but only Tmod3 sequesters β- and γcyto-actin monomers.
Conclusion: Isoform-specific actin monomer sequestration by Tmod3 may provide a mechanism for actin remodeling in TM-deficient regions of cells.
Significance: Defining the actin-regulatory activities of Tmods illuminates cytoskeletal dynamics.
Keywords: Actin, Cytoskeleton, Protein Chemistry, Protein-Protein Interactions, Tropomyosin, Actin Capping, Actin-binding Proteins, Tropomodulin
Abstract
Tropomodulins (Tmods) are F-actin pointed end capping proteins that interact with tropomyosins (TMs) and cap TM-coated filaments with higher affinity than TM-free filaments. Here, we tested whether differences in recognition of TM or actin isoforms by Tmod1 and Tmod3 contribute to the distinct cellular functions of these Tmods. We found that Tmod3 bound ∼5-fold more weakly than Tmod1 to α/βTM, TM5b, and TM5NM1. However, surprisingly, Tmod3 was as effective as Tmod1 at capping pointed ends of skeletal muscle α-actin (αsk-actin) filaments coated with α/βTM, TM5b, or TM5NM1. Tmod3 only capped TM-coated αsk-actin filaments more weakly than Tmod1 in the presence of recombinant αTM2, which is unacetylated at its NH2 terminus, binds F-actin weakly, and has a disabled Tmod-binding site. Moreover, both Tmod1 and Tmod3 were similarly effective at capping pointed ends of platelet β/cytoplasmic γ (γcyto)-actin filaments coated with TM5NM1. In the absence of TMs, both Tmod1 and Tmod3 had similarly weak abilities to nucleate β/γcyto-actin filament assembly, but only Tmod3 could sequester cytoplasmic β- and γcyto-actin (but not αsk-actin) monomers and prevent polymerization under physiological conditions. Thus, differences in TM binding by Tmod1 and Tmod3 do not appear to regulate the abilities of these Tmods to cap TM-αsk-actin or TM-β/γcyto-actin pointed ends and, thus, are unlikely to determine selective co-assembly of Tmod, TM, and actin isoforms in different cell types and cytoskeletal structures. The ability of Tmod3 to sequester β- and γcyto-actin (but not αsk-actin) monomers in the absence of TMs suggests a novel function for Tmod3 in regulating actin remodeling or turnover in cells.
Introduction
Actin filament assembly and turnover are essential for the mechanical strength and physiological function of virtually all eukaryotic cells and are regulated by a host of actin-binding proteins, including side-binding and capping proteins. Recently, members of the tropomodulin (Tmod)2 family of actin filament (F-actin) pointed end capping proteins have attracted attention as key regulators of actin filament architecture and organization (1, 2). Of the four known Tmod isoforms (Tmods1–4), the best characterized isoforms are Tmod1 and Tmod3, both of which are present in diverse cells and tissues (2). Tmod1 is predominantly expressed in postmitotic, terminally differentiated cells where it caps the pointed ends of actin filaments in stable, long lived cytoskeletal assemblies, such as the sarcomeres of striated muscle cells (3) and the short actin filaments of the spectrin-actin membrane skeleton in red blood cells, where it regulates precise actin filament lengths and stability (2). Unlike Tmod1, Tmod3 is predominantly found in more dynamic cellular contexts, such as the dendritic actin network in the leading lamellipodia of migrating endothelial cells where it acts as a negative regulator of F-actin assembly and cell migration rates (4). Tmod3 is also present in proliferating erythroblasts prior to enucleation with Tmod3 deficiency leading to impaired differentiation, aberrant F-actin organization, and reduced enucleation of mouse erythroblasts (5). However, in a subset of its contexts, Tmod3 can be incorporated into a stable F-actin membrane skeleton in a manner similar to Tmod1, such as on the lateral membranes of polarized epithelial cells (6) or in the sarcoplasmic reticulum (SR) of skeletal muscle fibers (7). In these contexts, Tmod3 depletion leads to F-actin cytoskeletal disruption and impaired membrane mechanics (6, 7) similar to Tmod1 depletion phenotypes in red blood cells (8) and ocular lens fibers (9). Moreover, Tmod3 levels decrease whereas Tmod1 levels increase as erythroid precursors prepare to enucleate and begin to synthesize membrane skeleton components during terminal erythroid differentiation (5), highlighting the divergent roles of Tmod1 versus Tmod3 in regulating dynamic versus stable actin filament architectures in proliferating versus terminally differentiated cells, respectively.
Tmods consist of an N-terminal tropomyosin (TM)/pointed end actin capping (TM-Cap) domain containing two TM-binding motifs and a TM-dependent actin pointed end capping motif followed by a C-terminal leucine-rich repeat/Cap domain containing five leucine-rich repeat motifs and a TM-independent actin pointed end capping motif (see Fig. 1A) (2). TMs are α-helical, rodlike F-actin side-binding proteins that bind to the TM-Cap domain of Tmods via Tmod recognition motifs located in the variable N-terminal exons of TM isoforms (see Fig. 1B) (10–17). Tmod1 caps the pointed ends of TM-decorated actin filaments with an affinity more than 1,000-fold greater than for bare actin pointed ends (Kd ∼0.2 μm for pure actin and in the nm-to-pm range for TM-actin filaments) (18–20). In addition, mutations in Tmod1 that disable striated muscle α/βTM binding lead to reduced Tmod1 capping of α/βTM-coated actin filaments in vitro (21–23) as well as ineffective targeting of Tmod1 to thin filament pointed ends in the sarcomeres of both cultured cardiac and skeletal myocytes (23, 24). Moreover, various combinations of Tmod and TM isoforms show distinct binding affinities (20, 25–27), and Tmod1 caps actin filaments coated with nonmuscle TM5a more strongly than filaments coated with striated muscle α/βTM in vitro (20), suggesting that TM isoforms may selectively modulate the pointed end capping activities of Tmods in cells. However, the abilities of different TM isoforms to regulate the pointed end capping of Tmod1 versus Tmod3 have not been studied.
FIGURE 1.

Schematics of the molecular layouts of Tmod and TM proteins used in this study. A, molecular layout of Tmod1 and Tmod3 with TM-binding and actin-binding α-helices labeled. The TM-Cap domain α1- and α3-helices are located within TM-binding Site 1 and Site 2, respectively. The α2-helix contains an actin-binding site, which binds monomers, nucleates filaments, and is required for TM-dependent pointed end capping. The leucine-rich repeat (LRR)-Cap domain α6-helix and the C-terminal tail contain a second actin-binding site important for actin nucleation and TM-independent pointed end capping. For a review, see Ref. 2. B, identities and exon usage of wild-type TM proteins (α/βTM, αTM2, TM5b, and TM5NM1) and the location of the Tmod-binding sites in exon 1a or 1b on TMs. For reviews, see Refs. 13 and 17. Note that native rabbit skeletal muscle α/βTM is acetylated at the N terminus, whereas recombinant αTM2, TM5b, and TM5NM1 expressed in E. coli are not.
Although it is known that TM isoforms can differentially govern the pointed end capping function of Tmod1 (20), it remains unknown whether different actin isoforms can govern the pointed end capping function of any Tmod. Although the six actin isoforms share at least 93% sequence identity, they show differential expression patterns and are associated with distinct cytoskeletal assemblies (28). In skeletal muscle fibers, Tmod1 caps the pointed ends of the skeletal muscle α-actin (αsk-actin) that polymerizes into the sarcomeric thin filaments, whereas Tmod3 is associated with the cytoplasmic γ-actin (γcyto-actin) that polymerizes into the SR-associated membrane skeleton (7). Skeletal muscle fibers also contain a costamere-associated cytoplasmic β-actin network (29), although which Tmod isoforms (if any) associate with this network remain unknown. The segregation of Tmod1 versus Tmod3 with sarcomeric αsk-actin filaments versus SR-associated γcyto-actin filaments, respectively, suggests that Tmod1 and Tmod3 may exhibit different capping activities for the pointed ends of TM-coated αsk-actin filaments as compared with β-actin or γcyto-actin filaments. On the other hand, there does not seem to be a simple one-to-one correspondence between Tmods and actin isoforms because Tmod1 is associated with the TM5b/TM5NM1-coated β-actin (not αsk-actin) filaments in the spectrin-actin membrane skeleton of red blood cells (30–34). In addition to capping pointed ends, in vitro biochemical studies have shown that, in the absence of TMs, Tmods can also bind αsk-actin monomers and weakly nucleate αsk-actin assembly (35, 36). Tmod1 and Tmod3 have similar abilities to nucleate αsk-actin polymerization (36), and both Tmod1 and Tmod3 can bind to αsk-actin monomers at low ionic strength (36). However, only Tmod3 has been shown to reduce αsk-actin polymerization at physiological ionic strength likely by sequestering αsk-actin monomers (35). Neither Tmod1 nor Tmod3 interactions with cytoplasmic β- and γcyto-actin have been studied either in the presence or in the absence of TMs.
Here, we compare Tmod1 and Tmod3 with respect to Tmod-TM binding, Tmod-TM-actin pointed end capping, and Tmod/actin interactions using various combinations of Tmod, TM, and actin isoforms. Tmod1 bound to muscle (α/βTMs) and nonmuscle TMs (TM5NM1 and TM5b) ∼5-fold better than Tmod3, but Tmod1 and Tmod3 showed no significant differences in pointed end capping of αsk-actin filaments coated with any of these TMs. Moreover, both Tmod1 and Tmod3 were equally effective at capping platelet β/γcyto-actin filaments coated with TM5NM1. Thus, Tmod1 and Tmod3 do not discriminate between TM isoforms or actin isoforms with respect to high affinity capping of pointed ends, and the TM-actin pointed end capping activities of Tmod1 and Tmod3 fail to explain the differential associations of these Tmods with diverse cytoskeletal structures in cells. However, in the absence of TMs, Tmod3 (but not Tmod1) sequestered cytoplasmic β- and γcyto-actin monomers and inhibited polymerization but had relatively little effect on steady state αsk-actin polymerization in a sedimentation assay. We propose that Tmod3 functions to sequester β- and γcyto-actin monomers and inhibit filament assembly in dynamic regions of cells with reduced levels of TMs, such as protruding lamellipodia (37, 38). This explains previous observations that Tmod3 levels are inversely correlated with lamellipodial F-actin levels, providing a mechanism for negative regulation of endothelial cell migration rates by Tmod3 (4). In other regions of cells containing TMs, TM binding to polymerizing β- and γcyto-actin filaments may switch Tmod3 from relatively low affinity (μm) actin monomer binding to high affinity (nm) pointed end capping, thereby stabilizing Tmod3·TM·actin filament complexes.
