

Abstract
The gut is anatomically positioned to play a critical role in the regulation of metabolic homeostasis, providing negative feedback via nutrient sensing and local hormonal signaling. Gut hormones, such as cholecystokinin (CCK) and glucagon-like peptide-1 (GLP-1), are released following a meal and act on local receptors to regulate glycemia via a neuronal gut-brain axis. Additionally, jejunal nutrient sensing and leptin action are demonstrated to suppress glucose production, and both are required for the rapid antidiabetic effect of duodenal jejunal bypass surgery. Strategies aimed at targeting local gut hormonal signaling pathways may prove to be efficacious therapeutic options to improve glucose control in diabetes.
Keywords: Diabetes, Fatty Acid, Glucose Metabolism, Hormones, Intestine, Bariatric Surgery, Food Intake
Introduction
Diabetes is now a worldwide epidemic affecting ∼350 million people worldwide (1). Almost 80% of type 2 diabetic individuals are overweight or obese (2) due in part to the interaction between genetics and the environment, such as overconsumption of a hypercaloric, western diet that is associated with a dysregulation in glucose homeostasis (3). As hyperglycemia is a primary cause of diabetic complications and accounts for the majority of economic costs associated with diabetes (4), current therapeutic approaches to treat diabetes and to lower the associated economic burden are focused on regulating glucose levels and hepatic glucose production (GP).6 Indeed, metformin is the most widely prescribed diabetic drug, normalizing glycemia through a reduction in GP (5). However, metformin has side effects, leading to the development of the recent oral hypoglycemic agents aimed at increasing circulating levels and action of gut-derived incretins, such as dipeptidyl peptidase-4 (DPP-4) inhibitors and glucagon-like peptide 1 receptor (GLP-1R) agonists (exenatide and liraglutide). The early success of these treatments highlights the importance of the gastrointestinal tract in regulating glucose homeostasis.
Indeed, the gastrointestinal tract is anatomically positioned as the first line of defense to prevent nutrient excess by initiating negative feedback mechanisms through nutrient-induced secretion of gastrointestinal hormones (Fig. 1). Starting in the stomach, ghrelin is an orexigenic peptide, with increased levels associated with the timing of a meal (6, 7), whereas intake of nutrients suppresses ghrelin secretion in both rodents and humans (8, 9). Although ghrelin may act in an endocrine fashion (10, 11), vagal afferents innervating the stomach express the ghrelin receptor, and in both rodents and humans, a vagotomy (which disrupts communication between the stomach and brain) abolishes the ability of ghrelin to increase food intake (12, 13), suggesting a local (paracrine) effect. In addition to its effects on food intake, ghrelin regulates glucose homeostasis by increasing the gastric emptying rate (14) and inhibiting glucose-stimulated insulin secretion (15). More distally to the site of ghrelin secretion, the small intestine contains a variety of regulatory signals (Fig. 1) including: (i) the more proximal hormones within the duodenum and jejunum, cholecystokinin (CCK) in I cells and glucose-dependent insulinotropic polypeptide (GIP) in K cells, and (ii) more distal hormones in the ileum and large intestine within L cells, glucagon-like peptide-1/2 (GLP-1/2), oxyntomodulin (OXN), and peptide YY (PYY). The secretion of these hormones is stimulated by nutrients within the intestine that then act on their respective receptors either centrally, or locally on vagal afferents that are in close proximity to enteroendocrine cells, to regulate metabolic homeostasis through various changes in food intake, gastric emptying, intestinal motility, and/or energy expenditure (see Ref. 16). In light of this evidence, we put forward a working hypothesis that nutrient-induced gut-derived hormones activate local gut signaling events to trigger the central nervous system via the vagus nerve to regulate glucose homeostasis.
FIGURE 1.
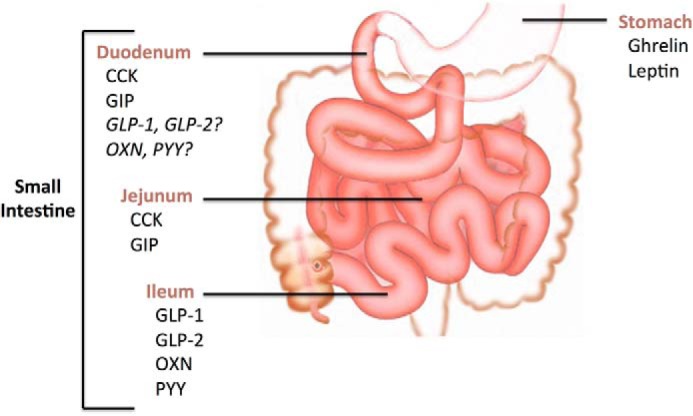
Gut peptide hormones and regulatory signals are released along the length of the gastrointestinal tract. Both ghrelin and leptin are secreted from the stomach, whereas CCK and GIP are secreted from I cells and K cells, respectively, in the duodenum. It is debated whether L cells, which secrete GLP-1, GLP-2, OXN, and PYY, are located in the duodenum. More distally secreted in the intestine are CCK and GIP from the jejunum, and GLP-1, GLP-2, OXN, and PYY from the ileum.
CCK Action in the Duodenum
CCK is secreted from enteroendocrine I-cells predominantly located in the proximal small intestine, mainly in response to fatty acids (Fig. 1) (17). Besides promoting satiation through a neuronal network, endogenous CCK also mediates the ability of intestinal lipids to lower GP via a gut-brain-liver neuronal axis (18). This glucose-lowering effect of duodenal lipids is dependent on intracellular esterification of long-chain fatty acids (LCFAs) to long-chain fatty acyl-CoA (LCFA-CoA) via acyl-CoA synthetase (ACS)-3 (Fig. 2), indicating the importance of both luminal lipid hydrolysis to LCFAs and subsequent absorption into intestinal cells possibly via fatty acid transporter cluster of differentiation 36 (CD36) (19). Although the role of CD36 in mediating the glucoregulatory role of lipids remains to be tested, it is interesting to note that CD36 knock-out mice exhibit reduced release of CCK both in vivo and in vitro (19). Furthermore, LCFA-induced secretion and the glucose-lowering effect of CCK are dependent upon protein kinase C-δ (PKC-δ) stimulation, which is localized to the duodenal mucosal layer (20, 21). However, the mechanistic link between PKC-δ signaling and CCK secretion remains to be investigated. Nevertheless, this LCFA → PKC-δ → CCK pathway requires activation of CCK-1 receptors (CCK-1R), located on the vagal afferent fibers innervating the small intestine (22). Binding of CCK to its receptor leads to protein kinase A (PKA) signaling, which is sufficient and necessary to enhance N-methyl-d-aspartate (NMDA) receptor-mediated neuronal transmission in the nucleus of the solitary tract to lower GP via the hepatic vagal branch (Fig. 2) (23). Taken together, these results suggest that paracrine CCK action mediates the ability of duodenal lipids to regulate glucose homeostasis by signaling to the central nervous system.
