

Background: Vitamin D insufficiency has been associated with chronic inflammatory diseases. However, the underlying mechanisms remain unclear.
Results: Vitamin D inhibits COX-2-mediated inflammatory response by modulating the Akt/NF-κB signaling pathway via direct up-regulation of thioesterase superfamily member 4.
Conclusion: Vitamin D plays novel roles in anti-inflammation.
Significance: Supplemental vitamin D could protect against chronic inflammatory diseases by targeting THEM4/Akt/NF-κB signaling.
Keywords: Cyclooxygenase (COX) Pathway, Lipopolysaccharide (LPS), Macrophages, NF-kappa B (NF-KB), Vitamin D, VDR
Abstract
Inadequate vitamin D status has been linked to increased risk of type 2 diabetes and cardiovascular disease. Inducible cyclooxygenase (COX) isoform COX-2 has been involved in the pathogenesis of such chronic inflammatory diseases. We found that the active form of vitamin D, 1,25(OH)2D produces dose-dependent inhibition of COX-2 expression in murine macrophages under both basal and LPS-stimulated conditions and suppresses proinflammatory mediators induced by LPS. Administration of 1,25(OH)2D significantly alleviated local inflammation in a carrageenan-induced paw edema mouse model. Strikingly, the phosphorylation of both Akt and its downstream target IκBα in macrophages were markedly suppressed by 1,25(OH)2D in the presence and absence of LPS stimulation through up-regulation of THEM4 (thioesterase superfamily member 4), an Akt modulator protein. Knockdown of both vitamin D receptor and THEM4 attenuated the inhibitory effect of 1,25(OH)2D on COX-2 expression in macrophages. A functional vitamin D-responsive element in the THEM4 promoter was identified by chromatin immunoprecipitation and luciferase reporter assay. Our results indicate that vitamin D restrains macrophage-mediated inflammatory processes by suppressing the Akt/NF-κB/COX-2 pathway, suggesting that vitamin D supplementation might be utilized for adjunctive therapy for inflammatory disease.
Introduction
Vitamin D deficiency is widespread, especially in adults from the middle East and Asia (1). It is believed that the main reason for vitamin D deficiency is lack of proper sun exposure, because many foods are naturally abundant in vitamin D (2). In the human body, vitamin D is enzymatically converted to 25-hydroxycholecalciferol or 25-hydroxyvitamin D3 in the liver and then transformed to 1,25-dihydroxy-vitamin D (1,25(OH)2D),3 the biologically active form, in the proximal tubule of the kidney (2). 1,25(OH)2D exerts physiological roles by acting on its receptor (vitamin D receptor (VDR)), which forms a heterodimer with the retinoid X receptor and regulates target gene expression by binding to specific hormone response elements (vitamin D-responsive elements (VDREs)) (3–5). 1,25(OH)2D stimulates intestinal calcium absorption and bone calcium mobilization; thus, insufficiency of vitamin D can lead to rickets in children and osteopenia/osteoporosis in adults (2). Cross-sectional studies have reported that vitamin D deficiency is associated with increased risk of chronic inflammatory disorders, including rheumatoid arthritis, cardiovascular disease, diabetes, and even cancer (6).
Prostaglandins (PGs) are a class of bioactive lipid mediators of an array of (patho)physiological conditions such as female reproduction, gastrointestinal ulcer, inflammation, and cancer (7, 8). Cyclooxygenase (COX), a membrane-bound protein, is the rate-limiting enzyme for PG biosynthesis. There are two isoforms encoded by distinct gene products: a constitutive COX-1 and an inducible COX-2 (9). COX-1 is broadly distributed to maintain physiological functions, whereas COX-2 can be induced rapidly in inflammatory cells by internal and external stimuli such as IL-1 and LPS (10). Traditional nonsteroidal anti-inflammatory drugs (NSAIDs) exhibit analgesic and antipyretic effects by blocking COX-derived PG formation (11). NSAIDs selective for inhibition of COX-2 retain anti-inflammatory efficacy with less adverse effects involving the gastrointestinal tract and reproductive system, but long term usage may predispose to cardiovascular events such as stroke (11). Vitamin D may regulate arachidonic acid release and modulate PGE2 production (12, 13) and displays antiproliferative and chemopreventive activities probably by suppressing COX-2-derived PG production in prostate and breast cancer cells (14, 15). Moreover, vitamin D exhibits robust immunomodulation effects on inflammatory cells including macrophages, which is consistent with the epidemiological associations between vitamin D deficiency and a large number of autoimmune and inflammatory diseases such as rheumatoid arthritis, lupus, inflammatory bowel disease, type 1 diabetes, infections, malignancies, transplant rejection, and cardiovascular diseases (16–18). These observations indicate vitamin D may play roles in anti-inflammation by regulating COX-2 expression and PG production, especially in inflammatory cells. However, the underlying mechanisms remain to be determined.
In this study, we found that vitamin D down-regulates expression of COX-2 and its PG products in macrophages and reduces inflammatory cytokine secretion by suppressing Akt phosphorylation and NF-κB signaling. Furthermore, vitamin D induces transcription of THEM4 (thioesterase superfamily member 4, a negative regulator of Akt) by activating VDR, which binds to a functional VDRE in the THEM4 promoter. Knockdown of either VDR or THEM4 attenuated vitamin D suppression of COX-2 expression. Administration of vitamin D also retards air pouch- and carrageenan-induced inflammation in mice. Thus, vitamin D modulates inflammatory responses through the VDR/THEM4/Akt/COX-2 pathway, which may contribute to the cardiovascular protective effect of vitamin D.
EXPERIMENTAL PROCEDURES
Animal Husbandry
COX-2 KO and COXNeo/Neo (also named COX-2 knockdown (KD)) mice were initially produced on a mixed C57BL/6 × Sv129 genetic background (50%:50%) and maintained on this hybrid C57BL/6/Sv129 background for over 20 generations. Littermates were used for control in all experiments. Age and sex were matched between experimental group and control group. All animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences of the Chinese Academy of Sciences.
Isolation of Peritoneal Macrophages
Mice were sacrificed by CO2 asphyxiation, and the abdominal skin was sterilized with 70% alcohol. Cold, sterile PBS (5 ml) was injected into the peritoneal cavity as a washing solution. Peritoneal macrophages were harvested with a syringe and stored in an ice bath. Harvested macrophages were washed twice with cold PBS and seeded at 1 × 107 to 2.5 × 107 per 60-mm dish in 10% FBS RPMI 1640 medium supplemented with a 1% mixture of penicillin/streptomycin solution.
Cell Culture and Drug Treatment
The RAW264.7 cell line was obtained from the American Type Culture Collection (Manassas, VA) and maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The cells were seeded onto 6-well culture plates at 5 × 106 cells/well unless otherwise specified. After seeding for 24 h, the cells were cultured in serum-free medium and incubated with different concentrations of 1,25-dihydroxy-vitamin D2 or D3 as indicted (Sigma-Aldrich) (final concentrations: 0, 10, 30, 50, and 100 nm) for 6 or 12 h. The final concentration of LPS was 5 μg/ml.
RNA Extraction, Semiquantitative RT-PCR, and Quantitative RT-PCR
Total RNAs were isolated from the macrophages using TRIzol reagent according to the standard procedure (Invitrogen) and then were reverse-transcribed with the Moloney murine leukemia virus reverse transcriptase (TaKaRa). The cDNAs were amplified by semiquantitative RT-PCR and real time RT-PCR with the SYBR Green PCR system (Applied Biosystems). Primers were designed with Primer Premier 5.0 and were as follows: COX-2-F, 5′-TGTGACTGTACCCGGACTGG-3′; COX-2-R, 5′-TGCACATTGTAAGTAGGTGGAC-3′; COX-1-F, 5′-GATTGTACTCGCACGGGCTAC-3′; COX-1-R, 5′-GGATAAGGTTGGACCGCACT-3′; TNF-α-F, 5′-CCCTCACACTCAGATCATCTTCT-3′; TNF-α-R, 5′-GCTACGACGTGGGCTACAG-3′; IL-6-F, 5′-ATGAAGTTCCTCTCTGCAAGAGACT-3′; IL-6-R, 5′-CACTAGGTTTGCCGAGTAGATCTC-3′; VDR-F, 5′-CGCTCCAACCAGTCTTTTACC-3′; VDR-R, 5′-AGCCCCACCTGGAACTTTATG-3′; c-Jun-F, 5′-TTCTACGACGATGCCCTCAAC-3′; c-Jun-R, 5′-CAGGTTCAAGGTCATGCTCTGTT-3′; THEM4-F, 5′-CAAATGTCAAAGGCCCAACA-3′; THEM4-R, 5′-GCACCTCCGTGAACAAATCC-3′; GAPDH-F, 5′-CGACTTCAACAGCGACACTCAC-3′; GAPDH-R, 5′-CCCTGTTGCTGTAGCCAAATTC-3′; β-actin-F, 5′-GGCTGTATTCCCCTCCATCG-3′; and β-actin-R, 5′-CCAGTTGGTAACAATGCCATGT-3′. The results were normalized with β-actin or GAPDH mRNA.
Western Blotting
Protein samples were separated by 10% SDS-PAGE and transferred onto PVDF membrane (Millipore) by electroblotting. The membranes were blocked in 5% milk in TBST (0.1% Tween 20, 150 mm NaCl, 50 mm Tris, pH 7.4) and then incubated with rabbit anti-COX-2 polyclonal antibody (1:1000; Cayman Chemical Co.), rabbit anti-COX-1 polyclonal antibody (1:1000; Cayman Chemical Co.), rabbit anti-phospho-Akt antibody (1:1000; Cell Signal Technology), rabbit anti-Akt antibody (1:1000; Cell Signaling Technology), rabbit anti-phospho-GSK-3β antibody (1:1000; Cell Signaling Technology), rabbit anti-GSK-3β antibody (1:1000; Cell Signaling Technology), rabbit anti-phospho-IκB antibody (1:1000; Cell Signaling Technology), rabbit anti-IκB antibody (1:1000; Cell Signaling Technology), rabbit anti-NF-κB (p65) antibody (1:1000; Cell Signaling Technology), rabbit anti-phospho-ERK1/2 MAPK antibody (1:1000; Cell Signaling Technology), mouse anti-ERK1/2 MAPK antibody (1:1000; Cell Signaling Technology), rabbit anti-phospho-p38 MAPK antibody (1:1000; Cell Signaling Technology), rabbit anti-HA-tag (1:1000; Cell Signaling Technology), and mouse anti-β-actin monoclonal antibody (1:5000; Cell Signaling Technology). Horseradish peroxidase-conjugated goat anti-mouse IgG and horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2000; Cell Signaling Technology) were used as secondary antibodies. Signals were detected by enhanced chemiluminescence (ECL; Pierce).