EXPERIMENTAL PROCEDURES
Proteins
αsk-Actin was prepared from rabbit muscle acetone powder as described previously (39). N-(1-Pyrene)iodoacetamide-labeled αsk-actin was prepared and stored as described previously (40). For nonmuscle actin capping and nucleation experiments, human platelet actin (∼85% β-actin, 15% γcyto-actin) and pyrene-labeled human platelet actin were obtained from Cytoskeleton, Inc. (Denver, CO), solubilized to prepare G-actin, and pyrene-labeled as described previously (40). For actin monomer sequestration experiments, β-, γcyto-, and insect actins were expressed using a baculovirus-driven expression system in insect cells and purified as described previously (41). Protein concentrations of actins were determined from the absorbance at 290 nm and an extinction coefficient of 0.63 m−1 cm−1 (41). Capping protein of chicken skeletal muscle (CapZ) was a gift from Dorothy A. Schafer (University of Virginia, Charlottesville, VA) and Sumiko Kimura (Chiba University, Japan). Rabbit skeletal muscle α/βTM was prepared as described previously (42). Recombinant rat TM5b (TM5b cDNA in the pJC20 vector), a gift from Michael A. Geeves (University of Kent, Canterbury, UK), was expressed in BL21 Escherichia coli and purified as described previously (43). A clone of human TM5NM1 was a gift from Andrea Bacconi (The Scripps Research Institute, La Jolla, CA) and amplified from a pEGFP-TM5NM1 expression vector using PCR primers designed to introduce NcoI and XhoI restriction sites for cloning into the bacterial pET-14b expression vector (Novagen). Recombinant human TM5NM1 was expressed in BL21 E. coli and purified in the same manner as for TM5b. Rat αTM2 was a gift from Sarah E. Hitchcock-DeGregori (Rutgers University, Piscataway, NJ) (44). TM protein concentrations were determined using a BCA Protein Assay kit (Pierce). Human Tmod1, mouse Tmod3, and their TM binding-incompetent mutants (Tmod1-L27G/I131D and Tmod3-L29G/L134D) were expressed as glutathione S-transferase fusion proteins in BL21 E. coli and purified by thrombin cleavage followed by affinity chromatography using glutathione-Sepharose and ion exchange chromatography as described previously (36). Concentrations of purified Tmods and mutants were determined spectroscopically as described previously (36).
TM-Tmod Binding Assays
We determined the relative strengths of Tmod/TM interactions by performing blot overlay assays as described previously (45, 46). Briefly, increasing amounts of purified wild-type or mutant Tmod1 and Tmod3 were electrophoretically separated by SDS-PAGE on 12% Laemmli minigels (47) and transferred to nitrocellulose in methanol-containing transfer buffer. Parallel gels were stained with Coomassie Blue to control for protein loading or transferred to nitrocellulose membranes to assess TM binding. Blots were incubated with 5 μg/ml purified TMs, and TMs bound to immobilized Tmods were then detected using anti-TM antibodies as follows: TM311 (1:1,000; Abcam) for rabbit skeletal muscle α/βTM, AB5441 (1:1,000; Millipore) for TM5b, and AB5447 (1:1,000; Millipore) for TM5NM1. After washing, blot overlays were incubated with HRP-conjugated secondary antibodies (1:10,000; Invitrogen). Bands were detected using an enhanced chemiluminescence kit (Pierce) according to the manufacturer's instructions and exposed to film. Background-corrected band intensities were quantified in NIH ImageJ.
To confirm that the Tmod-TM binding avidities determined in the blot overlay assays were not confounded by SDS-PAGE treatment and denaturation of Tmods, we also performed dot blot overlay assays in the absence of detergents. Briefly, 200 ng of wild-type Tmods were diluted in 400 μl of 5 μg/ml BSA in a Hepes-buffered salt solution and then directly spotted onto a nitrocellulose membrane using a Schleicher & Schuell dot blot apparatus. BSA alone was also spotted as a negative control for TM binding, and membranes were stained with Ponceau S to control for total protein loading. Incubation with TMs and detection with anti-TM antibodies were then performed as described in the previous paragraph. After washing, blots were incubated with 680LT-conjugated secondary antibodies (1:20,000; LI-COR Biosciences) and imaged on a LI-COR Odyssey infrared imaging system (resolution, 84 μm). Background-corrected dot intensities on blots were quantified in NIH ImageJ. For background correction, the intensity of the “BSA alone” dot was subtracted from the intensities of the Tmod1 and Tmod3 dots for each TM. The intensity of the BSA alone dot was never more than ∼2% of the total (uncorrected) signal intensity of TM bound to each Tmod.
Co-sedimentation Assay for TMs Binding to F-actin
The binding affinity of TMs to F-actin was measured by co-sedimentation as described previously (48) but with slight modifications. Stock solution of G-actin in buffer A (2 mm Tris, 0.2 mm CaCl2, 0.2 mm ATP, 1 mm DTT, pH 8.0) was polymerized into F-actin by addition of volume of 10× polymerizing buffer (200 mm Hepes, 1 m KCl, 20 mm MgCl2, 2 mm DTT, 5 mm ATP, 10 mm EGTA, pH 7.5). Binding of TMs to F-actin was performed in F-buffer (20 mm Hepes, 0.1 m KCl, 2 mm MgCl2, 1 mm DTT, pH 7.4) for 30 min at room temperature. Protein mixtures were ultracentrifuged in a Beckman TLA100 rotor at 150,000 × g for 20 min, and supernatants were removed. Supernatants and pellets were solubilized in SDS gel sample buffer, adjusted to the same volume, and analyzed by SDS-PAGE on 4–20% gradient minigels. Gels were stained with Coomassie Blue, and background-corrected band intensities were quantified by densitometry using NIH ImageJ.
Fluorescence Assays for Actin Polymerization
Elongation rates at actin filament pointed ends were measured as described previously (18, 49) but with slight modifications. Briefly, 1.8 μm G-actin (8% pyrene-labeled for αsk-actin or 10% pyrene-labeled for human platelet β/γcyto-actin) was converted to Mg2+-actin as described previously (50). CapZ-capped actin filament nuclei (CapZ:actin, 1:100) were prepared by mixing 10 μm G-actin and 100 nm CapZ in G-buffer followed by addition of volume 10× polymerization buffer and incubation overnight on ice. Seeds (final concentration, 1.6 nm CapZ, 160 nm actin) were incubated with the indicated concentrations of Tmods and TMs added to the indicated concentrations of G-actin for 5 min at 25 °C and polymerization was initiated immediately by addition of volume of 10× polymerization buffer (200 mm Hepes, pH 7.5, 1 m KCl, 20 mm MgCl2, 2 mm DTT, 5 mm ATP, 10 mm EGTA). Fluorescence measurements (excitation, 366.5 nm; emission, 407 nm) were performed using a Fluoromax 3 fluorometer (HORIBA Jobin Yvon, Edison, NJ) or an Envision 2103 Multilabel Reader (PerkinElmer Life Sciences). Capping activities for Tmod1 or Tmod3 were obtained from initial elongation rates over the first 1 min as determined from the slopes of the polymerization traces by linear regression analysis using Microsoft Excel. Rates in the presence of increasing concentrations of Tmods or TMs were divided by the rate in the absence of Tmod, yielding a series of rate/control rate ratios, which were converted into pointed end capping efficiencies using the following formula: Pointed end capping efficiency = (1 − Rate/Control rate) × 100 (%). Thus, when rate/control rate = 0, 100% of pointed ends are capped.
Nucleation of platelet β/γcyto-actin was measured as described previously for αsk-actin (36) but with the following modifications. 3.4 μm G-actin (5% pyrene-labeled) was converted to Mg2+-actin as described previously (36), and then the indicated concentrations of Tmod proteins were added and incubated for 1 min. Polymerization was then initiated as above. Fluorescence measurements were performed using an Envision 2103 Multilabel Reader as described above.
G-actin Binding Assays
Non-denaturing PAGE was performed as described by Safer (51). G-actin and increasing concentrations of either Tmod1 or Tmod3 were incubated in buffer A (2 mm Tris, pH 8.0, 0.2 mm CaCl2, 0.2 mm DTT, 0.2 mm ATP) for 20 min at 25 °C. Protein mixtures were then supplemented with 0.25 volume of loading buffer (50% glycerol, 0.05% bromphenol blue) and electrophoresed using a Bicine/triethanolamine buffer system. Proteins were visualized by staining with Coomassie Blue. In sedimentation assays for G-actin sequestration, G-actin was mixed with Tmod1 or Tmod3, and polymerization was initiated by adding 10× polymerization buffer. After polymerizing to steady state for 16 h at 25 °C, protein mixtures were separated by ultracentrifugation in a Beckman TLA100 rotor at 300,000 × g for 20 min. Supernatants and pellets were solubilized in SDS gel sample buffer, adjusted to the same volume, and analyzed by SDS-PAGE on 12% gels followed by Coomassie Blue staining. In both types of G-actin binding assays, background-corrected protein band intensities were quantified using NIH ImageJ.