FIGURE 2.

Gut nutrient-sensing mechanisms and subsequent peptide hormone release in normal and DJB surgery settings. Nutrient influx in both the duodenum and the jejunum triggers hormonal release and downstream signaling to lower GP through a neuronal network. In the duodenum, these mechanisms are disrupted upon high fat feeding. DJB surgery results in the influx of nutrients and hormones, such as leptin, directly into the jejunum to suppress GP through downstream mechanisms. ACS, acyl-CoA synthetase; NTS, nucleus of the solitary tract; SGLT1, sodium glucose luminal transporter-1.
Interestingly, this ability of CCK and intestinal lipids to reduce GP is diminished when rats are placed on a 3-day high fat (HF) diet (18, 24) that much resembles the western diet, highlighting the pathophysiological relevance of this pathway. Closer analysis indicates that this diet induces CCK resistance at the level of the CCK-1 receptor signaling by impairing PKA activation and subsequently, the gut-brain-liver axis (23). Thus, the ability to manipulate gut CCK signaling may prove an efficacious therapeutic option, evidenced by the fact that improvements in glycemic control were seen in humans with impaired glucose tolerance after 8-week treatment of the bile acid sequestrant, colesevelam. These improvements were the results of colesevelam preventing bile acid absorption and thus increasing local CCK secretion, which possibly activates the aforementioned glucoregulatory pathway (25).
GLP-1 Action in the Gut
Although CCK signaling appears to mediate the glucose-lowering effects of intestinal lipids, GLP-1, which is released predominately from L-cells of the distal intestine (Fig. 1), has been extensively studied for its incretin effect. GLP-1 secretion is biphasic, with the early peak (within minutes) attributed to a neurohormonal reflex (26), although recent studies argue for direct nutrient exposure of proximal L-cells (27, 28) and the later peak mediated by direct nutrient stimulation of distal intestinal L-cells (29). Interestingly, both glucose and fatty acids are potent stimulators of GLP-1 release; however, the receptors and intracellular mechanisms mediating GLP-1 secretion are controversial and remain to be clarified (see Ref. 30). The glucoregulatory action of GLP-1 has been widely established since the pioneering study demonstrated its potent incretin effect almost three decades ago (31). Indeed, GLP-1 acts on the GLP-1R localized on the outer membrane of pancreatic β-cells to promote insulin secretion via activation of PKA-dependent and -independent pathways. Additionally, GLP-1 action in the pancreas inhibits glucagon and stimulates somatostatin release, and also increases β-cell sensitivity to glucose, insulin biosynthesis, and proliferation (32). However, GLP-1 is rapidly degraded by DPP-4, resulting in a diminished concentration of GLP-1 in the hepatoportal vein (∼50%) and an even smaller amount entering systemic circulation (less than 10%) (33). As such, studies have hypothesized that insulin release, as well as other glucoregulatory effects such as glucose utilization, GP, and counterregulatory hormone production, are all mediated by a gut-brain-periphery axis (34), involving the enteric nervous system, especially in the hepatic portal region (35). Indeed, findings have demonstrated that glucose-induced GLP-1 secretion activate vagal afferent neurons of the common hepatic branch activating the nucleus of the solitary tract (34, 36, 37). This signal is then relayed to the hypothalamus, which generates efferent signals to control glucose flux (36). Studies demonstrate that both the sensory and the motor components of the vagus are essential because blockade of the afferent or efferent vagal relay attenuates the increase to GLP-1R-mediated insulin release (38). However, this GLP-1-dependent enteric glucose sensor is not entirely straightforward as GLP-1 alters glucose utilization only when the hepatoportal glucose gradient conditions mimic the postprandial state (39). Furthermore, selective surgical removal of the common hepatic branch of the vagus did not abolish the glycemic effects of endogenous GLP-1, indicating that vagal afferents innervating the intestinal tract may be more important in mediating GLP-1-induced incretin signaling (40).
Postprandial levels of active GLP-1 are diminished in obese and diabetic individuals (41), leading to the development of GLP-1R agonists (exenatide and liraglutide) and DPP-4 inhibitors as type 2 diabetic treatments (42), which are moderately successful in improving glycemic control. Interestingly, the success of these GLP-1-modifying treatments has been attributed to up-regulation of this local GLP-1 gut-brain-periphery axis. For example, oral administration of low doses of DPP-4 inhibitor, sitagliptin, aimed at selectively inhibiting intestinal, but not systemic, DPP-4 activity still resulted in enhanced glycemic control and plasma insulin levels through activation of local GLP-1R on the vagus nerve. This indicates a predominant role for enhancement of the GLP-1 gut-brain-periphery axis in the ability of sitagliptin to reduce local intestinal DPP4 activity and regulate glycemia (43). Furthermore, the rapid remission of type 2 diabetes following gastric bypass surgeries has been hypothesized to be mediated by GLP-1 (44, 45). Indeed, gastric bypass results in dramatically and rapidly enhanced meal-stimulated GLP-1, independent of weight loss and calorie restriction, likely from more direct nutrient exposure to intestinal L-cells (45). Furthermore, antagonism of the GLP-1R following Roux-en-Y gastric bypass (RYGB) worsens glucose tolerance and postprandial insulin secretion (46). Additionally, responsiveness to GLP-1R agonism in rodents was able to predict improvements in glycemic control following bariatric surgery, independent of weight loss (47). Despite this, both vertical sleeve gastrectomy and RYGB are effective in normalizing glycemia in several models of models of GLP-1R deficiency (48, 49), whereas duodenal-jejunal bypass (DJB) improves glucose levels despite nonelevated postsurgical GLP-1 levels in nonobese type 1 diabetic rats (50). Thus, the role of GLP-1 in mediating the antidiabetic effects of bariatric surgery still remains highly controversial (44, 45, 51–55).