Enzyme-linked Immunosorbent Assay
The supernatants collected from cell culture or exudates collected from air pouch lavage were centrifuged for 15 min at 12,000 × g at 4 °C. ELISA for TNF-α and IL-6 was performed according to the manufacturer's instructions (R&D Systems). Absorbance was measured with a microplate reader (Biotek), using a wavelength fixed at 450 nm. A standard curve for both cytokines was used to calculate protein levels.
PG Extraction and Analysis
Cell culture supernatant and exudates from the air pouch model were collected after centrifugation for 15 min at 12,000 × g at 4 °C. PGs (PGE2, PGD2, 6-keto-PGF1α, PGF2α, and thromboxane B2) were extracted as described previously (19) and quantified by LC/MS/MS (20).
siRNA Knockdown
VDR, THEM4, and scramble siRNA were obtained from GenePharma (VDR-siRNA-F-mus, 5′-GCAGCCAAGACUACAAAUATT-3′; VDR-siRNA-R-mus, 5′-UAUUUGUAGUCUUGGCUGCTT-3′; THEM4-siRNA-F-mus, 5′-AGUAUGCGAUGUUCUAUAAATT-3′; and THEM4-siRNA-R-mus, 5′-UUAUAGAACAUCGCAUACUTT-3′). RAW264.7 cells were seeded in 6-well plates in DMEM containing 10% FBS at of 1.5–3.0 × 105 cells/well (corresponding to a density of 40–60% at the time of transfection). After 12–24 h, transfection was performed using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions, using 2 pmol of total siRNA and 5 μl of RNAiMax reagent per well. Cells were harvested after 72 h and analyzed by Western blotting or quantitative RT-PCR to determine the efficiency of gene knockdown.
Free Fatty Acid Measurement
Free fatty acids (FFAs) in the RAW264.7 cells with or without 1,25(OH)2D3 treatment and plasma FFAs of vitamin D2-treated mice were measured using a free fatty acid quantification kit (BioVision) according to the manufacturer's instruction.
Prediction of Transcription Factor Binding Sites
The fragment between −2000 and ±500 bp of the transcription start site of the THEM4 gene from the RefSeq database was analyzed for putative VDRE motifs in TRANSFAC version 10.2 using PWMSCAN (21). A match score with a p value < 5 × 10−6 was considered to be a high confidence binding site prediction.
Chromatin Immunoprecipitation
RAW264.7 and HEK293-A cells were transfected with VDR expression plasmids (GeneCopoeia Inc., Rockville, MD) using the LipofectamineTM 2000 reagent (Invitrogen). At 24 h post-transfection, cells were treated with or without 1,25(OH)2D3 (100 nm) for 12 h. A ChIP assay was performed with a Magna ChIPTM A/G chromatin immunoprecipitation kit (Millipore) according to the manufacturer's protocol. Briefly, cells were cross-linked with formaldehyde. The cross-linking reaction was stopped by addition of glycine followed by a washing step with PBS. The pellet was lysed and sonicated to shear the chromatin into 200–1000-bp fragments. The chromatin extract was incubated with 10 μg of rat anti-VDR antibody (Abcom) or rat IgG (negative control) at 4 °C with rotation overnight, and the antibody-antigen-DNA complex was collected by protein G-agarose. The immunocomplexes were washed and the protein-DNA complexes were eluted, and then proteinase K was used to reverse the cross-linking of protein-DNA complexes to free the DNA. DNA was purified with the DNA purification kit (Promega), dissolved in elution buffer, and used for quantitative PCR analysis. The following primers were used for detection of VDR binding sites: forward 5′-TCTTGCAGCTATCCCTAGCC-3′ and reverse 5′-GGTGCCTGACATGAAGGAAT-3′ (for mRegion1); forward 5′-GTTTGAACCACCCACCTCTG-3′ and reverse 5′-GTCAAAGGGTGCACCAAGAT-3′ (for mRegion2); forward 5′-CCCGGATTTGCATTATCTGA-3′ and reverse 5′-TAGGCGCATACCTTCTGAGC-3′ (for hRegion1); and forward 5′-CAACCCAGTCCGATTTCAAG-3′ and reverse 5′-GCGCTTACGCCTTAAAAGACT-3′ (for hRegion2). Quantitative PCR products were analyzed by electrophoresis on agarose gels.
Luciferase Assay
HEK293-A cells were seeded in 48-well plates and grown to 70–80% confluence. Transfection was performed using the LipofectamineTM 2000 reagent (Invitrogen). Cells were co-transfected with 10 ng of the pRL-TK vector DNA (Promega) and 100 ng of VDR expression plasmids and either the empty pGL3-Basic plasmid (a promoterless control from Promega) or pGL3-Basic plasmid containing a VDR binding sequence in the THEM4 promoter region. The pRL-TK vector, which provided constitutive expression of Renilla luciferase, was co-transfected as an internal control to correct for differences in transfection and harvesting efficiency. After 24 h of incubation, cells were harvested and analyzed for luciferase activity using the dual luciferase reporter assay system (Promega). To characterize the dose-dependent effect, cells were treated with 1,25(OH)2D3 for another 12 h before harvesting. Promoter activity is reported in relative light units and normalized against the activity of the empty pGL3-Basic vector.
Carrageenan-induced Paw Edema Mouse Model
Mice (2–3 months old) were anesthetized by intraperitoneal injection of 20 mg/kg ketamine hydrochloride and subjected to subcutaneous injection of 25 μl of 1% λ-carrageenan (Sigma-Aldrich) in 0.9% saline into the plantar region of the left hind paw, whereas the right paw received the same amount of saline solution. Intraperitoneal injection of 2 mg/kg vitamin D2 per day or intraperitoneal injection of 250 μg/kg vitamin D3 per day for 3 days was administered before carrageenan injection. We assessed edema 4 h later by using calipers to measure paw thickness at the metatarsal level (22). After λ-carrageenan treatment, the animals were euthanized, and the left hind paws were collected for histopathology.
Carrageenan-induced Air Pouch Mouse Model
Mice (12 weeks old) were anesthetized by intraperitoneal injection of 20 mg/kg ketamine hydrochloride. Dorsolateral air pouches were inflated by subcutaneous injection of 3 ml air on days 1 and 4. On day 6, 100 μl of 1% λ-carrageenan (Sigma-Aldrich) diluted in 1 ml of PBS was injected into the pouch. At 24 h after λ-carrageenan treatment, the animals were euthanized. Vitamin D2 (2 mg/kg per day) was administered by intraperitoneal injection 3 days before dorsolateral air pouch. The air pouch lavage was washed by repeat injection/aspiration using 2 ml of PBS, and the exudates were collected by centrifugation for PG analysis and proinflammatory cytokine detection.
Flow Cytometry Analysis
The infiltrated inflammatory cells in pouch exudates were centrifuged and resuspended in PBS. Phycoerythrin-conjugated anti-mouse F4/80 (eBioscience) was used to label macrophages surface marker according to the manufacturer's instructions. The presence of F4/80+ cells were determined and collected by BD FACSAria cell sorter (BD Biosciences) and analyzed using FlowJo (version 7.6.1; Tree Star, Inc.).
Histopathology
The left hind paw in the carrageenan-induced paw edema model was fixed in 10% buffered formalin for 24 h, processed routinely, and embedded in paraffin for staining with hematoxylin and eosin.
Statistics
The data are expressed as means ± S.E.; analyses were performed with the Student's t test or analysis of variance analysis of variance test as appropriate; p < 0.05 was considered statistically significant. Prism 5.0 software (GraphPad InStat 3) was used for all calculations.
RESULTS
1,25(OH)D3 Inhibits COX-2 Expression and PG Production in Macrophages
We sought to determine whether vitamin D influences the inflammatory response. Macrophages were pretreated with the active form of vitamin D-1,25(OH)2D3, and COX-2 expression and PG production were examined in the presence and absence of LPS stimulation. Although COX-2 expression is relatively low in RAW264.7 without LPS stimulation (Fig. 1), 1,25(OH)2D3 provided dose-dependent suppression of COX-2 expression at the mRNA and protein levels (Fig. 1, A, C, and D) but had no effect on COX-1 expression (Fig. 1, B, C, and E). Similarly, induction of COX-2 mRNA (Fig. 2A) and protein (Fig. 2, B and D) in RAW264.7 upon LPS stimulation were markedly attenuated by pretreatment with 1,25(OH)2D3 (including 30 nm; data not shown), whereas COX-1 expression was unaltered (Fig. 2, B, C, and E). As such, we also observed marked depression of 12-O-tetradecanoylphorbol-13-acetate-induced COX-2 expression by 1,25(OH)2D3 (data not shown). Then we tested the effects of 1,25(OH)2D3 on PG generation (PGE2, PGD2, 6-keto-PGF1α, PGF2α, and thromboxane B2) by RAW264.7 macrophages. All PG products induced by LPS, as anticipated, were significantly repressed by 1,25(OH)2D3 in a dose-dependent fashion (Fig. 2, F–J); indeed, its inhibitory capability at 100 nm is comparable with the widely used anti-inflammatory glucocorticosteroid dexamethasone (Fig. 2, F–J).
FIGURE 1.