RESULTS
Sarcomeric and Cytoskeletal TMs Bind to αsk-Actin Filaments with Varying Affinities
TM isoforms bind to αsk-actin filaments with distinct affinities (13), affecting TM polymerization along F-actin and the probability that the terminal TM will associate with the pointed end (52, 53). In its pointed end position, TM influences the actin pointed end capping activities of Tmods (18–20, 25, 26). Therefore, we assembled a panel of muscle and cytoskeletal TM isoforms with varying affinities for F-actin, including native striated muscle α/βTM, recombinant rat αTM2, and two cytoskeletal TMs, recombinant TM5NM1 from the γTM gene and TM5b from the αTM gene (Fig. 1). We examined the F-actin binding affinity of these TM isoforms for αsk-actin by standard co-sedimentation assays followed by SDS-PAGE, Coomassie Blue staining, and densitometry. The data were fit to the Hill equation to determine the affinity (Kapp) of TMs for F-actin and their Hill coefficients, indicative of cooperativity. Binding of all TMs showed sigmoidal saturation curves, indicating their cooperative binding to F-actin, as expected (Fig. 2A) (52, 53). α/βTM and TM5b were the strongest F-actin-binding TMs, achieving saturation at 1.0–1.5 μm TMs, whereas TM5NM1 and recombinant αTM2 were weaker binding TMs, achieving saturation between 2.0 and 2.5 μm TMs. Their Kapp values ranged from the strongest at 2.20 ± 0.11 μm−1 for native α/βTM to the weakest at 0.88 ± 0.10 μm−1 for recombinant αTM2, whereas their Hill coefficients ranged from 2.12 ± 0.13 for TM5NM1 to 2.97 ± 0.31 for TM5b (Fig. 2B). The strong F-actin binding of native α/βΤΜ and TM5b is consistent with previous reports (54–56) as is the weak F-actin binding of bacterially expressed recombinant αTM2 (57). Previous studies have demonstrated that long, exon 1a-containing muscle TMs, such as αTM2, require N-terminal acetylation for their high affinity binding to F-actin (which is not present in the bacterially expressed protein), whereas the short, exon 1b-containing TMs, such as TM5b, do not (57, 58). Overall, the relative binding strengths of these TMs for αsk-actin were from strongest to weakest: α/βTM > TM5b > TM5NM1 > αTM2.
FIGURE 2.
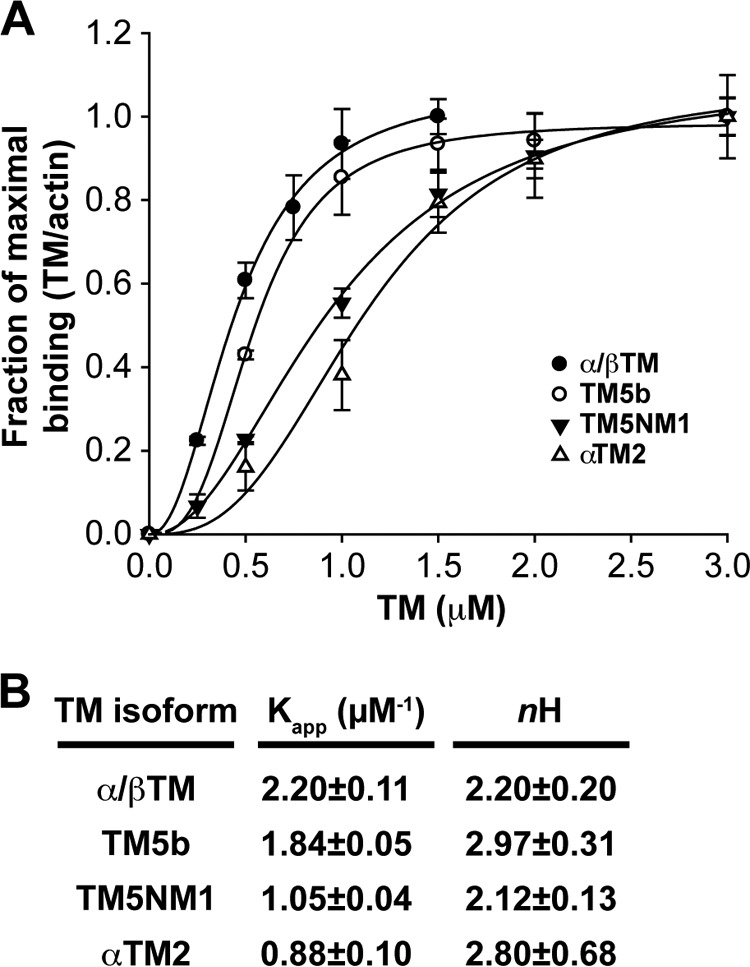
Binding of α/βTM, TM5b, TM5NM1, and αTM2 to skeletal muscle αsk-actin filaments as determined in a co-sedimentation assay. A, purified TMs at the indicated concentrations were mixed with 4 μm prepolymerized F-actin, incubated 30 min at room temperature, and ultracentrifuged to sediment F-actin with bound TMs. Equivalent volumes of supernatants and pellets were analyzed by SDS-PAGE and Coomassie Blue staining followed by densitometry to quantify the percentage of TM in the supernatant or pellet. After subtraction of the amount of TM sedimenting in the absence of F-actin, the amount of TM in the pellet was converted to mol of TM/mol of actin and plotted as a percentage of maximal TM binding to better compare the binding curves for each TM (which saturate F-actin at somewhat different molar ratios due to their different lengths). Note that α/βTM concentrations >1.5 μm were not tested because saturation had been achieved. Curves are drawn based on fitting to the Hill equation for cooperative binding using SigmaPlot 9.0. Data shown are mean ± S.D. (error bars) of three experiments. B, tabulation of the Kapp and Hill coefficient (nH) of each TM.
Tmod1 Interacts More Strongly with Both Muscle and Nonmuscle TMs than Does Tmod3
Similar to TM/αsk-actin interactions, TM/Tmod interactions are also isoform-specific (20, 25–27). Therefore, we compared the relative strengths of Tmod1 versus Tmod3 binding to α/βTM, TM5b, and TM5NM1. αTM2 was not included based on previous studies showing undetectable binding of Tmods to recombinant unacetylated αTM2 (59). Binding was evaluated semiquantitatively using blot overlay assays in which each TM was incubated with immobilized Tmods or Tmod mutants on nitrocellulose blots followed by antibodies to detect the bound TMs (45, 46). The amount of each TM bound was normalized to the level of Tmod or mutant protein by performing Coomassie Blue staining of a duplicate parallel gel (Fig. 3A) (45). These experiments showed that skeletal muscle α/βTM bound more strongly to Tmod1 than Tmod3 with a ∼5-fold difference in binding (Fig. 3, A and C), consistent with previous studies (45). TM5b and TM5NM1 also demonstrated stronger binding to Tmod1 as compared with Tmod3 with a similar ∼5-fold difference in avidity (Fig. 3, B and C). SDS-PAGE treatment and resultant denaturation of the immobilized Tmods are not the reason for the differential interactions of Tmod1 and Tmod3 with the various TMs because dot blot overlays in which wild-type Tmods were spotted onto nitrocellulose membranes in the absence of detergents and then incubated with TMs yielded TM-Tmod binding avidities similar to those obtained with the conventional blot overlays with all TMs binding ∼4–5-fold more strongly to Tmod1 than to Tmod3 (Fig. 3, D–G).
FIGURE 3.

Tmod1 binds more strongly than Tmod3 to α/βTM, TM5b, and TM5NM1. A and B, increasing amounts of Tmod proteins (WT Tmod1, Tmod1-L27G/I131D (double mutant (DM)), WT Tmod3, and Tmod3-L29G/L134D (double mutant (DM)); 100, 200, and 400 ng from the left) were separated by SDS-PAGE, transferred to nitrocellulose membranes, and overlaid with 5 μg/ml α/βTM (A), TM5b (B), or TM5NM1 (B). Bound TMs were detected using anti-TM antibodies. Parallel Coomassie-stained gels show Tmod loading amounts. Note that wild-type Tmod1 showed relatively strong TM-binding ability; thus, only one-fourth as much wild-type Tmod1 was loaded as compared with the other Tmod proteins (25, 50, and 100 ng from the left). C, relative TM-Tmod binding was quantified densitometrically by normalizing the band intensities from the overlays to their corresponding Coomassie-stained bands. Data shown are mean ± S.D. (error bars) of four lanes. D–F, wild-type Tmod1 or Tmod3 (200 ng each) diluted in 5 μg/ml BSA in a Hepes-buffered salt solution was spotted directly onto nitrocellulose membranes in the absence of detergents and overlaid with 5 μg/ml α/βTM (D), TM5b (E), or TM5NM1 (F). Bound TMs were detected using anti-TM antibodies. Ponceau S staining shows uniform loading amounts of Tmods and BSA. G, relative TM-Tmod binding was quantified densitometrically by normalizing the background-corrected dot intensities from the overlays to the background-corrected intensities of their corresponding Ponceau-stained dots. Data shown are mean ± S.D. (error bars) of three lanes.
To verify the specificity of TM binding in these assays, we also tested TM binding to Tmod1 and Tmod3 mutants in which both their first and second TM-binding sites were disabled by mutations in their TM-binding α1- and α3-helices (21, 22). As expected, all TMs exhibited significantly less binding to the double mutant Tmod1-L27G/I131D, demonstrating specificity of binding (Fig. 3, A--C). TM binding was also barely detectable with Tmod3-L29G/L134D, a mutant corresponding to Tmod1-L27G/I131D (Fig. 3, A–C), indicating that TMs recognize Tmod3 via TM-binding sites homologous to those in Tmod1 (Fig. 1A), as expected from previous studies with the TM-binding peptides of Tmods (60). All TMs tested displayed similar relative binding to the wild-type and mutant Tmod proteins with wild-type Tmod1 ≫ wild-type Tmod3 > Tmod1-L27G/I131D > Tmod3-L29G/L134D. Although these data do not address differences in affinities of Tmod1 and Tmod3 for α/βTM, TM5NM1, or TM5b (60), they do show that Tmod1 recognizes each TM better than does Tmod3. This is in agreement with measurements of Kd values for binding of Tmod1 and Tmod3 fragments containing TM-binding Site 1 or Site 2 (Fig. 1) to the N-terminal peptides αTM1a, αTM1b, and γTM1b corresponding to the α/βTM, TM5b, and TM5NM1 proteins, respectively (60).