Jejunal Nutrient Sensing
Although bariatric surgery is primarily used as a weight loss procedure for morbidly obese subjects, patients with type 2 diabetes very often exhibit rapid remission of diabetes. This led to the emergence of “metabolic surgery” as a new area of research and treatment (56). Indeed, studies in both rodents and humans demonstrate the glucose-lowering effects of RYGB surgery and sleeve gastrectomy (57, 58), the more common surgical procedures used today. Although these surgical techniques include a restrictive component (reduction of stomach size), the improvements in glucose levels are not correlated with changes in body weight (46, 59–61). To determine the effects of gastric restriction versus the involvement of the small intestine itself to regulate glucose control, an experimental surgical procedure was developed: DJB surgery. This surgical technique leaves the stomach intact but bypasses the proximal intestine, increasing the flow of nutrients directly into the jejunum and producing rapid antidiabetic effects. In Goto-Kakizaki rats, a rodent model of nonobese type 2 diabetes, DJB improved glycemic control in as little as 1 week postoperatively, with no changes in body weight (62). Rapid weight-independent improvements in glycemia are also consistent with DJB in nonobese or mild obese type 2 diabetic humans (63–66). These studies suggest that the rearrangement of the intestinal tract mediates the glucose-lowering effects of DJB. In light of the possibility that gut hormonal signaling contributes to the clinical and therapeutic antidiabetic outcomes of bariatric surgery as discussed above, this suggests that nutrient sensing, which mediates gut hormonal release, may play a role in the effect of bariatric surgery.
Nutrient sensing in the distal portions of the small intestine, from an increased flux of unabsorbed nutrients, could play a role in the antidiabetic effects of DJB. In rats, administration of both glucose and lipids into the jejunum decreases GP through a gut-brain-liver axis, likely via nutrient uptake and metabolism in intestinal cells and subsequent neuronal signaling (Fig. 2) (50). To determine the importance of nutrient sensing in DJB, this bariatric surgical technique was performed in two strains of insulin-deficient uncontrolled diabetic rodents, the diabetes-prone Biobreeding (BBdp) and nonobese uncontrolled streptozotocin (STZ)-induced rats. Interestingly, DJB resulted in a rapid reduction in plasma glucose levels (2 days postoperative) independent of weight loss (50). Although a physiological shunting of nutrients into the distal intestine achieved by refeeding activated jejunal nutrient sensing to maintain plasma glucose concentrations, blocking jejunal nutrient sensing reversed the beneficial glucose-lowering effects of DJB. Importantly, the rapid 2-day glucose-lowering effect of DJB is independent of insulin action in both models (50).
Traditionally, insulin action has been thought be an important mediator of glucose homeostasis through its actions on the liver, muscle, adipose tissue, and more recently, the brain. However, studies indicate that the hormone leptin can lower blood glucose concentrations in uncontrolled STZ-induced diabetic rodents independent of insulin action, described later, suggesting that insulin action may not be the sole mediator of glucose homeostasis. Contributing to this hypothesis, nutrient-sensing mechanisms also remain intact in this rodent model to lower glucose concentrations after DJB surgery independent of a rise in insulin levels (50). This is consistent with other studies demonstrating that DJB suppresses GP in type 2 diabetic rodents without improving insulin sensitivity (52). These findings suggest that although insulin mediates glucose suppression in some settings, other local intestinal signals are important contributors to the antidiabetic effects of DJB (Fig. 2).
Gastric Leptin
Gastric Leptin Release
As discussed above, nutrient sensing plays an important role in the antidiabetic effects of DJB. Although increased nutrient exposure to the distal small intestine likely results in increased release of CCK and GLP1, it is unlikely that these peptides play a role in the ability of DJB to lower GP as activation of jejunal PKA, a signaling pathway that mediates both CCK and GLP-1 receptor activation (67), has no effect on GP (23). However, CCK and GLP-1 receptors also signal through PKA-independent pathways in peripheral tissues (68–70), but whether these pathways exist in the intestine remains to be elucidated. Often disregarded, nutrient ingestion also results in the release of gastric leptin (71, 72), but its function in the intestine beyond control of food intake (73) remains largely unknown.
Gastric leptin is predominantly secreted from the fundic region of the stomach (71, 72) as well as in gastric epithelial cells (74). Although studies mostly focus on adipocyte-derived leptin and its central actions to control feeding, nutrient ingestion has been demonstrated to cause the release of gastric leptin (71, 72) directly into the gastric lumen (74, 75). Importantly, in order for gastric leptin to reach the intestine, it must withstand the harsh proteolytic environment of the stomach. It is postulated that leptin is secreted as a complex with the soluble leptin receptor (SLR), a peptide that resembles the extracellular domain of the leptin receptor (75). The SLR prevents the degradation of leptin and allows it to enter the intestine to subsequently bind and activate the functional long form leptin receptor (Ob-Rb) (76). In contrast, a study in humans suggests that gastric leptin is not bound to macromolecules such as the SLR (77). Nonetheless, leptin is unable to bind and activate the Ob-Rb when complexed to the SLR in vitro (78, 79). Although the effect of the SLR on leptin action remains unclear, leptin has been detected in duodenal juices and thus reaches the duodenum in its intact form (73). Furthermore, the Ob-Rb is found to be expressed on cells of, or vagal afferents innervating, the small intestine (76, 80–83), and activation of these receptors results in various functions, such as regulating nutrient transport (84). Notably, leptin administration in the intestinal lumen inhibits the recruitment of sodium-glucose transporters to the apical membrane, hindering intestinal glucose absorption, and demonstrating a role for leptin in regulating glucose control (85). However, given the well known glucoregulatory effects of central leptin, it remains possible that gastric leptin has a more extensive role via a gut-brain neuronal pathway.
Leptin Signaling and Action
The effects of leptin on glucose regulation have been extensively studied in the brain. Leptin binds to the hypothalamic Ob-Rb and activates downstream effectors STAT3 via Jak-2 (86, 87) and PI3K through phosphorylation of insulin receptor substrate (88) to regulate glucose homeostasis. Importantly, subcutaneous leptin administration in insulin-deficient diabetic rodents normalizes glucose levels (89–91). This effect is likely mediated by the CNS as intracerebroventricular leptin lowers plasma glucose levels in insulin-deficient rodents (92, 93), emphasizing the insulin-independent ability of leptin to lower glucose levels. Interestingly, both STAT3 (83, 84) and PI3K (94) are also present in the gut, and leptin has been shown to activate intestinal STAT3 to protect against infection (95). Therefore, it is possible that leptin acts on STAT3 and/or PI3K in the intestine to affect glucose homeostasis, as it does in the CNS. Moreover, similar to intracerebroventricular leptin, jejunal nutrient sensing lowers GP and mediates the antidiabetic effects of DJB surgery independent of insulin action. These data suggest a role for gastric leptin in mediating the antidiabetic effects of DJB. Thus, we aimed to dissect the role of leptin in the intestine to regulate glucose homeostasis and its potential in mediating the antidiabetic effect of DJB surgery.