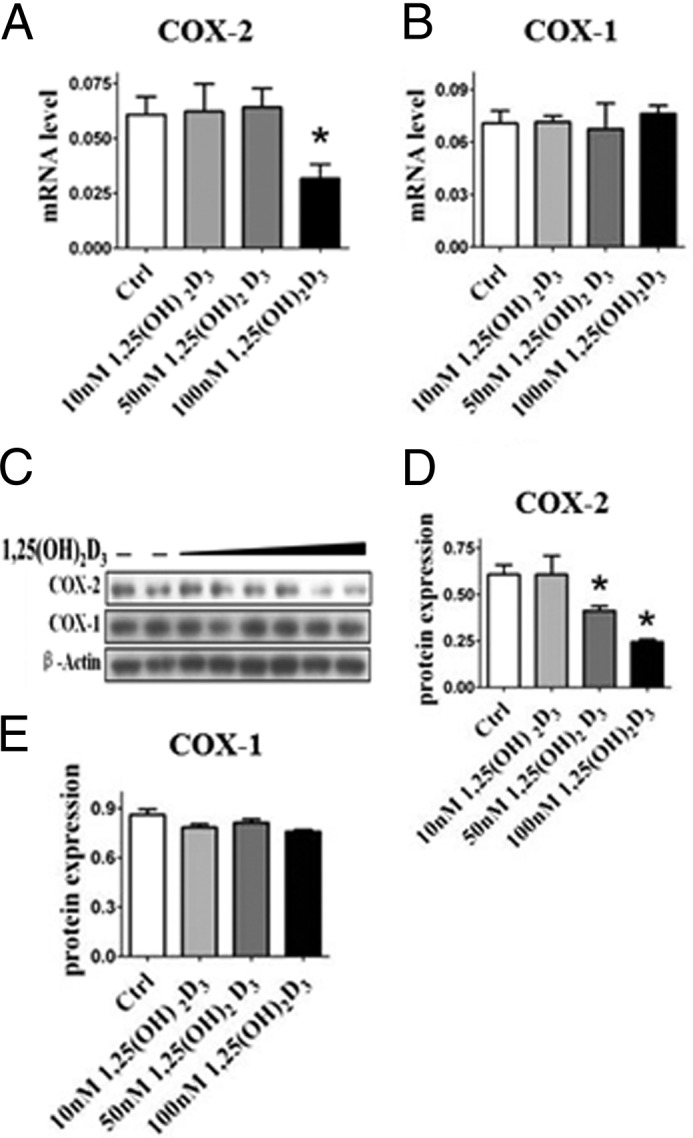
1,25(OH)2D3 inhibits COX-2 expression in RAW264.7 macrophages. RAW264.7 cells were treated with different dosages of 1,25(OH)2D3 (10 nm, 50 and 100 nm). A–C, the mRNA (A and B) and protein expression (C) of COX-2 and COX-1 were analyzed by quantitative RT-PCR and Western blotting. D and E, densitometric quantitation of COXs expression by comparison with β-actin as shown in C. *, p < 0.05; **, p < 0.01; n = 4. Ctrl, control.
FIGURE 2.

1,25(OH)2D3 inhibits COX-2 expression and PG production in macrophages. RAW264.7 cells were treated with different dosages of 1,25(OH)2D3 (10, 50, and 100 nm) in the absence and presence of LPS. A–C, the mRNA (A and C) and protein expressions (B) of COX-2 and COX-1 were analyzed by quantitative RT-PCR and Western blotting. D and E, densitometric quantitation of COXs expression by comparison with β-actin as shown in B. *, p < 0.05; **, p < 0.01; n = 4. F–J, effect of 1,25(OH)2D3 treatment on PG profile of RAW264.7 cell. *, p < 0.05; **, p < 0.01; n = 6. DXM, dexamethasone.
1,25(OH)2D3 Inhibits Secretion of Proinflammatory Cytokines in Macrophages
Along with COX-2 induction, large amounts of proinflammatory mediators are generated by inflammatory cells during the inflammatory process (23). We used semiquantitative RT-PCR and ELISA to determine whether 1,25(OH)2D3 may restrain the expression and secretion of proinflammatory cytokine in macrophages. 1,25(OH)2D3 treatment suppressed mRNA transcripts of both TNF-α and IL-6 in RAW264.7 cells in the absence and presence of LPS at dose-dependent mode, (Fig. 3, A and B), although their expression was up-regulated in response to LPS stimulation. Consistently, secretion of both TNF-α and IL-6 was also dose-dependently inhibited in supernatants of cultured macrophages by addition of 1,25(OH)2D3 (Fig. 3, C and D).
FIGURE 3.
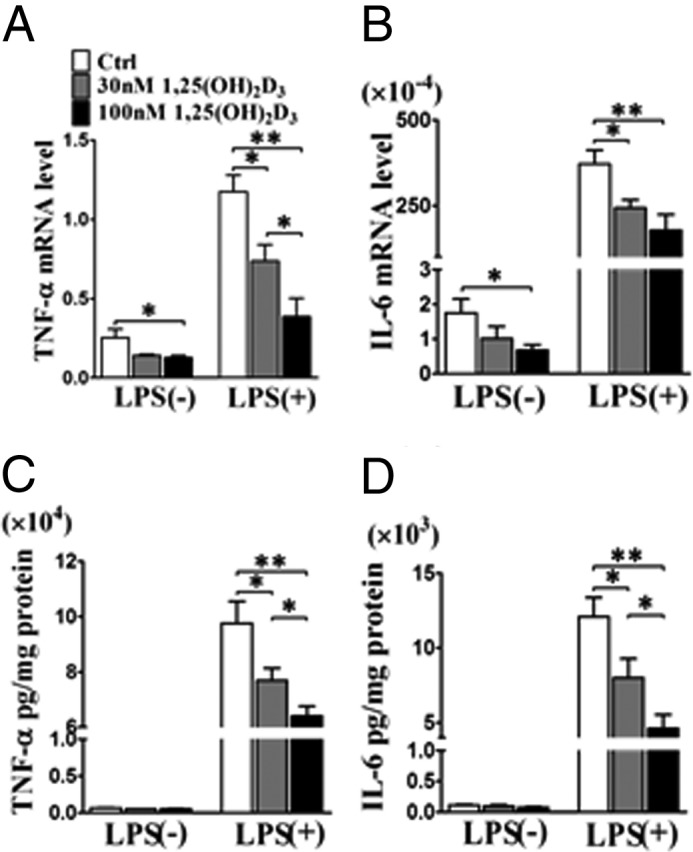
1,25(OH)2D3 inhibits the expression and secretion of proinflammatory cytokines in macrophages. A and B, effect of 1,25(OH)2D3 on mRNA expression of TNF-α and IL-6 in RAW264.7 cells in presence and absence of LPS stimulation. *, p < 0.05; n = 6. C and D, effect of 1,25(OH)2D3 on secretion of TNF-α and IL-6 in RAW264.7 cells in absence and presence of LPS stimulation. *, p < 0.05; n = 6. Ctrl, control.
COX-derived PGs, such as PGE2, regulate proinflammatory cytokine release (24). To determine whether suppression of TNF-α and IL-6 by 1,25(OH)2D3 is due to inhibition of COX-2 derived PGs, we analyzed the expression of TNF-α and IL-6 in primary peritoneal macrophages from COX-2 KO mice. Secretion of TNF-α and IL-6 from COX-2 KO peritoneal macrophages was much lower in comparison with wild type, although both proteins were clearly increased by LPS stimulation (Fig. 4, A and B). 1,25(OH)2D3 pretreatment attenuated LPS-induced secretion of TNF-α and IL-6 in peritoneal macrophages from WT mice (Fig. 4, A and B). However, we did not observe notable alterations of TNF-α and IL-6 secretion of peritoneal macrophages from COX-2 KO mice in response to 1,25(OH)2D3 even at 100 nm (Fig. 4, A and B). Similarly, COX-2 selective inhibitor NS398 terminated this inhibitory effect on secretion of proinflammatory cytokines (Fig. 4, C and D). Thus, inhibition of TNF-α and IL-6 secretion by 1,25(OH)2D3 in macrophages is mediated through suppression of COX-2.
FIGURE 4.
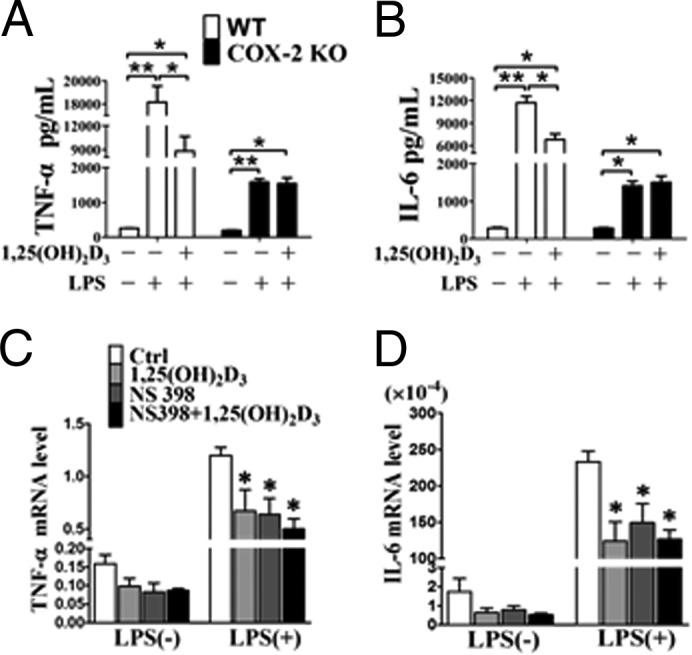
1,25(OH)2D3 inhibits secretion of proinflammatory cytokines by inhibiting of COX-2. A and B, effect of 1,25(OH)2D3 on secretion of TNF-α and IL-6 in peritoneal macrophages in the presence of LPS stimulation. *, p < 0.05; n = 6. C and D, inhibitory effect of 1,25(OH)2D3 on secretion of TNF-α and IL-6 vanished in NS398-pretreated RAW264.7 cells. *, p < 0.05; n = 6. Ctrl, control.