Tmod1 and Tmod3 Exhibit Similar Pointed End Capping Efficiencies for αsk-Actin and β/γcyto-Actin Filaments Regardless of the TM Isoform Present
To compare the pointed end capping efficiencies of Tmod1 versus Tmod3 with various TM isoforms, we first tested the abilities of Tmods to inhibit initial elongation rates from αsk-actin filament pointed ends. In these experiments, we measured pointed end elongation rates from CapZ-capped, TM-coated, pyrene-labeled αsk-actin filament seeds (Fig. 4, A–D) (18, 19, 49). Pointed end capping efficiencies were expressed as the percent inhibition of the initial elongation rate in the presence of Tmod with respect to the control rate without Tmod (Fig. 4, E–H). Surprisingly, despite substantial differences in TM binding (Fig. 3), both Tmod1 and Tmod3 displayed similar efficiencies of pointed end capping for αsk-actin filaments in the presence of α/βTM, TM5b, or TM5NM1 (Fig. 4, A–C and E–G). α/βTM- and TM5b-coated αsk-actin filaments were capped most strongly by Tmods with ∼40–50% capping efficiency at 2 nm Tmods, whereas TM5NM1-coated αsk-actin filaments were capped at ∼30% efficiency at 2 nm Tmods (Fig. 4, A–C and E–G). A difference between pointed end capping by Tmod1 and Tmod3 was only observed with recombinant αTM2-coated αsk-actin filaments with Tmod1 capping pointed ends more than twice as efficiently as Tmod3 (Fig. 4, D and H). Although Tmod1 was reasonably efficient at pointed end capping of αTM2-coated αsk-actin filaments (80% capping at 10 nm Tmod1), Tmod3 was only ∼30% efficient at 10 nm Tmod3 (Fig. 4H). Because all the TM concentrations in these assays were at or close to saturation for TM binding to actin (Fig. 2), the similarities or differences in Tmod1 and Tmod3 capping in the presence of the various TMs are most likely not directly related to their relative TM-actin binding affinities. Instead, differences in high affinity capping of TM-actin filament pointed ends by Tmod1 and Tmod3 may only be revealed in the case of an exceptionally weak Tmod/TM interaction as is the case for recombinant unacetylated αTM2 and Tmod1 (59). However, because muscle TMs are acetylated in vivo, differences in Tmod1 and Tmod3 capping of α/βTM-αsk-actin filaments are unlikely to explain selective assembly of Tmod1 with sarcomeric α/βTM-αsk-actin thin filaments (3).
FIGURE 4.

Tmod1 and Tmod3 cap αsk-actin filament pointed ends similarly with α/βTM, TM5b, and TM5NM1, but Tmod3 caps αsk-actin pointed ends more weakly than Tmod1 with αTM2. A–D, pyrene-actin polymerization kinetics depicting αsk-actin elongation from the pointed ends of CapZ-capped αsk-actin filaments in the presence of Tmod1 or Tmod3 and either α/βTM (A), TM5b (B), TM5NM1 (C), or αTM2 (D). Concentrations of proteins were: 1.8 μm G-actin (8% pyrene-labeled), 1.6 nm CapZ-F-actin seeds (1:100; 1.6 nm CapZ, 160 nm αsk-actin), and Tmods and TMs as indicated. E–H, the pointed (P) end capping activities of Tmod1 and Tmod3 calculated from initial rates of αsk-actin elongation from pointed ends of CapZ-capped actin filaments in the presence of Tmod1 or Tmod3 and either α/βTM (E), TM5b (F), TM5NM1 (G), or αTM2 (H). The pointed end capping efficiency is plotted as (1 − Rate/Control rate) × 100 (%). Thus, when rate/control rate = 0, 100% of pointed ends are capped. Data shown are mean ± S.D. (error bars) of three experiments. *, p < 0.05 (t test). AU, arbitrary units.
In another approach to compare capping of αsk-actin filaments by Tmod1 and Tmod3 in the presence of α/βTM, TM5b, or TM5NM1, we tested high affinity Tmod capping as a function of TM concentration. We reasoned that, at subsaturating TM concentrations, there would be a reduced probability that TM molecules would be located at the pointed end position to enhance the capping affinities of Tmods (18–20, 52, 53). Therefore, we compared the pointed end capping efficiency of Tmods over a range of TM concentrations with αsk-actin filaments. With the exception of the 0.5 μm concentration, the pointed end capping activities of Tmod1 and Tmod3 were indistinguishable for α/βTM (Fig. 5A). Furthermore, both Tmod1 and Tmod3 displayed similar extents of pointed end capping at all concentrations of TM5b and TM5NM1 (Fig. 5, B and C). It is likely that the high affinity interaction of Tmods with TM-actin pointed ends favors TM binding along the pointed end regions of actin filaments over all TM concentrations. In this experiment, both Tmod1 and Tmod3 achieved 70–80% capping with 2.5 nm Tmod at 0.5 μm TM5b and 1.0 μm TM5NM1, which are TM concentrations that half-maximally saturate binding to F-actin as measured in our co-sedimentation assays (Fig. 2). Thus, although the blot overlay assays indicated that Tmod1 binds ∼4–6-fold more strongly than Tmod3 to all TMs tested (Fig. 3), this is not reflected by similar differences in high affinity pointed end capping of Tmod1 and Tmod3 in the presence of these TMs. Collectively, these data fail to identify noteworthy differences in high affinity capping of αsk-actin filaments by Tmod1 and Tmod3 in the presence of different TMs. Therefore, the TM-dependent pointed end capping activities of Tmod1 and Tmod3 are unlikely to account for the preferential association of Tmod1 with αsk-actin structures, such as the thin filaments in skeletal muscle sarcomeres (3).
FIGURE 5.
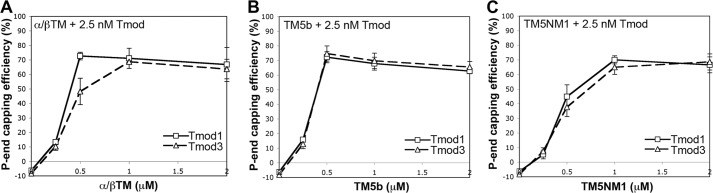
Tmod1 and Tmod3 cap αsk-actin pointed ends similarly over a range of TM concentrations for α/βTM, TM5NM1, and TM5b. A–C, the pointed (P) end capping activities of Tmod1 and Tmod3 were evaluated by measuring their ability to inhibit the initial rates of αsk-actin elongation from the pointed ends of CapZ-capped actin filaments in the presence of the increasing amounts of α/βTM (A), TM5b (B), or TM5NM1 (C). Concentrations of proteins were: 1.8 μm G-actin (8% pyrene-labeled), 1.6 nm CapZ-F-actin seeds (1:100; 1.6 nm CapZ, 160 nm αsk-actin), and Tmods and TMs as indicated. The pointed end capping efficiency is plotted as (1 − Rate/Control rate) × 100 (%). Thus, when rate/control rate = 0, 100% of pointed ends are capped. Data shown are mean ± S.D. (error bars) of three experiments.
Next, we tested whether Tmod1 or Tmod3 differentially capped β/γcyto-actin filaments coated with a nonmuscle TM. In these experiments, we used purified human platelet β/γcyto-actin. CapZ-capped β/γcyto-actin filament seeds were coated with TM5NM1, and pointed end elongation rates were measured by adding β/γ-actin monomers (10% pyrene-labeled) in the presence of saturating concentrations of TM5NM1. Results show that both Tmod1 and Tmod3 capped pointed ends of TM5NM1-coated β/γcyto-actin filaments with similar efficiencies at both 5 and 10 nm Tmod (Fig. 6). Based on these experiments with β/γcyto-actin and the experiments described above with αsk-actin, no significant differences were observed between Tmod1 and Tmod3 with respect to high affinity capping of TM-actin filaments. Thus, differences in Tmod1 and Tmod3 capping of actin filaments composed of β/γcyto-actin and nonmuscle TMs are unlikely to account for preferential Tmod3 assembly with γcyto-actin filaments (containing TM5NM1 and TM4) in the SR of striated muscle (7) or Tmod1 assembly with β-actin filaments (containing TM5NM1 and TM5b) in the erythrocyte membrane skeleton (8).
FIGURE 6.
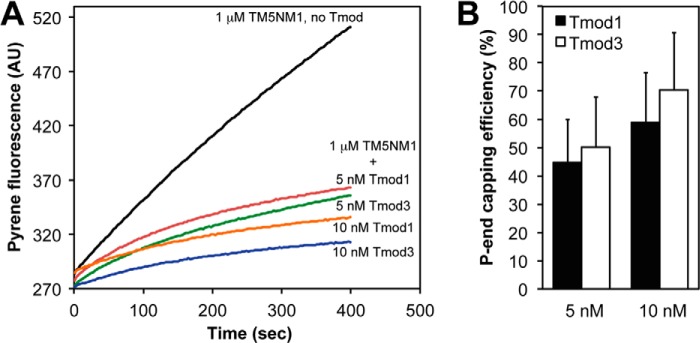
Tmod1 and Tmod3 cap platelet β/γcyto-actin pointed ends similarly with TM5NM1. A, pyrene-actin polymerization kinetics depicting platelet β/γcyto-actin elongation from the pointed ends of CapZ-capped β/γcyto-actin filaments in the presence of TM5NM1 and two different concentrations of Tmod1 or Tmod3. Concentrations of proteins were: 1.8 μm G-actin (8% pyrene-labeled), CapZ-F-actin seeds (1:100; 1.6 nm CapZ, 160 nm β/γcyto-actin), and Tmods and TMs as indicated. B, the pointed (P) end capping activities of Tmod1 and Tmod3 were evaluated by measuring their ability to inhibit the initial rates of β/γcyto-actin elongation from the pointed ends of CapZ-capped actin filaments in the presence of TM5NM1 and either Tmod1 or Tmod3. The pointed end capping efficiency is plotted as (1 − Rate/Control rate) × 100 (%). Thus, when rate/control rate = 0, 100% of pointed ends are capped. Data shown are mean ± S.D. (error bars) of three experiments. AU, arbitrary units.