Jejunal Leptin Lowers GP via a Neuronal Mediated Ob-Rb → PI3K Signaling Cascade
An intestinal leptin infusion was administered to determine its preabsorptive effects on glucose regulation. Although duodenal leptin did not affect glucose homeostasis, jejunal leptin increased the glucose infusion rate needed to maintain euglycemia, which was due to a decrease in GP, rather than an increase in peripheral glucose uptake during the clamps (96). This effect was mediated by the Ob-Rb as coinfusion of leptin with a chemical inhibitor or infusion of jejunal leptin in two leptin receptor-deficient rodent models, the Koletsky fak/fak rats and db/db mice, abolished the glucoregulatory effect of leptin. Furthermore, activation of PI3K, and not STAT3, was required for intrajejunal leptin to lower GP as infusion of the STAT3 inhibitor with leptin failed to negate its glucose-lowering effects, but inhibiting PI3K abolished the ability of leptin to lower GP (96). It is interesting to note that duodenal leptin was unable to lower GP and likewise failed to increase PI3K activity, indicating that PI3K activation may mediate the glucose-lowering effect of leptin. Given our previous work detailing a gut-brain-liver neuronal axis to lower GP (97, 98), we tested the involvement of a neuronal network in mediating the effects of leptin. The local anesthetic tetracaine was infused to block neuronal innervation of the jejunum, which consequently abolished the ability of leptin to increase the glucose infusion rate and suppress GP (96). Thus, jejunal leptin triggers a neuronal mediated Ob-Rb → PI3K signaling cascade to lower GP. Although untested, it is possible that similar to CCK acting on gut vagal afferents to lower GP, gastric leptin may activate Ob-Rb receptors on vagal afferents terminating in the jejunal lamina propria as the Ob-Rb receptor has been detected in the nodose ganglia (81, 99).
Jejunal Leptin Lowers GP in Diseased Models
As hypothalamic leptin is demonstrated to lower GP in rats fed a HF diet for 3 days, jejunal leptin action was tested in this same model. Rats develop duodenal CCK resistance after 3 days of hyperphagic HF feeding; however, jejunal leptin actions remained intact, and it was able to lower GP in this rodent model. Moreover, leptin also lowered GP and plasma glucose levels in uncontrolled STZ induced-diabetic rats with insulin levels reduced by ∼80%, independent of changes in plasma insulin (96). This indicates that although duodenal nutrient/CCK resistance may demonstrate a possible pathophysiological role in the development of diabetes, the success of jejunal leptin in diseased states demonstrates a potential role for gastric leptin in mediating the glucose-lowering effect of DJB.
Jejunal Leptin Mediates the Antidiabetic Effects of DJB
Therefore, to test the involvement of leptin in mediating the antidiabetic effects of DJB, we performed fasting-refeeding studies to stimulate physiological gastric leptin release in STZ-diabetic rats that had received DJB surgery, while disrupting leptin signaling. Although glucose control remained intact in rats with a jejunal saline infusion, it was disrupted by blocking leptin signaling. However, blockade of leptin signaling did not disrupt glucose homeostasis to the same extent as seen in STZ-diabetic rodents who had received sham surgery (96). Thus, other factors influencing glucose control could be at play, and it may be that nutrient-sensing mechanisms and leptin signaling in the jejunum converge to control glucose homeostasis after DJB surgery (Fig. 2).
Conclusion
In a recent review, Schwartz et al. (100) elegantly proposed a “revised” two-compartment model for the pathogenesis of type 2 diabetes. As opposed to the traditional islet malfunction-centered view, this model suggests that diabetes develops from a cooperative failure of glucose homeostasis from both pancreatic islets and a brain-centered glucoregulatory system. In light of accumulating research presented in this review, we argue that the gastrointestinal tract should also be recognized as a major player in this revised model of type 2 diabetes pathogenesis. The importance of this “second brain” in glucose homeostasis is often overlooked, despite its central location and highly intricate and bidirectional signaling pathways. Indeed, the gut represents the initial target site of nutrients, mediating the influx of glucose and other metabolic products. Furthermore, as highlighted in this review, the gut produces the initial meal-induced feedback signals to the rest of the body, via neuronal and hormonal signals. These gut-derived peptides, such as CCK and GLP-1, exhibit both insulin-dependent and insulin-independent glucoregulatory mechanisms, indicating a link from the gut to both the brain and the pancreas. Rapid failure of this gut-brain axis in mediating glucose homeostasis in diseased conditions (18, 23, 24) may both precede and contribute to the initial dysregulations of the brain and islets.
Thus, we propose that the gut lies upstream of this two-compartment model, where early impairments in gut function may be the initial stimuli that lead to initial damages of both the brain and the islet glucoregulatory systems, which subsequently worsen into the development of diabetes. The best support of this hypothesis comes from the fact that the most effective treatment of type 2 diabetes to date is from remodeling of the gut via bariatric surgery. Although the underlying mechanism(s) resulting in rapid normalization of glucose homeostasis after bariatric surgery is still unknown, it likely involves gut-initiated restoration of both insulin-dependent and neuronal mediated insulin-independent mechanisms. Indeed, evidence suggests that early changes in incretin levels may contribute to the rapid remission of types 2 diabetes, whereas we have demonstrated the importance of the insulin-independent nutrient and hormonal gut-brain-liver signaling axis in restoring glucose homeostasis. Therefore, the ability to manipulate this second brain may be an effective strategy to target both the islet-centered and the brain-centered glucose control systems, and improve the management of type 2 diabetes.
This work was supported by Canadian Institutes of Health Research (CIHR) Grant CIHR-MOP-82701 (to T. K. T. L.).
- GP
- glucose production
- DPP-4
- dipeptidyl peptidase-4
- GLP-1
- glucagon-like peptide 1
- GLP-1R
- GLP1 receptor
- CCK
- cholecystokinin
- GIP
- glucose-dependent insulinotropic polypeptide
- OXN
- oxyntomodulin
- PYY
- peptide YY
- LCFA
- long-chain fatty acid
- HF
- high fat
- RYGB
- Roux-en-Y gastric bypass
- DJB
- duodenal-jejunal bypass
- STZ
- streptozotocin
- SLR
- soluble leptin receptor.