1,25(OH)2D3 Inhibits Akt Phosphorylation and the NF-κB Pathway
LPS promotes robust COX-2 expression through activation of both Akt and MAPKs mediated by Toll-like receptor 4 (25). Activation of Akt promotes nuclear translocation of nuclear factor NF-κB p65 unit and drives target gene expression (i.e. COX-2) by degrading IκB (25). As expected, LPS treatment enhanced phosphorylation of Akt and its substrate GSK-3β and induced transcription of COX-2; and inhibitor Ly294002 inhibited LPS-induced COX-2 expression (Fig. 5A). Interestingly, 1,25(OH)2D3 inhibited Akt phosphorylation induced by LPS in RAW264.7 cells and suppressed IκB phosphorylation in the cytoplasm and nuclear translocation of NF-κB (p65) (Fig. 5, B–G). Knockdown of VDR by specific siRNA (Fig. 5E) blunted the inhibitory effect of 1,25(OH)2D3 on phosphorylation of Akt and IκB phosphorylation (Fig. 5, F–H) and subsequently restored the LPS-induced COX-2 expression (Fig. 5I). Thus, the suppression of COX-2 by 1,25(OH)2D3 may be mediated by the AKT/IκB/NF-κB pathway through activation of VDR.
FIGURE 5.

1,25(OH)2D3 inhibits COX-2 expression through suppression of Akt/NF-κB pathway macrophages. A, effect of Akt inhibitor Ly294002 on LPS-induced COX-2 expression and Akt/NF-κB signaling in RAW264.7 cells. B, effect of 1,25(OH)2D3 on phosphorylation of IκB (p-IκB) in cytoplasmic and nuclear translocation of NF-κB (p65) in RAW264.7 cells. C and D, densitometric quantitation of p-IκB and nuclear p65 expression by comparison with β-actin as shown in B. *, p < 0.05; **, p < 0.01; n = 4. E, knockdown of VDR by specific siRNA (si-VDR). **, p < 0.01; n = 3. F–I, effect of knocking down VDR on 1,25(OH)2D3-mediated suppression of Akt/NF-κB/COX-2 axis in macrophages. G–I, densitometric quantitation of the expression of p-Akt, p-IKBα, and COX-2 by comparison with Akt, IKBα, and β-actin, respectively. *, p < 0.05; **, p < 0.01; n = 3. Ctrl, control; Scram, scramble.
Effect of 1,25(OH)2D3 on MAPK Pathways in Macrophages
The classic MAPKs include well characterized ERK1/2, JNK, and p38 (26). As shown in Fig. 4, LPS induced phosphorylation of ERK1/2 and COX-2 expression; ERK inhibitor PD98059 MAPK (27), as expected, depressed phosphorylation of ERK1/2 and down-regulated its target gene—COX-2, not COX-1 (Fig. 6, A–C). In contrast to COX-2 expression with sharp reduction, phosphorylation of ERK1/2 in macrophages was unchanged by 1,25(OH)2D3 in the absence and presence of LPS (Fig. 6D). p38 MAPK regulates COX-2 expression by stabilizing the COX-2 mRNA (28), whereas JNK activation induces phosphorylation of its substrate c-Jun and promotes transcription of COX-2 gene through binding AP-1 (29). Thus, p38 MAPK and JNK inhibitors (SB203580 and SP600125, respectively) suppressed the induction of COX-2 protein by LPS (Fig. 7, A and B), although we failed to observe the visible influences of 1,25(OH)2D3 on phosphorylation of p38 (Fig. 7C) and JNK (Fig. 7D) and c-Jun mRNA expression levels (Fig. 7E). Taken together, the regulatory effect of 1,25(OH)2D3 on COX-2 expression is not dependent on the MAPK pathway.
FIGURE 6.
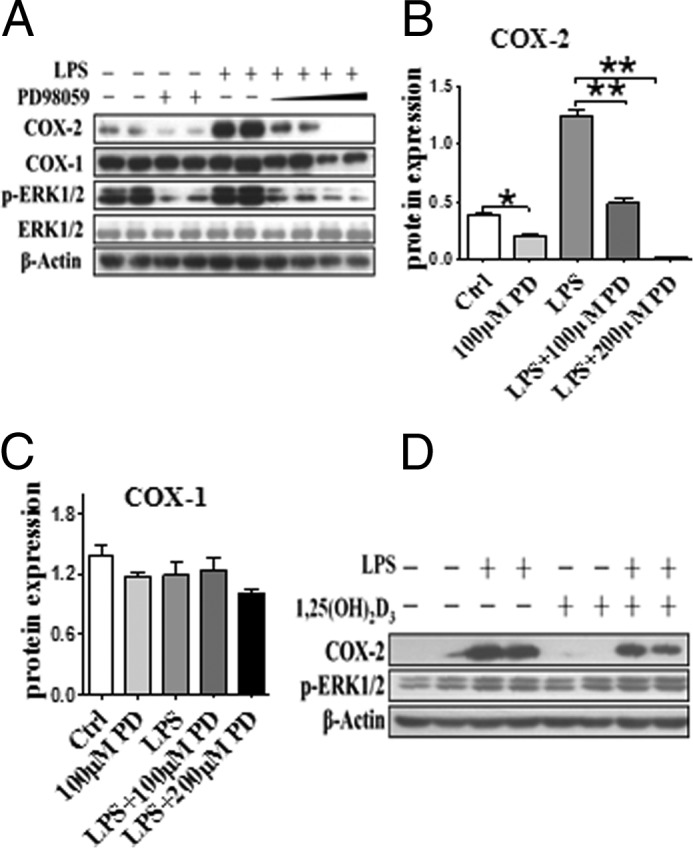
Effect of 1,25(OH)2D3 on ERK1/2 MAPK pathway in macrophages. A, effect of ERK1/2 MAPK inhibitor PD98059 (100 and 200 nm) on COX-2 expression and phosphorylation of ERK1/2 (p-ERK1/2) in RAW264.7 cells. B and C, densitometric quantitation of expression of COXs by comparison with β-actin as shown in A. *, p < 0.05; **, p < 0.01; n = 4. D, the effect of 1,25(OH)2D3 on p-ERK1/2 in macrophages. Ctrl, control.
FIGURE 7.

Impact of 1,25(OH)2D3 on p38 and JNK MAPK pathways in macrophages. A, effect of p38 MAPK inhibitor SB203580 on LPS-induced COX-2 expression in RAW264.7 cells. B, effect of JNK MAPK inhibitor SP600125 on LPS-induced COX-2 expression in RAW264.7 cells. C, effect of 1,25(OH)2D3 on phosphorylation of p38 (p-p38) in RAW264.7 cells. D, effect of 1,25(OH)2D3 on phosphorylation of JNK (p-JNK) in RAW264.7 cells. E, effect of 1,25(OH)2D3 on mRNA expression of c-Jun in RAW264.7 cells. p = NS; n = 6. Ctrl, control.
1,25(OH)2D3 Inhibits AKT Phosphorylation by Inducing THEM4 Transcription via VDR Activation
AKT activity can be modulated through its interaction with various binding partners (30). For example, AKT activity can also be down-regulated by the pseudokinase TRB3 (tribble3) (31) and CTMP (carboxyl-terminal modulator protein, also named THEM4), an negative regulatory component of AKT (32). We found that THEM4 expression was up-regulated in both RAW264.7 cells (Fig. 8, A and B) and primary peritoneal macrophages (Fig. 9A) by 1,25(OH)2D3 treatment at protein levels, which could be blunted by knockdown of VDR (Figs. 8, C and D, and 9B), suggesting that 1,25(OH)2D3 modulates THEM4 expression by activating VDR. Treatment with 1,25(OH)2D3 failed to influence PDK1 and TRB3 expression (data not shown). Interestingly, we observed that the suppression of AKT phosphorylation and COX-2 expression by 1,25(OH)2D3 was restored by THEM4 knockdown with specific siRNAs (Fig. 8E) in the presence and absence of LPS in both RAW264.7 (Fig. 8, F–H) and primary peritoneal macrophages (Fig. 9C). Given that THEM4 belongs to the acyl-CoA thioesterases of the hot dog fold family, which hydrolyze acyl-CoA esters to the FFA and coenzyme A (CoASH) (33), we examined whether 1,25(OH)2D3 influences the FFA generation in macrophages. Dose-dependent elevation of cellular FFA production by 1,25(OH)2D3 was observed (Fig. 8I). Thus, the inhibitory effect of 1,25(OH)2D3 on AKT activity and COX-2 transcription in macrophages is attributed to up-regulation of THEM4. One might speculate that VDR regulates THEM4 expression by direct binding to the VDRE in the THEM4 promoter. Indeed, three potential VDRE sites (one in mRegion 1, AAAGTCCCTAATCCCTTTCCT; and two in mRegion 2, CGCCGGCAATTGGGGTCAGG and TCACCACCTGTCCCC) were predicted in the murine THEM4 promoter by bioinformatics analysis (Fig. 10A). VDR binding at these sites of the murine THEM4 promoter were tested by ChIP assays. Treatment of RAW264.7 cells with 1,25(OH)2D3 significantly enhanced VDR binding at mRegion 1 (6-fold; p < 0.05), but not mRegion 2 versus IgG controls. (Fig. 10, B and C). To examine VDR-dependent promoter activation, we generated THEM4 promoter luciferase reporters by inserting both mRegion 1 and mRegion 2 into pGL3 vectors and co-transfected these reporters with a VDR expression plasmid into RAW264.7 cells. 1,25(OH)2D3 treatment induced robust luciferase activities of pGL3 + region 1 in a VDR- and 1,25(OH)2D3 dose-dependent manner (Fig. 10, D and E). Likewise, we found one VDRE located −450 to −435 of the transcriptional start site in human THEM4 gene promoter using ChIP assay (Fig. 11, A–C). Therefore, 1,25(OH)2D3 represses COX-2 expression through the VDR-mediated THEM4/AKT/NF-κB pathway (Fig. 10F).
FIGURE 8.