Tmod1 and Tmod3 Similarly Nucleate β/γcyto-Actin Filaments in the Absence of TMs
Tmods can promote αsk-actin polymerization in the absence of TMs by stabilizing spontaneously formed actin nuclei with activity at submicromolar concentrations of Tmods (35, 36, 49). Tmod1 and Tmod3 similarly promote αsk-actin filament assembly with a 200 nm concentration of either Tmod nucleating assembly of ∼3–4-fold increased numbers of barbed ends compared with αsk-actin alone (36). We tested whether Tmod1 and Tmod3 might be different at promoting F-actin assembly by comparing the kinetics of spontaneous β/γcyto-actin polymerization in the presence of increasing concentrations of Tmod1 or Tmod3. Using pyrene-actin polymerization assays, we observed that 50–300 nm Tmod1 or Tmod3 similarly enhanced the rate of 3.4 μm β/γcyto-actin polymerization in a dose-dependent fashion (Fig. 7, A and B). Although higher concentrations of Tmod3 (400 nm) promoted even higher rates of β/γcyto-actin polymerization, the final level of polymerized actin was lower (Fig. 7B) most likely due to β/γcyto-actin monomer sequestration by Tmod3 (see Figs. 8 and 9). This only becomes evident at later time points because of the slow on-rate of Tmod3 for G-actin (2.9 × 10−3 μm−1 s−1) compared with the faster on-rate of Tmod3 for the pointed ends of spontaneously formed actin filament nuclei (35, 36).
FIGURE 7.
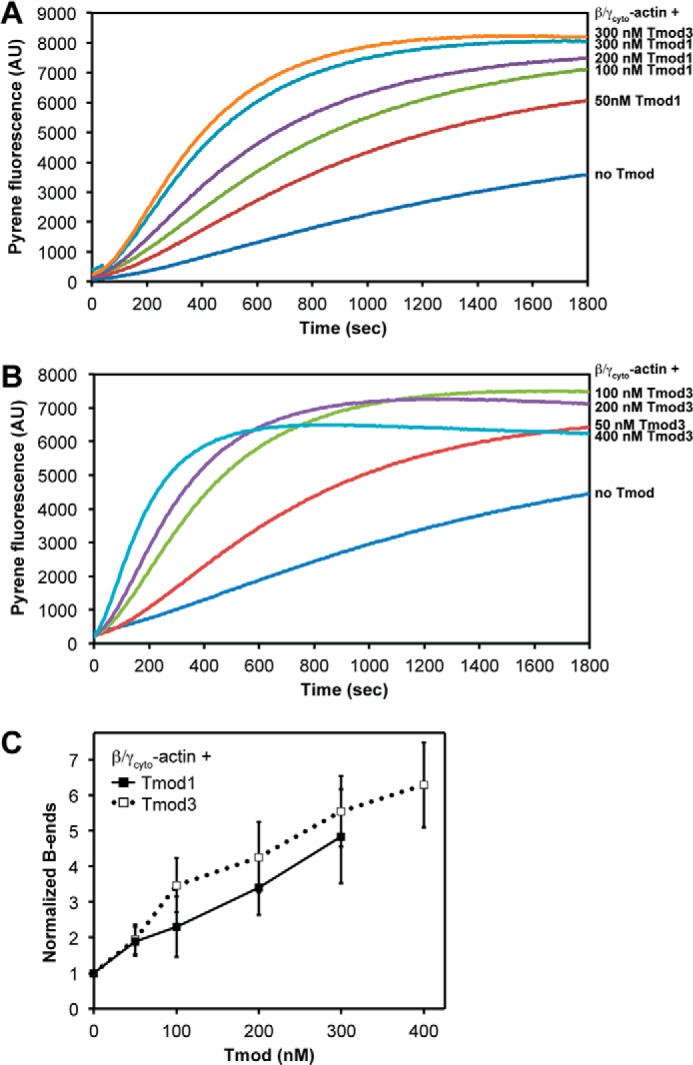
Tmod1 and Tmod3 similarly promote spontaneous platelet β/γcyto-actin polymerization. A and B, Tmod1 or Tmod3 was mixed with 3.4 μm platelet β/γcyto-G-actin (5% pyrene-labeled), and salts were added to initiate polymerization. Actin polymerization kinetics were monitored by increased pyrene-actin fluorescence as described under “Experimental Procedures.” AU, arbitrary units. In A, 300 nm Tmod3 was tested to allow direct comparison with 300 nm Tmod1 in a single experiment. C, concentration of β/γcyto-actin barbed (B) ends was calculated from the polymerization rate when 50% of monomers were polymerized and then normalized to the concentration of barbed ends in the “no Tmod” control. Data shown are mean ± S.D. (error bars) of three experiments.
FIGURE 8.

Tmod1 and Tmod3 interact similarly with αsk-actin, β-actin, and γcyto-actin monomers in a non-denaturing PAGE assay at low ionic strength. A and B, increasing concentrations of Tmod1 (A) or Tmod3 (B) were incubated with 4 μm αsk-actin, β-actin, or γcyto-actin monomers. The mixtures were then separated using non-denaturing PAGE. In the presence of both Tmod and actin, the actin band was depleted as the Tmod concentration increased, indicating formation of a complex. Because the recombinant cytoplasmic actins were produced using a baculovirus-driven expression system and contained 20–25% contaminating insect actin (41), we also tested pure insect actin as a control. C and D, densitometric quantification of the percentage of actin monomers complexed with Tmod1 (C) or Tmod3 (D). Note that the amounts of actin in Tmod1·actin complexes cannot be compared directly with the amounts of actin in Tmod3·actin complexes because different concentrations of Tmod1 and Tmod3 were used in their respective experiments, and the samples were electrophoresed on different gels.
FIGURE 9.

Tmod3 preferentially sequesters cytoplasmic β- and γcyto-actin monomers in the presence of physiological salt, but Tmod1 does not. A and B, effects of Tmod1 (A) and Tmod3 (B) on F-actin sequestration in a sedimentation assay. 4 μm αsk-actin, β-actin, or γcyto-actin was polymerized in the presence of increasing amounts of Tmod1 and Tmod3 and ultracentrifuged to sediment F-actin. Because the recombinant cytoplasmic actins were produced using a baculovirus-driven expression system and contained 20–25% contaminating insect actin (41), we also tested pure insect actin as a control. Supernatants (s) and pellets (p) were analyzed by SDS-PAGE and Coomassie Blue staining. C and D, densitometric quantification of the percentage of actin in the supernatant of mixtures of Tmod1 and actin isoforms (C) or mixtures of Tmod3 and actin isoforms (D). Data shown are mean ± S.D. (error bars) of three experiments.
Calculation of the concentrations of free barbed ends in these assays showed that Tmod1 and Tmod3 had similar dose-dependent actin-nucleating activities with 300 nm Tmods leading to ∼5–6-fold increases in the number of barbed ends over β/γcyto-actin alone (Fig. 7C). When we compared these data with previous data obtained for nucleation experiments using αsk-actin (36), we found that neither Tmod1 nor Tmod3 nucleated αsk-actin assembly as efficiently as β/γcyto-actin with only ∼3-fold increases in the number of barbed ends at 400 nm Tmods with αsk-actin (compare Fig. 7C and Ref. 36). For both αsk- and β/γcyto-actin, Tmod3 promoted assembly of slightly more barbed ends than Tmod1, but the differences were statistically insignificant. Thus, the similar nucleating activities of Tmod1 and Tmod3 with both αsk- and β/γcyto-actins indicate that differences in actin nucleation are unlikely to explain the selectivity of Tmod1 or Tmod3 for cytoskeletal actin filaments comprising different actin isoforms.
Tmod3 Sequesters β- and γcyto-Actin Monomers More Strongly than Does Tmod1
Similar to other actin-nucleating proteins, Tmods can also bind αsk-actin monomers with Tmod3 having greater monomer binding activity than Tmod1 (35, 36). However, the monomer binding interactions of Tmods with β- and γcyto-actin monomers have not been investigated. In these assays, we tested the binding of Tmods to native rabbit skeletal muscle αsk-actin as well as to human cytoplasmic β- and γcyto-actins obtained using a baculovirus-driven expression system in insect cells (41). The purified cytoplasmic actins were contaminated with 20–25% insect actin (41), so purified insect actin was also tested as an additional control. First, we examined the interaction of Tmods with actin monomers using non-denaturing PAGE at low ionic strength as described previously (36, 51). In these assays, Tmod co-migrates with the Tmod·G-actin complex, and formation of the Tmod·G-actin complex is reflected by a decreased intensity of the G-actin band as the Tmod concentration is increased (Fig. 8, A and B). Tmod1 and Tmod3 bound all G-actin isoforms (36) with Tmod3 displaying stronger overall binding than Tmod1 based on enhanced complex formation at lower concentrations of Tmod3 than Tmod1 (Fig. 8, A and B). Densitometric quantification of the percentage of actin complexed with Tmod at various Tmod concentrations did not reveal any dramatic differences between Tmod1 and Tmod3 in terms of their ability to bind the various G-actin isoforms (Fig. 8, C and D). However, non-denaturing PAGE is performed at low ionic strength, and assaying Tmod-G-actin binding with this methodology may not reflect physiological interactions.
To examine Tmod-G-actin binding in the presence of physiological salt, we performed G-actin sequestration assays. αsk-Actin, cytoplasmic β- and γcyto-actins, and insect actin were polymerized to steady state in the presence of increasing concentrations of Tmod1 or Tmod3, and the F-actin was then ultracentrifuged to separate actin monomers from filaments followed by SDS-PAGE and Coomassie Blue staining of supernatants and pellets. On these gels, Tmod1 migrates slightly above actin, whereas Tmod3 migrates slightly below actin. Results show that Tmod1 had little effect on the amount of G-actin in the supernatant regardless of which actin isoform was tested, indicating that Tmod1 does not significantly bind or sequester G-actin at physiological salt conditions (Fig. 9, A and C). This is in agreement with previous studies using pyrene-actin polymerization assays showing that Tmod1 does not sequester αsk-actin (18, 19, 35). By contrast, Tmod3 dramatically increased the amount of unpolymerized actin in the supernatant for cytoplasmic β- and γcyto-actins and insect actin in a Tmod3 concentration-dependent manner (Fig. 9, B and D). At the highest concentration of Tmod3 tested (6 μm) and 4 μm actins, nearly all of the β- and γcyto-actins and insect actin remained in the supernatant. By contrast, Tmod3 had only a relatively small effect on the amount of αsk-actin in the supernatant fraction. These results indicate that, first, Tmod3 binds to β- and γcyto-actins more strongly than Tmod1 and, second, Tmod3 binds to β- and γcyto-actins more strongly than to αsk-actin. Note that the monomer sequestration activity of Tmod3 observed here operates in the micromolar concentration range (Fig. 9D), well above the nanomolar concentrations of Tmod3 required for high affinity pointed end capping of TM-coated αsk- or β/γcyto-actin filaments (Figs. 4–6). Thus, Tmod3 sequestration of β- and/or γcyto-actin might provide a mechanism for dynamic regulation of β- and γcyto-actin monomer-polymer levels in regions of cells that lack TMs, such as protruding lamellipodia (37, 38).