REFERENCES
- 1. Scully T. (2012) Diabetes in numbers. Nature 485, S2–S3 [DOI] [PubMed] [Google Scholar]
- 2. Stagnitti M. N. (2001) Statistical Brief # 34 The Prevalence of Obesity and Other Chronic Health Conditions among Diabetic Adults in the U. S. Community Population, 2001, Medical Expenditure Panel Survey (MEPS), pp. 5–9, Agency for Healthcare Research and Quality, U.S. Department of Health and Human Services, Rockville, MD [Google Scholar]
- 3. Speakman J. R., O'Rahilly S. (2012) Fat: an evolving issue. Dis. Model. Mech. 5, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clark C. M., Jr. (1998) The burden of chronic hyperglycemia. Diabetes Care 21, Suppl. 3, C32–C34 [DOI] [PubMed] [Google Scholar]
- 5. Rojas L. B., Gomes M. B. (2013) Metformin: an old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drazen D. L., Vahl T. P., D'Alessio D. A., Seeley R. J., Woods S. C. (2006) Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 147, 23–30 [DOI] [PubMed] [Google Scholar]
- 7. Cummings D. E., Purnell J. Q., Frayo R. S., Schmidova K., Wisse B. E., Weigle D. S. (2001) A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719 [DOI] [PubMed] [Google Scholar]
- 8. Monteleone P., Bencivenga R., Longobardi N., Serritella C., Maj M. (2003) Differential responses of circulating ghrelin to high-fat or high-carbohydrate meal in healthy women. J. Clin. Endocrinol. Metab. 88, 5510–5514 [DOI] [PubMed] [Google Scholar]
- 9. Overduin J., Frayo R. S., Grill H. J., Kaplan J. M., Cummings D. E. (2005) Role of the duodenum and macronutrient type in ghrelin regulation. Endocrinology 146, 845–850 [DOI] [PubMed] [Google Scholar]
- 10. Tschöp M., Smiley D. L., Heiman M. L. (2000) Ghrelin induces adiposity in rodents. Nature 407, 908–913 [DOI] [PubMed] [Google Scholar]
- 11. Wren A. M., Seal L. J., Cohen M. A., Brynes A. E., Frost G. S., Murphy K. G., Dhillo W. S., Ghatei M. A., Bloom S. R. (2001) Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 86, 5992. [DOI] [PubMed] [Google Scholar]
- 12. Date Y., Murakami N., Toshinai K., Matsukura S., Niijima A., Matsuo H., Kangawa K., Nakazato M. (2002) The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123, 1120–1128 [DOI] [PubMed] [Google Scholar]
- 13. le Roux C. W., Neary N. M., Halsey T. J., Small C. J., Martinez-Isla A. M., Ghatei M. A., Theodorou N. A., Bloom S. R. (2005) Ghrelin does not stimulate food intake in patients with surgical procedures involving vagotomy. J. Clin. Endocrinol. Metab. 90, 4521–4524 [DOI] [PubMed] [Google Scholar]
- 14. Falkén Y., Hellström P. M., Sanger G. J., Dewit O., Dukes G., Grybäck P., Holst J. J., Näslund E. (2010) Actions of prolonged ghrelin infusion on gastrointestinal transit and glucose homeostasis in humans. Neurogastroenterol. Motil. 22, e192–e200 [DOI] [PubMed] [Google Scholar]
- 15. Reimer M. K., Pacini G., Ahrén B. (2003) Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 144, 916–921 [DOI] [PubMed] [Google Scholar]
- 16. Duca F. A., Covasa M. (2012) Current and emerging concepts on the role of peripheral signals in the control of food intake and development of obesity. Br. J. Nutr. 108, 778–793 [DOI] [PubMed] [Google Scholar]
- 17. Williams J. A., Blevins G. T. (1993) Cholecystokinin and regulation of pancreatic acinar cell function. Physiol. Rev. 73, 701–723 [DOI] [PubMed] [Google Scholar]
- 18. Cheung G. W., Kokorovic A., Lam C. K., Chari M., Lam T. K. (2009) Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab. 10, 99–109 [DOI] [PubMed] [Google Scholar]
- 19. Sundaresan S., Shahid R., Riehl T. E., Chandra R., Nassir F., Stenson W. F., Liddle R. A., Abumrad N. A. (2013) CD36-dependent signaling mediates fatty acid-induced gut release of secretin and cholecystokinin. FASEB J. 27, 1191–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Breen D. M., Yue J. T., Rasmussen B. A., Kokorovic A., Cheung G. W., Lam T. K. (2011) Duodenal PKC-δ and cholecystokinin signaling axis regulates glucose production. Diabetes 60, 3148–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kokorovic A., Cheung G. W., Breen D. M., Chari M., Lam C. K., Lam T. K. (2011) Duodenal mucosal protein kinase C-δ regulates glucose production in rats. Gastroenterology 141, 1720–1727 [DOI] [PubMed] [Google Scholar]
- 22. Raybould H. E., Gayton R. J., Dockray G. J. (1988) Mechanisms of action of peripherally administered cholecystokinin octapeptide on brain stem neurons in the rat. J. Neurosci. 8, 3018–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rasmussen B. A., Breen D. M., Luo P., Cheung G. W. C., Yang C. S., Sun B., Kokorovic A., Rong W., Lam T. K. (2012) Duodenal activation of cAMP-dependent protein kinase induces vagal afferent firing and lowers glucose production in rats. Gastroenterology 142, 834–843.e3 [DOI] [PubMed] [Google Scholar]
- 24. Wang P. Y., Caspi L., Lam C. K., Chari M., Li X., Light P. E., Gutierrez-Juarez R., Ang M., Schwartz G. J., Lam T. K. (2008) Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 452, 1012–1016 [DOI] [PubMed] [Google Scholar]
- 25. Marina A. L., Utzschneider K. M., Wright L. A., Montgomery B. K., Marcovina S. M., Kahn S. E. (2012) Colesevelam improves oral but not intravenous glucose tolerance by a mechanism independent of insulin sensitivity and β-cell function. Diabetes Care 35, 1119–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rocca A. S., Brubaker P. L. (1999) Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology 140, 1687–1694 [DOI] [PubMed] [Google Scholar]
- 27. Theodorakis M. J., Carlson O., Michopoulos S., Doyle M. E., Juhaszova M., Petraki K., Egan J. M. (2006) Human duodenal enteroendocrine cells: source of both incretin peptides, GLP-1 and GIP. Am. J. Physiol. Endocrinol. Metab. 290, E550–E559 [DOI] [PubMed] [Google Scholar]
- 28. Habib A. M., Richards P., Cairns L. S., Rogers G. J., Bannon C. A., Parker H. E., Morley T. C., Yeo G. S., Reimann F., Gribble F. M. (2012) Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153, 3054–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Layer P., Holst J. J., Grandt D., Goebell H. (1995) Ileal release of glucagon-like peptide-1 (GLP-1). Association with inhibition of gastric acid secretion in humans. Dig. Dis. Sci. 40, 1074–1082 [DOI] [PubMed] [Google Scholar]