1,25(OH)2D3 inhibits COX-2 expression by up-regulating THEM4 in RAW 264.7 cells. A, effect of 1,25(OH)2D3 on THEM4 expression in RAW264.7 cells. B, densitometric quantitation of THEM4 expression as shown in A. #, p < 0.05 versus nontreatment of 1,25(OH)2D3; n = 6. C, effect of VDR knockdown (si-VDR) on 1,25(OH)2D3-mediated up-regulation of THEM4 in RAW 264.7 cells. D, densitometric quantitation of THEM4 expression as shown in C. #, p < 0.05 versus nontreatment of 1,25(OH)2D3; *, p < 0.05 versus treatment of scramble siRNA; n = 6. E, knockdown of THEM4 by specific siRNA (siRNA). *, p < 0.05; n = 3. F, effect of knocking down THEM4 on 1,25(OH)2D3-mediated inhibition of COX-2 expression and p-Akt in RAW264.7 cells. G, densitometric quantitation of COX-2 as shown in F. *, p < 0.05 versus LPS control; #, p < 0.05 versus nontreatment with 1,25(OH)2D3; &, p < 0.05 versus scramble siRNA; n = 6. H, densitometric quantitation of p-Akt as shown in F. *, p < 0.05 versus LPS control; #, p < 0.05 versus nontreatment with 1,25(OH)2D3; &, p < 0.05 versus scramble siRNA; n = 6. I, effect of 1,25(OH)2D3 on cellular FFA production in RAW264.7 cells. *, p < 0.05; n = 6. Ctrl, control; Scram, scramble.
FIGURE 9.
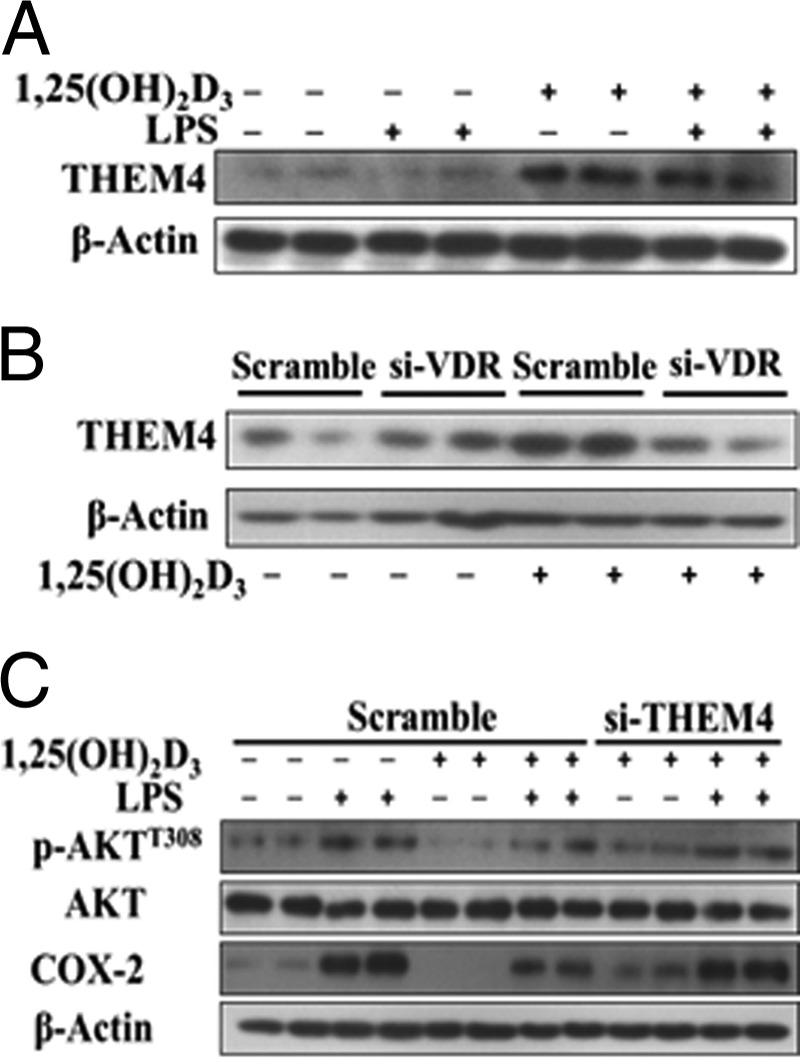
1,25(OH)2D3 inhibits COX-2 expression by up-regulating THEM4 in primary peritoneal macrophages. A, effect of 1,25(OH)2D3 on THEM4 expression in primary peritoneal macrophages. B, effect of knocking down VDR knockdown (si-VDR) on 1,25(OH)2D3-mediated up-regulation of THEM4 in primary peritoneal macrophages. C, effect of knocking down THEM4 on 1,25(OH)2D3-mediated inhibition of COX-2 expression and p-Akt in primary peritoneal macrophages. The experiments were repeated three times.
FIGURE 10.

VDR regulates THEM4 expression by direct binding to the VDRE in the THEM4 promoter. A, schematic illustration of the mouse THEM4 promoter and three predicted VDREs. B, ChIP analysis of the predicted VDREs in the mouse THEM4 promoter. *, p < 0.05 versus IgG control; n = 6. C, gel electrophoresis of PCR-amplified VDRE-containing fragments. D, the impact of VDR on the functional VDRE in the THEM4 promoter. *, p < 0.05 versus pGL3; #, p < 0.05 versus 100 ng of VDR plasmid; &, p < 0.05; versus 200 ng of VDR plasmid. E, the impact of 1,25(OH)2D3 on the functional VDRE in the THEM4 promoter. *, p < 0.05 versus pGL3; #, p < 0.05 versus 1 nm 1,25(OH)2D3; &, p < 0.05; versus 10 nm 1,25(OH)2D3. F, schematic diagram of proposed molecular mechanisms by which vitamin D attenuates LPS-induced COX-2 expression in macrophages. IKK, IκB kinase; TLR4, Toll-like receptor 4.
FIGURE 11.
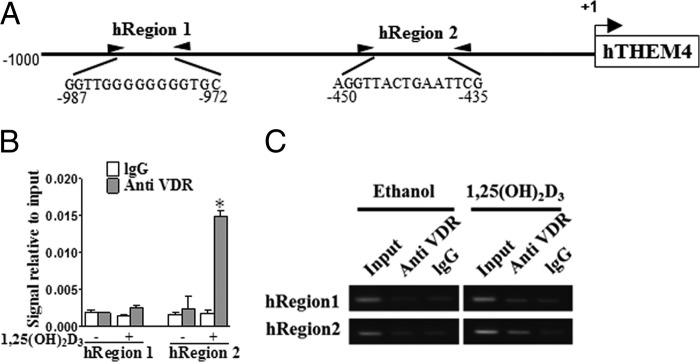
Functional VDREs in human THEM4 promoter. A, two predicted VDREs (hRegion 1 and hRegion 2) in the human THEM4 promoter. B, ChIP analysis of two VDREs in the human THEM4 promoter. *, p < 0.05 versus IgG control; n = 6. C, gel electrophoresis of PCR-amplified VDRE-containing fragments.
Vitamin D Inhibited Air Pouch-induced Inflammation in Mouse
Vitamin D3 is mainly produced in the skin by sun exposure, accounting for more than 90% of the vitamin D requirement of the body (34), whereas vitamin D2 is mainly obtained from dietary supplements (35). Both forms share similar structure and biological activity and are further metabolized in vivo (36). To determine whether vitamin D2 has similar effect on COX-2 expression, macrophages were pretreated with 1,25(OH)2D2. The elevated phosphorylation of AKT and expression of COX-2 after LPS stimulation were both suppressed by 1,25(OH)2D2 and restored by transfection of VDR siRNA (Fig. 12), indicating that 1,25(OH)2D2 modulates COX-2 expression by activating VDR. Then the role of vitamin D2 on air pouch-induced inflammation was examined, and pouch exudates after 24 h carrageenan injection were collected to assess infiltrated inflammatory cells, cytokines, and PG products. Carrageenan injection triggered acute inflammatory response with massive inflammatory cell infiltration, elevated proinflammatory cytokines, and PGs in pouch exudates compared with saline control (data not shown). Strikingly, vitamin D2 administration in WT mice significantly reduced infiltrated cells by 48% (p < 0.05; Fig. 13A), secretion of TNF-α and IL-6 by 29 and 38%, respectively (p < 0.05; Fig. 13, B and C), and PGD2, PGE2, and PGF2α production by 70, 78, and 61%, respectively (Fig. 14, A–E) in pouch exudates. These cytokine reductions caused by vitamin D2 were completely vanished in COX-2 KO mice (Fig. 13, B and C), although weaker inflammatory reactions compared with the WT controls. To further confirm the role of macrophage in pouch-induced inflammation model, infiltrated macrophages were sorted from pouch exudates by flow cytometry and then subjected to semiquantitative RT-PCR analysis Interestingly, vitamin D2 reduced total infiltrated macrophages (Fig. 14F) and also suppressed expression of COX-2, IL-6, and TNF-α in macrophages (Fig. 14, G–I). Again, THEM4 expression level in the infiltrated macrophages was doubled by vitamin D2 treatment (Fig. 14J). Thus, vitamin D2 ameliorated the air pouch-induced inflammatory response probably by suppressing macrophage COX-2.
FIGURE 12.
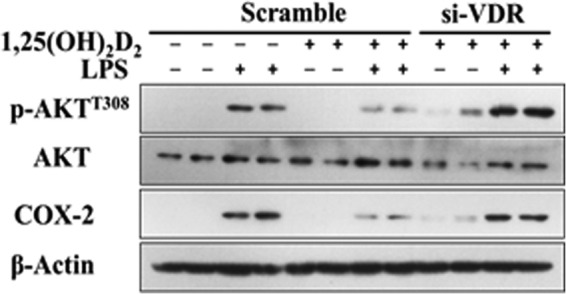
1,25(OH)2D2 inhibits LPS-induced COX-2 and phospho-AKTT308 in the RAW264. 7 cells. RAW264.7 cells were exposed to 250 nm 1,25(OH)2D2 or VDR siRNAs (si-VDR), and cell lysates were analyzed to Western blotting. The experiments were repeated three times.
FIGURE 13.