DISCUSSION
Tmod-TM Isoform Binding and Capping of TM-coated Actin Filaments
This study revealed differences between Tmod1 and Tmod3 in terms of their TM binding avidities for a diverse assortment of TM isoforms with Tmod1 binding α/βTM, TM5NM1, and TM5b ∼5-fold more strongly than Tmod3. The conclusion that Tmod1 is a stronger TM binder than Tmod3 is consistent with published measurements of Kd values for the TM-binding Site 1 and Site 2 fragments of Tmod1 and Tmod3 binding to N-terminal αTM1a, αTM1b, and γTM1b synthetic peptides corresponding to the α/βTM, TM5b, and TM5NM1 proteins, respectively, tested in our study (see Ref. 60). However, the differences that we observed for Tmod1 and Tmod3 binding to TMs are not directly comparable with those measured by Uversky et al. (60) because of the different assays as well as the fact that Uversky et al. (60) measured TM binding for the Tmod Site 1 and Site 2 fragments independently, whereas we studied TM binding to full-length Tmod1 and Tmod3.
In the case of Tmod1, a large body of evidence indicates that interaction of Tmod1 with TMs via the two TM-binding sites in the N-terminal domain of Tmod1 promotes its high affinity pointed end capping of TM-actin filaments (nm-pm Kd; Refs. 16, 18, 19, and 22–26). It has been proposed that the binding of the α1- and α3-helices of Tmod1 to the terminal TMs at the actin filament pointed end may enhance the interaction of the actin-capping site of Tmod1 with actin by providing two additional Tmod-binding sites at the pointed end (16, 21). This is supported by experiments using various N-terminal Tmod1 fragments containing helix-disrupting or charge-altering mutations leading to concurrent loss of TM binding and TM-dependent capping activity (15, 21, 22). Moreover, a Tmod1-R11K/D12N/Q144K triple mutant has greatly weakened binding to skeletal muscle α/βTM along with severalfold reduced Tmod1 capping of α/βTM-coated actin filaments (23, 61). Although the TM-dependent actin pointed end capping of Tmod3 has not been studied previously, the sequence conservation observed between Tmod1 and Tmod3 in their TM-binding Sites 1 and 2 (60) predicts that the TM-dependent high affinity capping of Tmod3 utilizes similar sites and mechanisms as does that of Tmod1. Yet, surprisingly, despite the differences between Tmod1 and Tmod3 in TM binding (Fig. 3 and Ref. 60), we did not observe differences in capping of TM-actin pointed ends for Tmod1 and Tmod3 either for either αsk-actin filaments coated with skeletal muscle α/βTM or cytoskeletal TM5NM1 and TM5b or for β/γcyto-actin filaments coated with TM5NM1. Only in the case of the bacterially expressed rat αTM2 was a difference in pointed end capping between Tmod1 and Tmod3 observed with Tmod3 roughly one-third as effective at capping αTM2-actin pointed ends as Tmod1 (Fig. 4). Recombinant muscle αTM2 is unacetylated with undetectable Tmod binding in binary assays (59), suggesting that severe attenuation of Tmod-TM binding may be required to detect differences in TM-actin filament pointed end capping by Tmods. Nevertheless, because this study shows that recombinant αTM2 still promotes relatively high affinity pointed end capping by Tmod1 (80% capping at 10 nm Tmod1), αTM2 may bind Tmod1 better than Tmod3 in the context of the actin filament pointed end. Alternatively, the presence of αTM2 may lead to a conformational change in the actin filament that enhances the interaction of Tmod1 more than that of Tmod3 with actin filament pointed ends.
The fact that Tmod1 and Tmod3 do not display significant differences in terms of their high affinity TM-actin capping activities (that are correlated with differences in TM binding) raises the possibility that Tmod/TM interactions may have additional functions at the actin filament pointed end. Using skinned muscle fiber mechanics and x-ray diffraction approaches, we recently showed that substitution of Tmod3 for Tmod1 at the thin filament pointed end in Tmod1−/− mouse skeletal muscle impairs α/βTM strand movement across the thin filament and reduces recruitment of myosin cross-bridges (62). It is possible that Tmod1 (but not Tmod3) interactions with α/βTM and αsk-actin at the pointed end induce long range conformational changes in α/βTM and/or the actin filament that facilitate the cooperative azimuthal movement of α/βTM during thin filament Ca2+ activation (62). Future structural and biochemical studies of thin filament regulation with purified proteins in reconstituted systems will be required to evaluate this possibility. It is also known that myosin cross-bridge cycling can induce actin polymerization (63, 64), pointing toward a novel (but untested) mechanism of isoform-specific regulation of actin dynamics by Tmod1 and Tmod3 via modulation of TM strand movement and myosin head activity.
The biochemical data generated in this study provide clues to how in vitro Tmod/TM/actin isoform interactions may underlie the in vivo characteristics of diverse Tmod-TM-actin structures in cells. For example, the similar pointed end capping activities of Tmod1 and Tmod3 with various TM and actin isoforms imply that, in skeletal muscle, TM and actin isoform differences per se are unlikely to account for Tmod1 selectively capping α/βTM-coated αsk-actin thin filaments and Tmod3 selectively capping SR-associated, TM5NM1/TM4-coated γcyto-actin filaments (7, 45). Instead, it is more likely that the association of Tmod3 with the SR is driven by its interaction with the membrane-associated protein small ankyrin 1.5 (sAnk1.5) at the M-line (7), whereas as-yet-unknown factors are involved in targeting and assembly of Tmod1 to the thin filaments. Such a mechanism is supported by investigations of developing skeletal muscle where Tmod1 is observed on pointed ends of the thin filaments during the assembly of myofibrils relatively early in skeletal muscle development (45, 65), whereas Tmod3 remains cytoplasmic prior to its later adoption of a striated staining pattern signifying assembly into the SR (7, 45). By contrast, in red blood cells where Tmod1 and Tmod3 are co-expressed late in differentiation (5), the stronger binding of Tmod1 to TM5NM1 and TM5b as compared with Tmod3 could account for the preferential assembly of Tmod1 with TMs on the β-actin filaments of the membrane skeleton (although a caveat is that their TM-β-actin capping affinities are similar; Fig. 6) (8). Notably, Tmod3 does assemble with the β-actin filaments in Tmod1−/− red blood cells but at reduced (one-fifth) levels with respect to Tmod1 in wild-type red blood cells (8), which may also reflect the weaker binding of Tmod3 to TM5NM1 and TM5b. Such differences in Tmod assembly could be exacerbated if Tmod concentrations were limiting during TM-actin filament assembly in vivo. Additional studies are needed to explain how or why the unique intermolecular affinities of various combinations of Tmod, TM, and actin isoforms contribute to the co-assembly of these isoforms in some cell types but not in others.
Tmod/Actin Isoform Interactions
We found that Tmod3 interacts with and sequesters cytoplasmic β- and γcyto-actin monomers more strongly than αsk-actin monomers, reducing steady state β- and γcyto-actin polymerization. We also show that Tmod1 lacks a similar actin monomer-sequestering ability for any actin isoform in agreement with previous studies on Tmod1 with αsk-actin (35). However, our previous experiments showed that Tmod3 also reduced pyrene-labeled αsk-actin polymerization at steady state (35), which we did not observe here in co-sedimentation assays measuring αsk-actin polymerization (Fig. 9). The reason for these differences is unclear, although one possibility is that pyrene-iodoacetamide labeling of αsk-actin on Cys-373 (40) may have enhanced the interaction of Tmod3 with αsk-actin, promoting monomer sequestration by Tmod3. Alternatively, the αsk-actin used in the previous study (35) could have been contaminated with β- and γcyto-actins, which are also present in skeletal muscle tissues.
Thus, Tmod3 (but not Tmod1) is a new member of a growing subset of actin-binding proteins that are capable of discriminating among actin isoforms, a group that includes annexin-5a (66), profilin (67), cofilin (68), thymosin-β4 (69), L-plastin (70), ezrin (71), and βCap73 (72) (for a review, see Ref. 28). Tmod3 does not discriminate between actin isoforms in nucleation assays or in pointed end capping assays (this study and Refs. 35 and 36), suggesting that the monomer-sequestering activity of Tmod3 may rely on divergent actin isoform residues that are exposed in the monomer but not at the polymer pointed end. Our previous mutagenesis study showed that mouse Tmod3 binds to αsk-actin monomers via the TM-Cap domain (36). Chemical cross-linking and LC-MS/MS experiments have indicated that Tmod3 residues Glu-74 and Glu-79 cross-link to αsk-actin subdomain 2 (actin Lys-61), whereas Lys-53, Lys-94, Lys-96, and Lys-169 cross-link to actin subdomain 1 (actin Asp-24, Asp-25, Glu-99, and Glu-100) (36). All of these residues are conserved among all Tmod and actin isoforms, suggesting that our studies did not detect the critical interacting residues that could confer selective recognition between Tmod3 and β- and γcyto-actins. Notably, skeletal muscle αsk-actin and γcyto-actin share 93% identity with their principal sequence differences residing in their ∼10 N-terminal residues (73). The N-terminal ends of actins are located in actin subdomain 1 where Tmod3-interacting residues are located (36), suggesting that differences in the N-terminal sequences of actin isoforms may directly affect isoform-specific Tmod/actin interactions. Additionally, mutagenesis of N-terminal residues in yeast actin to muscle actin residues leads to allosteric changes in subdomain 2 (74, 75), another Tmod3-binding site (36). Future biochemical and structural studies will be required to identify the interaction sites for Tmod3 on cytoskeletal β- and γcyto-actins.