- 30. Tolhurst G., Reimann F., Gribble F. M. (2012) Intestinal sensing of nutrients. Handb. Exp. Pharmacol. 209, 309–335 [DOI] [PubMed] [Google Scholar]
- 31. Kreymann B., Williams G., Ghatei M. A., Bloom S. R. (1987) Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet 2, 1300–1304 [DOI] [PubMed] [Google Scholar]
- 32. Drucker D. J. (2006) The biology of incretin hormones. Cell Metab. 3, 153–165 [DOI] [PubMed] [Google Scholar]
- 33. Holst J. J., Deacon C. F. (2005) Glucagon-like peptide-1 mediates the therapeutic actions of DPP-IV inhibitors. Diabetologia 48, 612–615 [DOI] [PubMed] [Google Scholar]
- 34. Burcelin R., Serino M., Cabou C. (2009) A role for the gut-to-brain GLP-1-dependent axis in the control of metabolism. Curr. Opin. Pharmacol. 9, 744–752 [DOI] [PubMed] [Google Scholar]
- 35. Burcelin R., Da Costa A., Drucker D., Thorens B. (2001) Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 50, 1720–1728 [DOI] [PubMed] [Google Scholar]
- 36. Knauf C., Cani P. D., Kim D. H., Iglesias M. A., Chabo C., Waget A., Colom A., Rastrelli S., Delzenne N. M., Drucker D. J., Seeley R. J., Burcelin R. (2008) Role of central nervous system glucagon-like Peptide-1 receptors in enteric glucose sensing. Diabetes 57, 2603–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishizawa M., Nakabayashi H., Uchida K., Nakagawa A., Niijima A. (1996) The hepatic vagal nerve is receptive to incretin hormone glucagon-like peptide-l, but not to glucose-dependent insulinotropic polypeptide, in the portal vein. J. Auton. Nerv. Syst. 61, 149–154 [DOI] [PubMed] [Google Scholar]
- 38. Balkan B., Li X. (2000) Portal GLP-1 administration in rats augments the insulin response to glucose via neuronal mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1449–R1454 [DOI] [PubMed] [Google Scholar]
- 39. Johnson K. M., Edgerton D. S., Rodewald T., Scott M., Farmer B., Neal D., Cherrington A. D. (2008) Intraportally delivered GLP-1, in the presence of hyperglycemia induced via peripheral glucose infusion, does not change whole body glucose utilization. Am. J. Physiol. Endocrinol. Metab. 294, E380–E384 [DOI] [PubMed] [Google Scholar]
- 40. Hayes M. R., Kanoski S. E., De Jonghe B. C., Leichner T. M., Alhadeff A. L., Fortin S. M., Arnold M., Langhans W., Grill H. J. (2011) The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. Am. J. Physiol. Regul Integr. Comp. Physiol. 301, R1479–R1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vollmer K., Holst J. J., Baller B., Ellrichmann M., Nauck M. A., Schmidt W. E., Meier J. J. (2008) Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 57, 678–687 [DOI] [PubMed] [Google Scholar]
- 42. Lovshin J. A., Drucker D. J. (2009) Incretin-based therapies for type 2 diabetes mellitus. Nat. Rev. Endocrinol. 5, 262–269 [DOI] [PubMed] [Google Scholar]
- 43. Waget A., Cabou C., Masseboeuf M., Cattan P., Armanet M., Karaca M., Castel J., Garret C., Payros G., Maida A., Sulpice T., Holst J. J., Drucker D. J., Magnan C., Burcelin R. (2011) Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 152, 3018–3029 [DOI] [PubMed] [Google Scholar]
- 44. Mingrone G., Castagneto-Gissey L. (2009) Mechanisms of early improvement/resolution of type 2 diabetes after bariatric surgery. Diabetes Metab. 35, 518–523 [DOI] [PubMed] [Google Scholar]
- 45. Rodieux F., Giusti V., D'Alessio D. A., Suter M., Tappy L. (2008) Effects of gastric bypass and gastric banding on glucose kinetics and gut hormone release. Obesity (Silver Spring). 16, 298–305 [DOI] [PubMed] [Google Scholar]
- 46. Chambers A. P., Jessen L., Ryan K. K., Sisley S., Wilson-Pérez H. E., Stefater M. A., Gaitonde S. G., Sorrell J. E., Toure M., Berger J., D'Alessio D. A., Woods S. C., Seeley R. J., Sandoval D. A. (2011) Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats. Gastroenterology 141, 950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Habegger K. M., Heppner K. M., Amburgy S. E., Ottaway N., Holland J., Raver C., Bartley E., Müller T. D., Pfluger P. T., Berger J., Toure M., Benoit S. C., Dimarchi R. D., Perez-Tilve D., D'Alessio D. A., Seeley R. J., Tschöp M. H. (2014) GLP-1R responsiveness predicts individual gastric bypass efficacy on glucose tolerance in rats. Diabetes 63, 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson-Pérez H. E., Chambers A. P., Ryan K. K., Li B., Sandoval D. A., Stoffers D., Drucker D. J., Pérez-Tilve D., Seeley R. J. (2013) Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like peptide 1 receptor deficiency. Diabetes 62, 2380–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mokadem M., Zechner J. F., Margolskee R. F., Drucker D. J., Aguirre V. (2013) Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency. Mol. Metab. 10.1016/j.molmet.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Breen D. M., Rasmussen B. A., Kokorovic A., Wang R., Cheung G. W., Lam T. K. (2012) Jejunal nutrient sensing is required for duodenal-jejunal bypass surgery to rapidly lower glucose concentrations in uncontrolled diabetes. Nat. Med. 18, 950–955 [DOI] [PubMed] [Google Scholar]
- 51. Pacheco D., de Luis D. A., Romero A., González Sagrado M., Conde R., Izaola O., Aller R., Delgado A. (2007) The effects of duodenal-jejunal exclusion on hormonal regulation of glucose metabolism in Goto-Kakizaki rats. Am. J. Surg. 194, 221–224 [DOI] [PubMed] [Google Scholar]
- 52. Jiao J., Bae E. J., Bandyopadhyay G., Oliver J., Marathe C., Chen M., Hsu J. Y., Chen Y., Tian H., Olefsky J. M., Saberi M. (2013) Restoration of euglycemia after duodenal bypass surgery is reliant on central and peripheral inputs in Zucker fa/fa rats. Diabetes 62, 1074–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bojsen-Møller K. N., Dirksen C., Jørgensen N. B., Jacobsen S. H., Serup A. K., Albers P. H., Hansen D. L., Worm D., Naver L., Kristiansen V. B., Wojtaszewski J. F., Kiens B., Holst J. J., Richter E. A., Madsbad S. (2013) Early enhancements of hepatic and later of peripheral insulin sensitivity combined with increased postprandial insulin secretion contribute to improved glycemic control after Roux-en-Y gastric bypass. Diabetes 10.2337/db13-1307 [DOI] [PubMed] [Google Scholar]