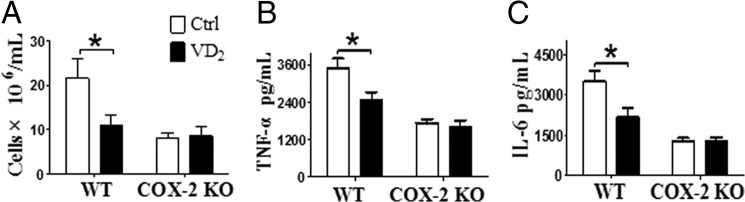
Vitamin D2 exhibits anti-inflammatory effects by suppressing macrophage COX-2 in a carrageenan-induced air pouch mouse model. Vitamin D2 (VD2, 2 mg/kg per day) was administrated by intraperitoneal injection 3 days before dorsolateral air pouch. Pouch exudates after 24 h of carrageenan injection were collected. The infiltrated inflammatory cells (A) were counted, and the secretion of TNF-α (B) and IL-6 (C) was analyzed by ELISA. *, p < 0.05; n = 6.
FIGURE 14.
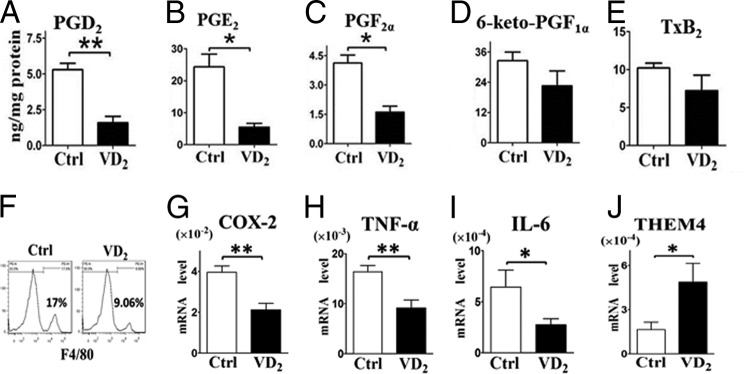
Vitamin D2 suppresses PG secretion and expression of proinflammatory cytokines in macrophages in carrageenan-induced air pouch mouse model. A–E, PGs (PGE2, PGD2, 6-keto-PGF1α, PGF2α, and thromboxane B2 (TxB2)) in the cell exudates were analyzed by LC/MS/MS. *, p < 0.05; **, p < 0.01; n = 6. F, F4/80 positive cells were sorted from pouch exudates by flow cytometry after 24 h of carrageenan injection. G–J, effect of vitamin D2 on expression of COX-2, TNF-α, IL-6, and THEM4 in phycoerythrin-conjugated F4/80 positive cells. *, p < 0.05; **, p < 0.01; n = 6. VD2, vitamin D2; Ctrl, control.
Vitamin D Suppressed Carrageenan-induced Paw Edema in Mice
The carrageenan-induced paw edema mediated by PGs and histamine represents a classical model of edema formation and hyperalgesia (22). Edema measured as increased paw thickness at the metatarsal level was significantly lower in COX-2 KD mice than in WT controls challenged with Carrageenan (36% decrease, p < 0.01; Fig. 15A). Vitamin D3 pretreatment provided significant protection with a 31% decrease of paw thickness in mice (0.92 ± 0.11 mm versus 1.35 ± 0.08 mm, p < 0.05; Fig. 15A). However, we failed to detect overt protection of COX-2 KD mice by vitamin D3 (0.83 ± 0.15 mm versus 0.84 ± 0.18 mm; Fig. 15A). Hematoxylin and eosin staining of inflamed paw tissue conferred the analogous effects of vitamin D treatment on local inflammation response (the thickness of epidermis and dermis of vitamin D3/WT group, 221.8 pixels ± 16.6 versus nontreated WT group, 304.4 pixels ± 25.9. p < 0.05 (Fig. 15, B and C). Consistently, we observed the protective effect of vitamin D2 against carrageenan-induced paw edema in WT mice (0.93 ± 0.11 mm versus 1.27 ± 0.12 mm, p < 0.05; Fig. 16), but not in COX-2 KD mice.
FIGURE 15.
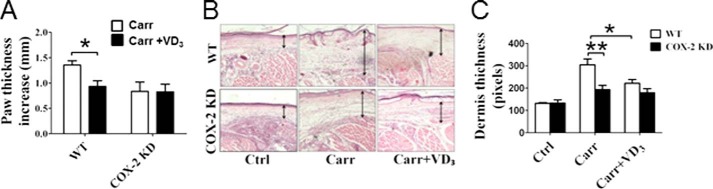
Vitamin D3 exhibits anti-inflammatory effects in carrageenan-induced paw edema mouse model. Vitamin D3 (250 μg/kg per day) was administrated by intraperitoneal injection 3 days before carrageenan injection. A, carrageenan (Carr)-induced paw edema was measured using calipers in WT mice and COX-2 KD mice. The results are displayed as increases in paw thickness. *, p < 0.05 versus WT; n = 8–10. B, representative hematoxylin and eosin staining of carrageenan-challenged paw tissue in WT mice and COX-2 KD mice. C, thickness of epidermis and dermis was calculated under a microscope. *, p < 0.05 versus WT; **, p < 0.01 versus WT; n = 8–10. VD3, vitamin D3; Ctrl, control.
FIGURE 16.
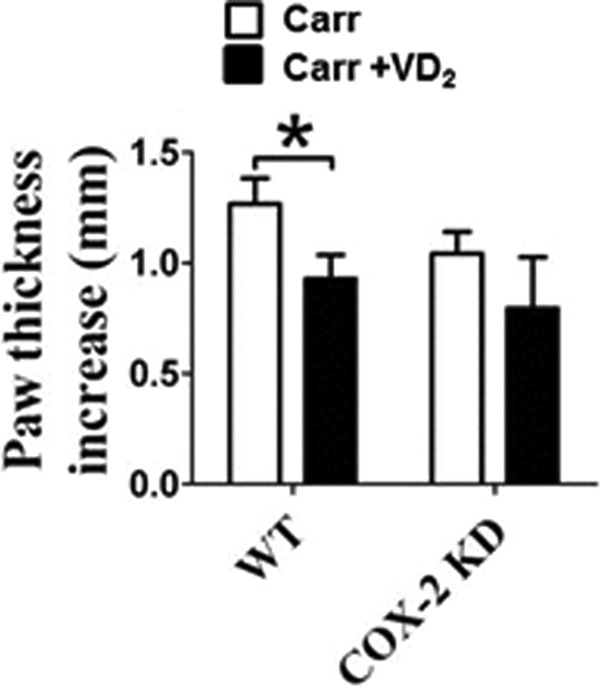
Vitamin D2 exhibits anti-inflammatory effects in carrageenan-induced paw edema mouse model. Vitamin D2 (2 mg/kg per day) was administered by intraperitoneal injection 3 days before carrageenan injection. Carrageenan-induced paw edema was measured using calipers in WT mice and COX-2 KD mice. The results are displayed as increases in paw thickness. *, p < 0.05 versus WT; n = 8. VD2, vitamin D2; Carr, carrageenan.
DISCUSSION
Vitamin D plays a vital role in calcium homeostasis through its action in the kidney, intestine, bone, and parathyroid glands. It is also involved in many noncalcemic activities including antiproliferative, prodifferentiative, and immunomodulatory effects (37). Thus, sufficient vitamin D supply has been linked to decreased risks of many inflammation-related diseases such as Crohn disease, asthma, type 2 diabetes, cardiovascular diseases, and even cancers (6). In this study, we found (i) 1,25(OH)2D (including both 1,25(OH)2D2 and 1,25(OH)2D3) suppresses secretion of proinflammatory cytokines in macrophages through down-regulation of inducible COX-2; (ii) 1,25(OH)2D inhibits Akt/NF-κB signaling and transcription of its targeting gene-COX-2 in macrophages through activation of VDR; (iii) 1,25(OH)2D treatment increases VDR binding to putative VDRE in the promoter region of an Akt endogenous modulator-THEM4 and enhances its expression; and (iv) vitamin D also inhibits carrageenan-induced inflammatory responses in air pouch and paw edema models in a COX-2-dependent fashion. Therefore, vitamin D inhibits Akt/NF-κB/COX-2 axis-mediated proinflammatory cytokines by targeting THEM4, which contributes to the effect of vitamin D in inflammatory-related diseases.
Macrophages play a dominant role in the regulation of inflammatory and immune responses and activated macrophages can produce and secrete proinflammatory cytokines and lipid mediators such as PGs. Macrophages participate in local conversion of 25(OH)2D into an active 1,25(OH)2D (38). We observed that the active form of vitamin D-1,25(OH)2D repressed LPS-induced COX-2 expression and decreased COX-2-derived PG production and the expression of IL-6 and TNFα in macrophages; thus, vitamin D administration decreased inflammatory cell infiltration in the air pouch model of inflammation and alleviated carrageenan-induced hind paw inflammation in mice. Similar reductions of COX-2 expression by 1,25(OH)2D were seen in cancer cells such as prostate and breast cancers (15, 39) and in human myometrial cells (40). 1,25(OH)2D could also retard PG-mediated signaling in cancer cells by increasing the expression of 15-hydroxyprostaglandin dehydrogenase (39), which degrades PG and suppresses PG receptor expression, which in turn inhibits positive feedback signaling to increase COX-2 expression (41). We failed to detect alternations of PG receptors in macrophages by 1,25(OH)2D treatment. Surprisingly, 1,25(OH)2D3 and its derivative 1,25(OH)2-16-ene-23-yne-D3 (IC50 at 5.8 nm) selectively inhibit COX-2 activity with no effect on COX-1 activity (42). However, 1,25(OH)2D3 also modulates COX-1 expression in osteoclast-supporting stromal cells (43); this effect may be involved in vitamin D-induced proliferation and differentiation of growth plate chondrocytes (44–46).