Could Tmod3 act as a monomer-sequestering protein in vivo? Immunoprecipitation of FLAG-tagged Tmod3 from HEK293 cells in the presence of latrunculin A has demonstrated that Tmod3 can interact with β/γcyto-actin monomers in a cellular context (35). Moreover, Tmod3 is enriched in the leading lamellipodia of migrating human microvascular endothelial cells where F-actin levels are inversely proportional to Tmod3 levels in GFP-Tmod3 overexpression and Tmod3 siRNA depletion experiments (4). This is as expected from a monomer-sequestering protein and provides a simple explanation for reduced rates of cell migration resulting from Tmod3 overexpression; namely, increased actin monomer sequestration by Tmod3 would reduce the supply of actin available for polymerization and protrusion of lamellipodia, thereby reducing cell migration rates (4, 76). Such a mechanism is expected to depend on reduced TM levels as TM binding to actin filaments would otherwise provide high affinity Tmod3-binding sites, leading to Tmod3 capping and stabilization of the filaments and more F-actin, which was not observed. Indeed, previous studies have reported reduced levels of TM in lamellipodia of migrating cells (37, 38), although this phenomenon may depend on cell type or lamellipodial properties. Note that bulk actin sequestration in cells by Tmod3 is unlikely based on a global Tmod3 concentration of ∼0.5 μm in migrating human microvascular endothelial cells (4) as compared with a significantly higher concentration of actin, which constitutes 5–15% of the total protein content of endothelial cells (77). However, Tmod3 may be present at a higher local concentration in the lamellipodia of migrating cells based on immunolocalization indicating enrichment of Tmod3 in this region (4). By comparison, in Tmod1−/− red blood cells where Tmod3 is increased and present at one-fifth normal Tmod1 levels, there is no decrease in F-actin in the membrane skeleton or increase in β-actin monomers in the cytosol (8). In this markedly different cellular context (i.e. a highly stable actin cytoskeleton), the β-actin filaments in the membrane skeleton of the Tmod1−/− red cells are associated with TM5NM1 and TM5b, which would favor high affinity Tmod3 capping of pointed ends and thereby preclude monomer sequestration by Tmod3. Thus, we propose that Tmod3 operates as a low affinity (μm) monomer-sequestering protein in the case of TM-free actin filaments in dynamic cellular contexts but switches to a high affinity (nm) pointed end capping protein when associated with TM-coated actin filaments in stable cellular contexts.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-HL083464 (to V. M. F.) and R01-DC008803 (to P. A. R.). This work was also supported by a development grant from the Muscular Dystrophy Association (to D. S. G.) and grants-in-aid for young scientists and for scientific research on innovative areas from the Japan Society for the Promotion of Science (to S. Y.).
- Tmod
- tropomodulin
- TM
- tropomyosin
- SR
- sarcoplasmic reticulum
- Cap
- pointed end actin capping
- αsk-actin
- skeletal muscle α-actin
- γcyto-actin
- cytoplasmic γ-actin
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
REFERENCES
- 1. Fischer R. S., Fowler V. M. (2003) Tropomodulins: life at the slow end. Trends Cell Biol. 13, 593–601 [DOI] [PubMed] [Google Scholar]
- 2. Yamashiro S., Gokhin D. S., Kimura S., Nowak R. B., Fowler V. M. (2012) Tropomodulins: pointed-end capping proteins that regulate actin filament architecture in diverse cell types. Cytoskeleton 69, 337–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gokhin D. S., Fowler V. M. (2011) Tropomodulin capping of actin filaments in striated muscle development and physiology. J. Biomed. Biotechnol. 2011, 103069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer R. S., Fritz-Six K. L., Fowler V. M. (2003) Pointed-end capping by tropomodulin3 negatively regulates endothelial cell motility. J. Cell Biol. 161, 371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sui Z., Nowak R. B., Bacconi A., Kim N. E., Liu H., Li J., Wickrema A., An X. L., Fowler V. M. (2014) Tropomodulin3-null mice are embryonic lethal with anemia due to impaired erythroid terminal differentiation in the fetal liver. Blood 123, 758–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber K. L., Fischer R. S., Fowler V. M. (2007) Tmod3 regulates polarized epithelial cell morphology. J. Cell Sci. 120, 3625–3632 [DOI] [PubMed] [Google Scholar]
- 7. Gokhin D. S., Fowler V. M. (2011) Cytoplasmic γ-actin and tropomodulin isoforms link to the sarcoplasmic reticulum in skeletal muscle fibers. J. Cell Biol. 194, 105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moyer J. D., Nowak R. B., Kim N. E., Larkin S. K., Peters L. L., Hartwig J., Kuypers F. A., Fowler V. M. (2010) Tropomodulin 1-null mice have a mild spherocytic elliptocytosis with appearance of tropomodulin 3 in red blood cells and disruption of the membrane skeleton. Blood 116, 2590–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nowak R. B., Fischer R. S., Zoltoski R. K., Kuszak J. R., Fowler V. M. (2009) Tropomodulin1 is required for membrane skeleton organization and hexagonal geometry of fiber cells in the mouse lens. J. Cell Biol. 186, 915–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fowler V. M. (1987) Identification and purification of a novel Mr 43,000 tropomyosin-binding protein from human erythrocyte membranes. J. Biol. Chem. 262, 12792–12800 [PubMed] [Google Scholar]
- 11. Sung L. A., Lin J. J. (1994) Erythrocyte tropomodulin binds to the N-terminus of hTM5, a tropomyosin isoform encoded by the γ-tropomyosin gene. Biochem. Biophys. Res. Commun. 201, 627–634 [DOI] [PubMed] [Google Scholar]
- 12. Vera C., Sood A., Gao K. M., Yee L. J., Lin J. J., Sung L. A. (2000) Tropomodulin-binding site mapped to residues 7–14 at the N-terminal heptad repeats of tropomyosin isoform 5. Arch. Biochem. Biophys. 378, 16–24 [DOI] [PubMed] [Google Scholar]
- 13. Gunning P., O'Neill G., Hardeman E. (2008) Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 88, 1–35 [DOI] [PubMed] [Google Scholar]
- 14. Fowler V. M. (1990) Tropomodulin: a cytoskeletal protein that binds to the end of erythrocyte tropomyosin and inhibits tropomyosin binding to actin. J. Cell Biol. 111, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greenfield N. J., Kostyukova A. S., Hitchcock-DeGregori S. E. (2005) Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophys. J. 88, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kostyukova A. S., Hitchcock-Degregori S. E., Greenfield N. J. (2007) Molecular basis of tropomyosin binding to tropomodulin, an actin-capping protein. J. Mol. Biol. 372, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gunning P. W., Schevzov G., Kee A. J., Hardeman E. C. (2005) Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 15, 333–341 [DOI] [PubMed] [Google Scholar]
- 18. Weber A., Pennise C. R., Babcock G. G., Fowler V. M. (1994) Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol. 127, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weber A., Pennise C. R., Fowler V. M. (1999) Tropomodulin increases the critical concentration of barbed end-capped actin filaments by converting ADP·Pi-actin to ADP-actin at all pointed filament ends. J. Biol. Chem. 274, 34637–34645 [DOI] [PubMed] [Google Scholar]
- 20. Kostyukova A. S., Hitchcock-DeGregori S. E. (2004) Effect of the structure of the N terminus of tropomyosin on tropomodulin function. J. Biol. Chem. 279, 5066–5071 [DOI] [PubMed] [Google Scholar]
- 21. Kostyukova A. S., Choy A., Rapp B. A. (2006) Tropomodulin binds two tropomyosins: a novel model for actin filament capping. Biochemistry 45, 12068–12075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kostyukova A. S., Rapp B. A., Choy A., Greenfield N. J., Hitchcock-DeGregori S. E. (2005) Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry 44, 4905–4910 [DOI] [PubMed] [Google Scholar]
- 23. Moroz N. A., Novak S. M., Azevedo R., Colpan M., Uversky V. N., Gregorio C. C., Kostyukova A. S. (2013) Alteration of tropomyosin-binding properties of tropomodulin-1 affects its capping ability and localization in skeletal myocytes. J. Biol. Chem. 288, 4899–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsukada T., Kotlyanskaya L., Huynh R., Desai B., Novak S. M., Kajava A. V., Gregorio C. C., Kostyukova A. S. (2011) Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. J. Biol. Chem. 286, 2194–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kostyukova A. S. (2008) Tropomodulin/tropomyosin interactions regulate actin pointed end dynamics. Adv. Exp. Med. Biol. 644, 283–292 [DOI] [PubMed] [Google Scholar]
- 26. Kostyukova A. S. (2008) Tropomodulins and tropomodulin/tropomyosin interactions. Cell. Mol. Life Sci. 65, 563–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Babcock G. G., Fowler V. M. (1994) Isoform-specific interaction of tropomodulin with skeletal muscle and erythrocyte tropomyosins. J. Biol. Chem. 269, 27510–27518 [PubMed] [Google Scholar]
- 28. Perrin B. J., Ervasti J. M. (2010) The actin gene family: function follows isoform. Cytoskeleton 67, 630–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prins K. W., Call J. A., Lowe D. A., Ervasti J. M. (2011) Quadriceps myopathy caused by skeletal muscle-specific ablation of βcyto-actin. J. Cell Sci. 124, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinder J. C., Gratzer W. B. (1983) Structural and dynamic states of actin in the erythrocyte. J. Cell Biol. 96, 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pinder J. C., Ungewickell E., Bray D., Gratzer W. B. (1978) The spectrin-actin complex and erythrocyte shape. J. Supramol. Struct. 8, 439–445 [DOI] [PubMed] [Google Scholar]