- 54. Zhang S. Y., Sun X. J., Zheng J. B., Wang W., Liu D., Chen N. Z., He S., Huo X. W., Smith W. (2014) Preserve common limb in duodenal-jejunal bypass surgery benefits rats with type 2-like diabetes. Obes. Surg. 24, 405–411 [DOI] [PubMed] [Google Scholar]
- 55. Salinari S., le Roux C. W., Bertuzzi A., Rubino F., Mingrone G. (2014) Duodenal-jejunal bypass and jejunectomy improve insulin sensitivity in Goto-Kakizaki diabetic rats without changes in incretins or insulin secretion. Diabetes 63, 1069–1078 [DOI] [PubMed] [Google Scholar]
- 56. Rubino F., Moo T. A., Rosen D. J., Dakin G. F., Pomp A. (2009) Diabetes surgery: a new approach to an old disease. Diabetes Care 32, Suppl. 2, S368–S372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bradley D., Magkos F., Klein S. (2012) Effects of bariatric surgery on glucose homeostasis and type 2 diabetes. Gastroenterology 143, 897–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rubino F., R'bibo S. L., del Genio F., Mazumdar M., McGraw T. E. (2010) Metabolic surgery: the role of the gastrointestinal tract in diabetes mellitus. Nat. Rev. Endocrinol. 6, 102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cohen R. V., Pinheiro J. C., Schiavon C. A., Salles J. E., Wajchenberg B. L., Cummings D. E. (2012) Effects of gastric bypass surgery in patients with type 2 diabetes and only mild obesity. Diabetes Care 35, 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schauer P. R., Kashyap S. R., Wolski K., Brethauer S. A., Kirwan J. P., Pothier C. E., Thomas S., Abood B., Nissen S. E., Bhatt D. L. (2012) Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N. Engl. J. Med. 366, 1567–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mingrone G., Panunzi S., De Gaetano A., Guidone C., Iaconelli A., Leccesi L., Nanni G., Pomp A., Castagneto M., Ghirlanda G., Rubino F. (2012) Bariatric surgery versus conventional medical therapy for type 2 diabetes. N. Engl. J. Med. 366, 1577–1585 [DOI] [PubMed] [Google Scholar]
- 62. Rubino F., Marescaux J. (2004) Effect of duodenal-jejunal exclusion in a non-obese animal model of type 2 diabetes: a new perspective for an old disease. Ann. Surg. 239, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Geloneze B., Geloneze S. R., Chaim E., Hirsch F. F., Felici A. C., Lambert G., Tambascia M. A., Pareja J. C. (2012) Metabolic surgery for non-obese type 2 diabetes: incretins, adipocytokines, and insulin secretion/resistance changes in a 1-year interventional clinical controlled study. Ann. Surg. 256, 72–78 [DOI] [PubMed] [Google Scholar]
- 64. Cohen R. V., Schiavon C. A., Pinheiro J. S., Correa J. L., Rubino F. (2007) Duodenal-jejunal bypass for the treatment of type 2 diabetes in patients with body mass index of 22–34 kg/m2: a report of 2 cases. Surg. Obes. Relat. Dis. 3, 195–197 [DOI] [PubMed] [Google Scholar]
- 65. Cohen R., Caravatto P. P., Correa J. L., Noujaim P., Petry T. Z., Salles J. E., Schiavon C. A. (2012) Glycemic control after stomach-sparing duodenal-jejunal bypass surgery in diabetic patients with low body mass index. Surg. Obes. Relat. Dis. 8, 375–380 [DOI] [PubMed] [Google Scholar]
- 66. Lee H. C., Kim M. K., Kwon H. S., Kim E., Song K. H. (2010) Early changes in incretin secretion after laparoscopic duodenal-jejunal bypass surgery in type 2 diabetic patients. Obes. Surg. 20, 1530–1535 [DOI] [PubMed] [Google Scholar]
- 67. Light P. E., Manning Fox J. E., Riedel M. J., Wheeler M. B. (2002) Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol. Endocrinol. 16, 2135–2144 [DOI] [PubMed] [Google Scholar]
- 68. Rupprecht L. E., Mietlicki-Baase E. G., Zimmer D. J., McGrath L. E., Olivos D. R., Hayes M. R. (2013) Hindbrain GLP-1 receptor-mediated suppression of food intake requires a PI3K-dependent decrease in phosphorylation of membrane-bound Akt. Am. J. Physiol. Endocrinol. Metab. 305, E751–E759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sutton G. M., Patterson L. M., Berthoud H.-R. (2004) Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J. Neurosci. 24, 10240–10247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Williams J. A., Sans M. D., Tashiro M., Schäfer C., Bragado M. J., Dabrowski A. (2002) Cholecystokinin activates a variety of intracellular signal transduction mechanisms in rodent pancreatic acinar cells. Pharmacol. Toxicol. 91, 297–303 [DOI] [PubMed] [Google Scholar]
- 71. Bado A., Levasseur S., Attoub S., Kermorgant S., Laigneau J. P., Bortoluzzi M. N., Moizo L., Lehy T., Guerre-Millo M., Le Marchand-Brustel Y., Lewin M. J. (1998) The stomach is a source of leptin. Nature 394, 790–793 [DOI] [PubMed] [Google Scholar]
- 72. Cinti S., Matteis R. D., Picó C., Ceresi E., Obrador A., Maffeis C., Oliver J., Palou A. (2000) Secretory granules of endocrine and chief cells of human stomach mucosa contain leptin. Int. J. Obes. Relat. Metab. Disord. 24, 789–793 [DOI] [PubMed] [Google Scholar]
- 73. Guilmeau S., Buyse M., Tsocas A., Laigneau J. P., Bado A. (2003) Duodenal leptin stimulates cholecystokinin secretion: evidence of a positive leptin-cholecystokinin feedback loop. Diabetes 52, 1664–1672 [DOI] [PubMed] [Google Scholar]
- 74. Cammisotto P. G., Renaud C., Gingras D., Delvin E., Levy E., Bendayan M. (2005) Endocrine and exocrine secretion of leptin by the gastric mucosa. J. Histochem. Cytochem. 53, 851–860 [DOI] [PubMed] [Google Scholar]
- 75. Cammisotto P. G., Gingras D., Renaud C., Levy E., Bendayan M. (2006) Secretion of soluble leptin receptors by exocrine and endocrine cells of the gastric mucosa. Am. J. Physiol. Gastrointest. Liver. Physiol. 290, G242–G249 [DOI] [PubMed] [Google Scholar]