COX-2 expression is induced by various mitogenic and proinflammatory stimuli and is regulated by multiple pathways such as MAPKs and NF-κB in many different cell types (47). In mammalian cells, three subfamilies of MAPKs have been well characterized: ERK1/2, JNKs, and p38 MAPKs (48). JNKs and ERK1/2 govern COX-2 mRNA transcription by binding the AP-1 site of its promoter and modulating co-activator CBP/p300, respectively (49). ERK1/2 maybe also mediates activation of NF-κB (50), whereas p38 MAPK regulates COX-2 mRNA stabilization (28). 1,25(OH)2D treatment has no notable effect on phosphorylation of p38, JNK, and ERK1/2 in macrophages, indicating that 1,25(OH)2D down-regulates COX-2 through a MAPK-independent pathway. In this study, 1,25(OH)2D suppressed LPS-triggered activation of Akt, leading to a reduction in IκB phosphorylation mediated by IKK (51) and COX-2 expression by restraining nuclear translocation of the p65 subunit. siRNA knockdown of VDR restored LPS-induced activation of the Akt/NF-κB axis and its mediated COX-2 translation in macrophages, further confirming that 1,25(OH)2D attenuates COX-2 expression and the secretion of proinflammatory cytokines by suppressing Akt/NF-κB signaling. Consistently, 1,25(OH)2D also modulates basal and cytokine-induced NF-κB activity in cells such as fibroblasts (52) and human lymphocytes (53). Likewise, a significant increase of NF-κB activity was reported in mice lacking the VDR (54), and addition of a specific VDR blocker to prostate cancer up-regulates NF-κB signaling (55). Recently, 1,25(OH)2D has also been shown to interfere with p38 MAPK signaling by targeting MAPK phosphatase-1 in peripheral blood mononuclear cells (56).
Akt, one of essential components in the PI3K signal pathway, plays a critical role in the regulation of many (physio)pathological processes including inflammatory responses (57). Generally, Akt is activated by phosphorylation at Thr308 and Ser473 by pleckstrin homology domain containing serine/threonine kinases-PDK1 and PDK2, respectively, and inactivated by an endogenous AKT inhibitor the pseudokinase TRB3 (31) and CTMP (also named THEM4) (32), which bind to AKT and prevent its phosphorylation and downstream signaling. We found that 1,25(OH)2D induces THEM4 expression in both basal and LPS-stimulated macrophages without influencing other regulatory genes, and this vitamin D-dependent induction of THEM4 could be abolished by silencing VDR. Binding of VDR to the THEM4 promoter induces recruitment of relevant co-activators and mediator proteins (58), leading to THEM4 transcriptional activation. ChIP and luciferase promoter assays identified a functional VDRE in mouse and human THEM4 promoters. Thus, vitamin D modulates COX-2 expression in macrophages by targeting THEM4. The amino-terminal domain of THEM4 could bind to Akt (59, 60), which restrains Akt activity and COX-2 expression as observed in macrophages (Fig. 17), and this binding does not influence its acyl-CoA thioesterase activity (60). Likewise, THEM4 prevents Akt phosphorylation and blocks downstream signaling through physical bindings (30), suggesting that the effect of THEM4 on Akt may not be dependent on its acyl-CoA thioesterase activity but requires further investigation.
FIGURE 17.
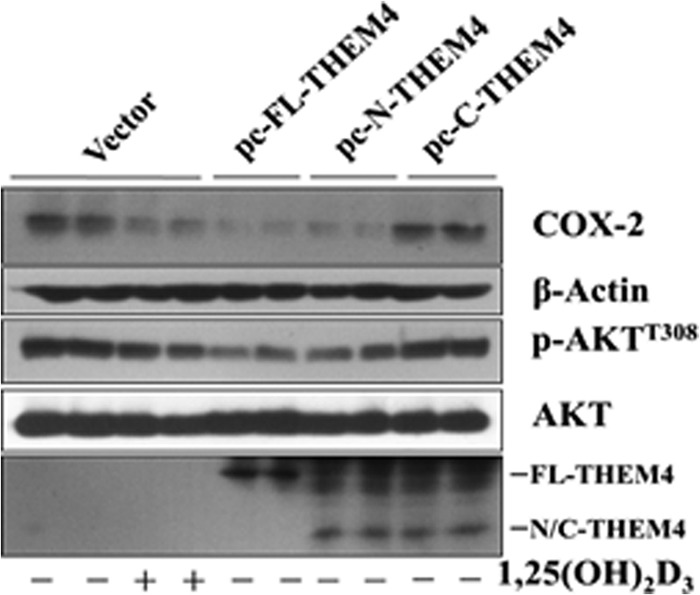
Effect of forced expression of amino-terminal domain of THEM4 on COX-2 expression and Akt phosphorylation in macrophages. The full length (FL-THEM4), the amino-terminal domain (1–109 amino acids, N-THEM4), and the C-terminal domain (110–230 amino acids, C-THEM4) were subcloned into pcDNA3.1, and a HA tag was added at the amino terminus. RAW264.7 cells were transiently transfected with each pcDNA3/THEM4 variant or pcDNA3 empty vector, and cell lysates were then subjected to Western blot analysis for COX-2, Akt phosphorylation, and HA tag.
Epidemiologic studies demonstrate that vitamin D deficiency increases the risk of a variety of inflammation related diseases including cancers, and higher consumption of vitamin D is associated with better prognosis. We showed that vitamin D suppresses the inflammatory reaction in mice by suppressing COX-2-dependent prostanoids and proinflammatory cytokine production and found that THEM4 is up-regulated by vitamin D, a novel mechanism by which vitamin D suppresses the Akt/NF-κB pathway-mediated inflammatory response in macrophages. These results suggest the therapeutic potential of vitamin D as adjuvant therapy for inflammatory diseases and even cancer.
This work was supported by Grants 2012CB945100, 2011CB503906, 2011ZX09307-302-01, and 2012BAK01B00 from the Ministry of Science and Technology of China and National Natural Science Foundation of China Grants 81030004 and 31200860, Natural Science Foundation of China-Canadian Institutes of Health Research Joint Grants NSFC81161120538 and CIHR-CCI117951, Grant KSCX2-EW-R-09 from the Knowledge Innovation Program of the Chinese Academy of Sciences, funds from the Clinic Research Center at Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences Grant CRC2010007, the Postdoctoral Fellowship Program of Shanghai Institutes for Biological Sciences, and Chinese Academy of Sciences Grants 2012KIP514 and 2013KIP312.
- 1,25(OH)2D
- 1,25-dihydroxy-vitamin D
- VDR
- vitamin D receptor
- VDRE
- vitamin D-responsive element
- COX
- cyclooxygenase
- PG
- prostaglandin
- THEM4
- thioesterase superfamily member 4
- NSAID
- nonsteroidal anti-inflammatory drug
- KD
- knockdown
- FFA
- free fatty acid.
REFERENCES
- 1. van Schoor N. M., Lips P. (2011) Worldwide vitamin D status. Best Pract. Res. Clin. Endocrinol. Metab. 25, 671–680 [DOI] [PubMed] [Google Scholar]
- 2. Holick M. F., Chen T. C. (2008) Vitamin D deficiency: a worldwide problem with health consequences. Am. J. Clin. Nutr. 87, 1080S–1086S [DOI] [PubMed] [Google Scholar]
- 3. Mangelsdorf D. J., Evans R. M. (1995) The RXR heterodimers and orphan receptors. Cell 83, 841–850 [DOI] [PubMed] [Google Scholar]
- 4. Malloy P. J., Pike J. W., Feldman D. (1999) The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr. Rev. 20, 156–188 [DOI] [PubMed] [Google Scholar]
- 5. Jurutka P. W., Whitfield G. K., Hsieh J. C., Thompson P. D., Haussler C. A., Haussler M. R. (2001) Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Rev. Endocr. Metab. Disord. 2, 203–216 [DOI] [PubMed] [Google Scholar]
- 6. Glade M. J. (2013) Vitamin D: health panacea or false prophet? Nutrition 29, 37–41 [DOI] [PubMed] [Google Scholar]
- 7. Smyth E. M., Grosser T., Wang M., Yu Y., FitzGerald G. A. (2009) Prostanoids in health and disease. J. Lipid Res. 50, (suppl.) S423–S428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ricciotti E., FitzGerald G. A. (2011) Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith W. L., DeWitt D. L., Garavito R. M. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 10. Ermert L., Ermert M., Goppelt-Struebe M., Walmrath D., Grimminger F., Steudel W., Ghofrani H. A., Homberger C., Duncker H., Seeger W. (1998) Cyclooxygenase isoenzyme localization and mRNA expression in rat lungs. Am. J. Respir. Cell Mol. Biol. 18, 479–488 [DOI] [PubMed] [Google Scholar]
- 11. Hinz B., Brune K. (2007) Antipyretic analgesics: nonsteroidal antiinflammatory drugs, selective COX-2 inhibitors, paracetamol and pyrazolinones. Handb. Exp. Pharmacol. 177, 65–93 [DOI] [PubMed] [Google Scholar]
- 12. Boyan B. D., Sylvia V. L., Dean D. D., Del Toro F., Schwartz Z. (2002) Differential regulation of growth plate chondrocytes by 1,25-(Oh)2d3 and 24r,25-(Oh)2d3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit. Rev. Oral Biol. Med. 13, 143–154 [DOI] [PubMed] [Google Scholar]
- 13. Liu X., Nelson A., Wang X., Farid M., Gunji Y., Ikari J., Iwasawa S., Basma H., Feghali-Bostwick C., Rennard S. I. (2014) Vitamin D modulates PGE2 synthesis and degradation in human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 50, 40–50 [DOI] [PubMed] [Google Scholar]
- 14. Krishnan A. V., Moreno J., Nonn L., Swami S., Peehl D. M., Feldman D. (2007) Calcitriol as a chemopreventive and therapeutic agent in prostate cancer: role of anti-inflammatory activity. J. Bone Miner. Res. 22, V74–V80 [DOI] [PubMed] [Google Scholar]
- 15. Krishnan A. V., Swami S., Peng L., Wang J., Moreno J., Feldman D. (2010) Tissue-selective regulation of aromatase expression by calcitriol: implications for breast cancer therapy. Endocrinology 151, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guillot X., Semerano L., Saidenberg-Kermanac'h N., Falgarone G., Boissier M. C. (2010) Vitamin D and inflammation. Joint Bone Spine 77, 552–557 [DOI] [PubMed] [Google Scholar]