- 32. Sung L. A., Gao K. M., Yee L. J., Temm-Grove C. J., Helfman D. M., Lin J. J., Mehrpouryan M. (2000) Tropomyosin isoform 5b is expressed in human erythrocytes: implications of tropomodulin-TM5 or tropomodulin-TM5b complexes in the protofilament and hexagonal organization of membrane skeletons. Blood 95, 1473–1480 [PubMed] [Google Scholar]
- 33. Ursitti J. A., Fowler V. M. (1994) Immunolocalization of tropomodulin, tropomyosin and actin in spread human erythrocyte skeletons. J. Cell Sci. 107, 1633–1639 [DOI] [PubMed] [Google Scholar]
- 34. Fowler V. M., Davis J. Q., Bennett V. (1985) Human erythrocyte myosin: identification and purification. J. Cell Biol. 100, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fischer R. S., Yarmola E. G., Weber K. L., Speicher K. D., Speicher D. W., Bubb M. R., Fowler V. M. (2006) Tropomodulin 3 binds to actin monomers. J. Biol. Chem. 281, 36454–36465 [DOI] [PubMed] [Google Scholar]
- 36. Yamashiro S., Speicher K. D., Speicher D. W., Fowler V. M. (2010) Mammalian tropomodulins nucleate actin polymerization via their actin monomer binding and filament pointed end-capping activities. J. Biol. Chem. 285, 33265–33280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gupton S. L., Anderson K. L., Kole T. P., Fischer R. S., Ponti A., Hitchcock-DeGregori S. E., Danuser G., Fowler V. M., Wirtz D., Hanein D., Waterman-Storer C. M. (2005) Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J. Cell Biol. 168, 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DesMarais V., Ichetovkin I., Condeelis J., Hitchcock-DeGregori S. E. (2002) Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J. Cell Sci. 115, 4649–4660 [DOI] [PubMed] [Google Scholar]
- 39. Pardee J. D., Spudich J. A. (1982) Purification of muscle actin. Methods Cell Biol. 24, 271–289 [DOI] [PubMed] [Google Scholar]
- 40. Kouyama T., Mihashi K. (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 114, 33–38 [PubMed] [Google Scholar]
- 41. Bergeron S. E., Zhu M., Thiem S. M., Friderici K. H., Rubenstein P. A. (2010) Ion-dependent polymerization differences between mammalian β- and γ-nonmuscle actin isoforms. J. Biol. Chem. 285, 16087–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smillie L. B. (1982) Preparation and identification of α- and β-tropomyosins. Methods Enzymol. 85, 234–241 [DOI] [PubMed] [Google Scholar]
- 43. Maytum R., Bathe F., Konrad M., Geeves M. A. (2004) Tropomyosin exon 6b is troponin-specific and required for correct acto-myosin regulation. J. Biol. Chem. 279, 18203–18209 [DOI] [PubMed] [Google Scholar]
- 44. Hitchcock-DeGregori S. E., Song Y., Moraczewska J. (2001) Importance of internal regions and the overall length of tropomyosin for actin binding and regulatory function. Biochemistry 40, 2104–2112 [DOI] [PubMed] [Google Scholar]
- 45. Gokhin D. S., Lewis R. A., McKeown C. R., Nowak R. B., Kim N. E., Littlefield R. S., Lieber R. L., Fowler V. M. (2010) Tropomodulin isoforms regulate thin filament pointed-end capping and skeletal muscle physiology. J. Cell Biol. 189, 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ilkovski B., Mokbel N., Lewis R. A., Walker K., Nowak K. J., Domazetovska A., Laing N. G., Fowler V. M., North K. N., Cooper S. T. (2008) Disease severity and thin filament regulation in M9R TPM3 nemaline myopathy. J. Neuropathol. Exp. Neurol. 67, 867–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 48. Hitchcock-DeGregori S. E., Heald R. W. (1987) Altered actin and troponin binding of amino-terminal variants of chicken striated muscle α-tropomyosin expressed in Escherichia coli. J. Biol. Chem. 262, 9730–9735 [PubMed] [Google Scholar]
- 49. Fowler V. M., Greenfield N. J., Moyer J. (2003) Tropomodulin contains two actin filament pointed end-capping domains. J. Biol. Chem. 278, 40000–40009 [DOI] [PubMed] [Google Scholar]
- 50. Young C. L., Southwick F. S., Weber A. (1990) Kinetics of the interaction of a 41-kilodalton macrophage capping protein with actin: promotion of nucleation during prolongation of the lag period. Biochemistry 29, 2232–2240 [DOI] [PubMed] [Google Scholar]
- 51. Safer D. (1989) An electrophoretic procedure for detecting proteins that bind actin monomers. Anal. Biochem. 178, 32–37 [DOI] [PubMed] [Google Scholar]
- 52. Wegner A. (1979) Equilibrium of the actin-tropomyosin interaction. J. Mol. Biol. 131, 839–853 [DOI] [PubMed] [Google Scholar]
- 53. Yang Y. Z., Korn E. D., Eisenberg E. (1979) Cooperative binding of tropomyosin to muscle and Acanthamoeba actin. J. Biol. Chem. 254, 7137–7140 [PubMed] [Google Scholar]
- 54. Pittenger M. F., Helfman D. M. (1992) In vitro and in vivo characterization of four fibroblast tropomyosins produced in bacteria: TM-2, TM-3, TM-5a, and TM-5b are co-localized in interphase fibroblasts. J. Cell Biol. 118, 841–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Temm-Grove C. J., Guo W., Helfman D. M. (1996) Low molecular weight rat fibroblast tropomyosin 5 (TM-5): cDNA cloning, actin-binding, localization, and coiled-coil interactions. Cell Motil. Cytoskeleton 33, 223–240 [DOI] [PubMed] [Google Scholar]
- 56. Maytum R., Konrad M., Lehrer S. S., Geeves M. A. (2001) Regulatory properties of tropomyosin effects of length, isoform, and N-terminal sequence. Biochemistry 40, 7334–7341 [DOI] [PubMed] [Google Scholar]
- 57. Urbancikova M., Hitchcock-DeGregori S. E. (1994) Requirement of amino-terminal modification for striated muscle α-tropomyosin function. J. Biol. Chem. 269, 24310–24315 [PubMed] [Google Scholar]
- 58. Skoumpla K., Coulton A. T., Lehman W., Geeves M. A., Mulvihill D. P. (2007) Acetylation regulates tropomyosin function in the fission yeast Schizosaccharomyces pombe. J. Cell Sci. 120, 1635–1645 [DOI] [PubMed] [Google Scholar]
- 59. Greenfield N. J., Fowler V. M. (2002) Tropomyosin requires an intact N-terminal coiled coil to interact with tropomodulin. Biophys. J. 82, 2580–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uversky V. N., Shah S. P., Gritsyna Y., Hitchcock-DeGregori S. E., Kostyukova A. S. (2011) Systematic analysis of tropomodulin/tropomyosin interactions uncovers fine-tuned binding specificity of intrinsically disordered proteins. J. Mol. Recognit. 24, 647–655 [DOI] [PubMed] [Google Scholar]
- 61. Kostyukova A., Maeda K., Yamauchi E., Krieger I., Maéda Y. (2000) Domain structure of tropomodulin: distinct properties of the N-terminal and C-terminal halves. Eur. J. Biochem. 267, 6470–6475 [DOI] [PubMed] [Google Scholar]
- 62. Ochala J., Gokhin D. S., Iwamoto H., Fowler V. M. (2014) Pointed-end capping by tropomodulin modulates actomyosin crossbridge formation in skeletal muscle fibers. FASEB J. 28, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Carlier M. F., Valentin-Ranc C., Combeau C., Fievez S., Pantoloni D. (1994) Actin polymerization: regulation by divalent metal ion and nucleotide binding, ATP hydrolysis and binding of myosin. Adv. Exp. Med. Biol. 358, 71–81 [DOI] [PubMed] [Google Scholar]
- 64. Haviv L., Gillo D., Backouche F., Bernheim-Groswasser A. (2008) A cytoskeletal demolition worker: myosin II acts as an actin depolymerization agent. J. Mol. Biol. 375, 325–330 [DOI] [PubMed] [Google Scholar]
- 65. Almenar-Queralt A., Gregorio C. C., Fowler V. M. (1999) Tropomodulin assembles early in myofibrillogenesis in chick skeletal muscle: evidence that thin filaments rearrange to form striated myofibrils. J. Cell Sci. 112, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 66. Tzima E., Trotter P. J., Orchard M. A., Walker J. H. (2000) Annexin V relocates to the platelet cytoskeleton upon activation and binds to a specific isoform of actin. Eur. J. Biochem. 267, 4720–4730 [DOI] [PubMed] [Google Scholar]
- 67. Larsson H., Lindberg U. (1988) The effect of divalent cations on the interaction between calf spleen profilin and different actins. Biochim. Biophys. Acta 953, 95–105 [DOI] [PubMed] [Google Scholar]
- 68. De La Cruz E. M. (2005) Cofilin binding to muscle and non-muscle actin filaments: isoform-dependent cooperative interactions. J. Mol. Biol. 346, 557–564 [DOI] [PubMed] [Google Scholar]
- 69. Weber A., Nachmias V. T., Pennise C. R., Pring M., Safer D. (1992) Interaction of thymosin β4 with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry 31, 6179–6185 [DOI] [PubMed] [Google Scholar]
- 70. Namba Y., Ito M., Zu Y., Shigesada K., Maruyama K. (1992) Human T cell L-plastin bundles actin filaments in a calcium-dependent manner. J. Biochem. 112, 503–507 [DOI] [PubMed] [Google Scholar]
- 71. Yao X., Cheng L., Forte J. G. (1996) Biochemical characterization of ezrin-actin interaction. J. Biol. Chem. 271, 7224–7229 [DOI] [PubMed] [Google Scholar]
- 72. Shuster C. B., Lin A. Y., Nayak R., Herman I. M. (1996) βCap73: a novel β actin-specific binding protein. Cell Motil. Cytoskeleton 35, 175–187 [DOI] [PubMed] [Google Scholar]
- 73. Bunnell T. M., Ervasti J. M. (2011) Structural and functional properties of the actin gene family. Crit. Rev. Eukaryot. Gene. Expr. 21, 255–266 [DOI] [PubMed] [Google Scholar]
- 74. McKane M., Wen K. K., Boldogh I. R., Ramcharan S., Pon L. A., Rubenstein P. A. (2005) A mammalian actin substitution in yeast actin (H372R) causes a suppressible mitochondria/vacuole phenotype. J. Biol. Chem. 280, 36494–36501 [DOI] [PubMed] [Google Scholar]
- 75. McKane M., Wen K. K., Meyer A., Rubenstein P. A. (2006) Effect of the substitution of muscle actin-specific subdomain 1 and 2 residues in yeast actin on actin function. J. Biol. Chem. 281, 29916–29928 [DOI] [PubMed] [Google Scholar]
- 76. Small J. V., Stradal T., Vignal E., Rottner K. (2002) The lamellipodium: where motility begins. Trends Cell Biol. 12, 112–120 [DOI] [PubMed] [Google Scholar]
- 77. Prasain N., Stevens T. (2009) The actin cytoskeleton in endothelial cell phenotypes. Microvasc. Res. 77, 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]