- 76. Cammisotto P., Bendayan M. (2012) A review on gastric leptin: the exocrine secretion of a gastric hormone. Anat. Cell Biol. 45, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sobhani I., Bado A., Vissuzaine C., Buyse M., Kermorgant S., Laigneau J. P., Attoub S., Lehy T., Henin D., Mignon M., Lewin M. J. (2000) Leptin secretion and leptin receptor in the human stomach. Gut 47, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zastrow O., Seidel B., Kiess W., Thiery J., Keller E., Böttner A., Kratzsch J. (2003) The soluble leptin receptor is crucial for leptin action: evidence from clinical and experimental data. Int. J. Obes. Relat. Metab. Disord. 27, 1472–1478 [DOI] [PubMed] [Google Scholar]
- 79. Yang G., Ge H., Boucher A., Yu X., Li C. (2004) Modulation of direct leptin signaling by soluble leptin receptor. Mol. Endocrinol. 18, 1354–1362 [DOI] [PubMed] [Google Scholar]
- 80. Mix H., Widjaja A., Jandl O., Cornberg M., Kaul A., Göke M., Beil W., Kuske M., Brabant G., Manns M. P., Wagner S. (2000) Expression of leptin and leptin receptor isoforms in the human stomach. Gut 47, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Buyse M., Ovesjö M. L., Goïot H., Guilmeau S., Péranzi G., Moizo L., Walker F., Lewin M. J., Meister B., Bado A. (2001) Expression and regulation of leptin receptor proteins in afferent and efferent neurons of the vagus nerve. Eur. J. Neurosci. 14, 64–72 [DOI] [PubMed] [Google Scholar]
- 82. Barrenetxe J., Villaro A. C., Guembe L., Pascual I., Muñoz-Navas M., Barber A., Lostao M. P. (2002) Distribution of the long leptin receptor isoform in brush border, basolateral membrane, and cytoplasm of enterocytes. Gut 50, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morton N. M., Emilsson V., Liu Y. L., Cawthorne M. A. (1998) Leptin action in intestinal cells. J. Biol. Chem. 273, 26194–26201 [DOI] [PubMed] [Google Scholar]
- 84. Iqbal J., Li X., Chang B. H. J., Chan L., Schwartz G. J., Chua S. C., Jr., Hussain M. M. (2010) An intrinsic gut leptin-melanocortin pathway modulates intestinal microsomal triglyceride transfer protein and lipid absorption. J. Lipid Res. 51, 1929–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ducroc R., Guilmeau S., Akasbi K., Devaud H., Buyse M., Bado A. (2005) Luminal leptin induces rapid inhibition of active intestinal absorption of glucose mediated by sodium-glucose cotransporter 1. Diabetes 54, 348–354 [DOI] [PubMed] [Google Scholar]
- 86. Buettner C., Pocai A., Muse E. D., Etgen A. M., Myers M. G., Jr., Rossetti L. (2006) Critical role of STAT3 in leptin's metabolic actions. Cell Metab. 4, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kievit P., Howard J. K., Badman M. K., Balthasar N., Coppari R., Mori H., Lee C. E., Elmquist J. K., Yoshimura A., Flier J. S. (2006) Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab 4, 123–132 [DOI] [PubMed] [Google Scholar]
- 88. Morton G. J., Gelling R. W., Niswender K. D., Morrison C. D., Rhodes C. J., Schwartz M. W. (2005) Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2, 411–420 [DOI] [PubMed] [Google Scholar]
- 89. Wang M. Y., Chen L., Clark G. O., Lee Y., Stevens R. D., Ilkayeva O. R., Wenner B. R., Bain J. R., Charron M. J., Newgard C. B., Unger R. H. (2010) Leptin therapy in insulin-deficient type I diabetes. Proc. Natl. Acad. Sci. U.S.A. 107, 4813–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yu X., Park B. H., Wang M. Y., Wang Z. V., Unger R. H. (2008) Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc. Natl. Acad. Sci. U.S.A. 105, 14070–14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chinookoswong N., Wang J. L., Shi Z. Q. (1999) Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes 48, 1487–1492 [DOI] [PubMed] [Google Scholar]
- 92. German J. P., Thaler J. P., Wisse B. E., Oh-I S., Sarruf D. A., Matsen M. E., Fischer J. D., Taborsky G. J., Jr., Schwartz M. W., Morton G. J. (2011) Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 152, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fujikawa T., Berglund E. D., Patel V. R., Ramadori G., Vianna C. R., Vong L., Thorel F., Chera S., Herrera P. L., Lowell B. B., Elmquist J. K., Baldi P., Coppari R. (2013) Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell Metab. 18, 431–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. El Homsi M., Ducroc R., Claustre J., Jourdan G., Gertler A., Estienne M., Bado A., Scoazec J. Y., Plaisancié P. (2007) Leptin modulates the expression of secreted and membrane-associated mucins in colonic epithelial cells by targeting PKC, PI3K, and MAPK pathways. Am. J. Physiol. Gastrointest Liver Physiol. 293, G365–G373 [DOI] [PubMed] [Google Scholar]
- 95. Guo X., Roberts M. R., Becker S. M., Podd B., Zhang Y., Chua S. C., Jr., Myers M. G., Jr., Duggal P., Houpt E. R., Petri W. A., Jr. (2011) Leptin signaling in intestinal epithelium mediates resistance to enteric infection by Entamoeba histolytica. Mucosal Immunol. 4, 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rasmussen B. A., Breen D. M., Duca F. A., Côté C. D., Zadeh-Tahmasebi M., Filippi B. M., Lam T. K. (2014) Jejunal leptin-PI3K signaling lowers glucose production. Cell Metab. 19, 155–161 [DOI] [PubMed] [Google Scholar]
- 97. Rasmussen B. A., Breen D. M., Lam T. K. (2012) Lipid sensing in the gut, brain and liver. Trends Endocrinol. Metab. 23, 49–55 [DOI] [PubMed] [Google Scholar]
- 98. Breen D. M., Rasmussen B. A., Côté C. D., Jackson V. M., Lam T. K. (2013) Nutrient-sensing mechanisms in the gut as therapeutic targets for diabetes. Diabetes 62, 3005–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li Y., Wu X., Zhou S., Owyang C. (2011) Low-affinity CCK-A receptors are coexpressed with leptin receptors in rat nodose ganglia: implications for leptin as a regulator of short-term satiety. Am. J. Gastrointest Liver Physiol. 300, G217-G227 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100. Schwartz M. W., Seeley R. J., Tschöp M. H., Woods S. C., Morton G. J., Myers M. G., D'Alessio D. (2013) Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503, 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]