- 17. Adams J. S., Hewison M. (2008) Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat. Clin. Pract. Endocrinol. Metab. 4, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu S., Sun J. (2011) Vitamin D, vitamin D receptor, and macroautophagy in inflammation and infection. Discov. Med. 11, 325–335 [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Zou F., Tang J., Zhang Q., Gong Y., Wang Q., Shen Y., Xiong L., Breyer R. M., Lazarus M., Funk C. D., Yu Y. (2013) Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ. Res. 113, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song W. L., Lawson J. A., Wang M., Zou H., FitzGerald G. A. (2007) Noninvasive assessment of the role of cyclooxygenases in cardiovascular health: a detailed HPLC/MS/MS method. Methods Enzymol. 433, 51–72 [DOI] [PubMed] [Google Scholar]
- 21. Levy S., Hannenhalli S. (2002) Identification of transcription factor binding sites in the human genome sequence. Mamm. Genome 13, 510–514 [DOI] [PubMed] [Google Scholar]
- 22. Winyard P. G., Willoughby D. A. (2003) Carageenan-induced paw edema in the rat and mouse. In Inflammation Protocols, Humana Press, Totowa, NJ [Google Scholar]
- 23. Heller A., Koch T., Schmeck J., van Ackern K. (1998) Lipid mediators in inflammatory disorders. Drugs 55, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nilsberth C., Elander L., Hamzic N., Norell M., Lönn J., Engström L., Blomqvist A. (2009) The role of interleukin-6 in lipopolysaccharide-induced fever by mechanisms independent of prostaglandin E2. Endocrinology 150, 1850–1860 [DOI] [PubMed] [Google Scholar]
- 25. Cerella C., Sobolewski C., Dicato M., Diederich M. (2010) Targeting COX-2 expression by natural compounds: a promising alternative strategy to synthetic COX-2 inhibitors for cancer chemoprevention and therapy. Biochem. Pharmacol. 80, 1801–1815 [DOI] [PubMed] [Google Scholar]
- 26. Wada T., Penninger J. M. (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23, 2838–2849 [DOI] [PubMed] [Google Scholar]
- 27. Pang L., Sawada T., Decker S. J., Saltiel A. R. (1995) Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J. Biol. Chem. 270, 13585–13588 [DOI] [PubMed] [Google Scholar]
- 28. Dean J. L., Brook M., Clark A. R., Saklatvala J. (1999) p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J. Biol. Chem. 274, 264–269 [DOI] [PubMed] [Google Scholar]
- 29. Waetzig V., Czeloth K., Hidding U., Mielke K., Kanzow M., Brecht S., Goetz M., Lucius R., Herdegen T., Hanisch U. K. (2005) c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 50, 235–246 [DOI] [PubMed] [Google Scholar]
- 30. Vivanco I., Sawyers C. L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2, 489–501 [DOI] [PubMed] [Google Scholar]
- 31. Du K., Herzig S., Kulkarni R. N., Montminy M. (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 32. Maira S.-M., Galetic I., Brazil D. P., Kaech S., Ingley E., Thelen M., Hemmings B. A. (2001) Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science 294, 374–380 [DOI] [PubMed] [Google Scholar]
- 33. Hunt M. C., Alexson S. E. (2002) The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog. Lipid Res. 41, 99–130 [DOI] [PubMed] [Google Scholar]
- 34. Holick M. F. (2005) Vitamin D for health and in chronic kidney disease. Semin. Dial. 18, 266–275 [DOI] [PubMed] [Google Scholar]
- 35. Holick M. F. (2007) Vitamin D deficiency. N. Engl. J. Med. 357, 266–281 [DOI] [PubMed] [Google Scholar]
- 36. Tsugawa N., Nakagawa K., Kawamoto Y., Tachibana Y., Hayashi T., Ozono K., Okano T. (1999) Biological activity profiles of 1alpha, 25-dihydroxyvitamin D2, D3, D4, D7, and 24-epi-1α, 25-dihydroxyvitamin D2. Biol. Pharm. Bull. 22, 371–377 [DOI] [PubMed] [Google Scholar]
- 37. Muszkat P., Camargo M. B., Griz L. H., Lazaretti-Castro M. (2010) Evidence-based non-skeletal actions of vitamin D. Arq. Bras. Endocrinol. Metabol. 54, 110–117 [DOI] [PubMed] [Google Scholar]
- 38. Hewison M. (2011) Antibacterial effects of vitamin D. Nat. Rev. Endocrinol. 7, 337–345 [DOI] [PubMed] [Google Scholar]
- 39. Moreno J., Krishnan A. V., Swami S., Nonn L., Peehl D. M., Feldman D. (2005) Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 65, 7917–7925 [DOI] [PubMed] [Google Scholar]
- 40. Thota C., Farmer T., Garfield R. E., Menon R., Al-Hendy A. (2013) Vitamin D elicits anti-inflammatory response, inhibits contractile-associated proteins, and modulates Toll-like receptors in human myometrial cells. Reprod. Sci. 20, 463–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tjandrawinata R. R., Hughes-Fulford M. (1997) Up-regulation of cyclooxygenase-2 by product-prostaglandin E2. Adv. Exp. Med. Biol. 407, 163–170 [DOI] [PubMed] [Google Scholar]
- 42. Aparna R., Subhashini J., Roy K. R., Reddy G. S., Robinson M., Uskokovic M. R., Venkateswara Reddy G., Reddanna P. (2008) Selective inhibition of cyclooxygenase-2 (COX-2) by 1α,25-dihydroxy-16-ene-23-yne-vitamin D3, a less calcemic vitamin D analog. J. Cell. Biochem. 104, 1832–1842 [DOI] [PubMed] [Google Scholar]
- 43. Adams A. E., Abu-Amer Y., Chappel J., Stueckle S., Ross F. P., Teitelbaum S. L., Suva L. J. (1999) 1,25 dihydroxyvitamin D3 and dexamethasone induce the cyclooxygenase 1 gene in osteoclast-supporting stromal cells. J. Cell. Biochem. 74, 587–595 [PubMed] [Google Scholar]
- 44. Sylvia V. L., Del Toro F., Dean D. D., Hardin R. R., Schwartz Z., Boyan B. D. (2001) Effects of 1α,25-(OH)2D3 on rat growth zone chondrocytes are mediated via cyclooxygenase-1 and phospholipase A2. J. Cell. Biochem. Suppl. 36, 32–45 [DOI] [PubMed] [Google Scholar]
- 45. Schwartz Z., Sylvia V. L., Del Toro F., Hardin R. R., Dean D. D., Boyan B. D. (2000) 24R,25-(OH)2D3 mediates its membrane receptor-dependent effects on protein kinase C and alkaline phosphatase via phospholipase A2 and cyclooxygenase-1 but not cyclooxygenase-2 in growth plate chondrocytes. J. Cell. Physiol. 182, 390–401 [DOI] [PubMed] [Google Scholar]
- 46. Boyan B. D., Sylvia V. L., Dean D. D., Pedrozo H., Del Toro F., Nemere I., Posner G. H., Schwartz Z. (1999) 1,25-(OH)2D3 modulates growth plate chondrocytes via membrane receptor-mediated protein kinase C by a mechanism that involves changes in phospholipid metabolism and the action of arachidonic acid and PGE2. Steroids 64, 129–136 [DOI] [PubMed] [Google Scholar]
- 47. Singer C. A., Baker K. J., McCaffrey A., AuCoin D. P., Dechert M. A., Gerthoffer W. T. (2003) p38 MAPK and NF-κB mediate COX-2 expression in human airway myocytes. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L1087–L1098 [DOI] [PubMed] [Google Scholar]
- 48. Dong C., Davis R. J., Flavell R. A. (2002) MAP kinases in the immune response. Annu. Rev. Immunol. 20, 55–72 [DOI] [PubMed] [Google Scholar]
- 49. Chun K. S., Surh Y. J. (2004) Signal transduction pathways regulating cyclooxygenase-2 expression: potential molecular targets for chemoprevention. Biochem. Pharmacol. 68, 1089–1100 [DOI] [PubMed] [Google Scholar]
- 50. Zhang L., Ma Y., Zhang J., Cheng J., Du J. (2005) A new cellular signaling mechanism for angiotensin II activation of NF-κB: An IκB-independent, RSK-mediated phosphorylation of p65. Arterioscler. Thromb. Vasc. Biol. 25, 1148–1153 [DOI] [PubMed] [Google Scholar]
- 51. O'Connell M. A., Bennett B. L., Mercurio F., Manning A. M., Mackman N. (1998) Role of IKK1 and IKK2 in lipopolysaccharide signaling in human monocytic cells. J. Biol. Chem. 273, 30410–30414 [DOI] [PubMed] [Google Scholar]
- 52. Harant H., Wolff B., Lindley I. J. (1998) 1α,25-dihydroxyvitamin D3 decreases DNA binding of nuclear factor-κB in human fibroblasts. FEBS Lett. 436, 329–334 [DOI] [PubMed] [Google Scholar]
- 53. Yu X. P., Bellido T., Manolagas S. C. (1995) Down-regulation of NF-κB protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. U.S.A. 92, 10990–10994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun J., Kong J., Duan Y., Szeto F. L., Liao A., Madara J. L., Li Y. C. (2006) Increased NF-κB activity in fibroblasts lacking the vitamin D receptor. Am. J. Physiol. Endocrinol. Metab. 291, E315–E322 [DOI] [PubMed] [Google Scholar]
- 55. Schwab M., Reynders V., Loitsch S., Steinhilber D., Stein J., Schröder O. (2007) Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NF κB signalling. Mol. Immunol. 44, 3625–3632 [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y., Leung D. Y., Richers B. N., Liu Y., Remigio L. K., Riches D. W., Goleva E. (2012) Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 188, 2127–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cantley L. C. (2002) The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 58. Kim S., Shevde N. K., Pike J. W. (2005) 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J. Bone Miner. Res. 20, 305–317 [DOI] [PubMed] [Google Scholar]
- 59. Cao J., Xu H., Zhao H., Gong W., Dunaway-Mariano D. (2009) The mechanisms of human hotdog-fold thioesterase 2 (hTHEM2) substrate recognition and catalysis illuminated by a structure and function based analysis. Biochemistry 48, 1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao H., Martin B. M., Bisoffi M., Dunaway-Mariano D. (2009) The Akt C-terminal modulator protein is an acyl-CoA thioesterase of the Hotdog-Fold family. Biochemistry 48, 5507–5509 [DOI] [PMC free article] [PubMed] [Google Scholar]