

Background: Motor function and emotions are regulated by dopamine and dopaminergic receptors.
Results: Pharmacological inhibition of PPM/PP2C phosphatases prevents dopamine D2 receptor-mediated dephosphorylation of the huntingtin protein.
Conclusion: A previously unappreciated signaling pathway exists downstream of dopamine D2 receptor activation.
Significance: This pathway may have therapeutic potential in dopamine-related disorders.
Keywords: Akt, Arrestin, Dopamine Receptors, G Protein-coupled Receptors (GPCR), G Proteins, Phosphatase, PP2C, Dopamine D2 Receptor, Huntingtin
Abstract
Striatal dopamine D2 receptor (D2R) relies upon G protein- and β-arrestin-dependent signaling pathways to convey its action on motor control and behavior. Considering that D2R activation inhibits Akt in the striatum and that huntingtin physiological functions are affected by Akt phosphorylation, we sought to investigate whether D2R-mediated signaling could regulate huntingtin phosphorylation. We demonstrate that D2R activation decreases huntingtin phosphorylation on its Akt site. This dephosphorylation event depends upon the Gαi-dependent engagement of specific members of the protein phosphatase metallo-dependent (PPM/PP2C) family and is independent of β-arrestin 2. These observations identify the PPM/PP2C family as a mediator of G protein-coupled receptor signaling and thereby suggest a novel mechanism of dopaminergic signaling.
Introduction
Several observations indicate that dopamine might contribute to the deleterious effects of the expanded/mutated huntingtin protein (mHTT)3 on striatal function. mHTT protein aggregates are predominantly observed in brain regions innervated by dopamine (1), and elevated extracellular dopamine in the striatum enhances formation of mHTT aggregates (2). Increased dopamine levels are sufficient to cause sporadic Huntington disease (HD)-like symptoms in dopamine transporter knock-out mice accompanied by a loss of striatal medium spiny neurons (3). On the other hand, dopamine D2 receptor (D2R) antagonism reduces mHTT toxicity and neuronal cell death both in cultured cells and in HD mouse models (4, 5). Conversely, tetrabenazine, a vesicular monoamine transporter 2 (VMAT2) inhibitor that essentially acts as a monoaminergic antagonist/depleting agent, was approved in 2008 by the Food and Drug Administration (FDA) for use in HD patients due to its ability to reduce hyperkinetic symptoms (6–8). Therefore, dopamine receptor signaling might play an important role in HD progression.
Over the course of the past 10 years, it has become evident that many phosphorylation events can modulate the proteolysis and toxicity of the mHTT (9–13). Although the various kinases responsible for these events have been intensely studied, their phosphatase counterparts and the signaling pathways leading to HTT dephosphorylation have only been scarcely investigated. The human genome encodes 211 protein phosphatases divided into six families according to the sequence homology of their catalytic domains (see Ref. 14 for review). Phospho-serine and phospho-threonine can be dephosphorylated by three distinct families of phosphatases, i.e. the serine/threonine-specific protein phosphatases (PPP) family (including PP1, PP2A, and PPP3CA/calcineurin), the protein phosphatase metallo-dependent (PPM/PP2C) family, and the dual specificity phosphatases (DUSPs) members of the protein-tyrosine phosphatase (PTP) family.
In neurons, the phosphorylation status of HTT Ser-421 4 affects the anterograde vesicular transport function of the wild type HTT (15) and the toxicity of the mutated HTT (16–18). Because evidence from cells and animal models suggests that HTT Ser-421 phosphorylation plays a neuroprotective role in suppressing mHTT-induced toxicity in HD (16–18), defining the mechanisms regulating HTT Ser-421 phosphorylation remains of significant importance. In addition, sustained D2R activation results in the deactivation of Akt by protein phosphatase 2A (PP2A) in mouse striatum (19), which could mediate a reduction in HTT phosphorylation at Ser-421 by Akt. Consequently, this study was undertaken to assess whether, and by what mechanism, dopamine D2 receptor can modulate the Akt-dependent phosphorylation of HTT. Here we provide evidence that D2R activation dynamically promotes HTT dephosphorylation on Ser-421, dephosphorylation that evidently does not require the β-arrestin 2-PP2A tandem. On the contrary, our results indicate that this dephosphorylation event is Gαi-mediated and point toward the PPM/PP2C phosphatases PPM1A and/or PPM1B, acting downstream of D2R activation to promote HTT dephosphorylation of Ser-421, therefore unveiling a new signaling paradigm for this receptor.
EXPERIMENTAL PROCEDURES
Materials
R(−)Apomorphine hydrochloride hemihydrate, 2-bromo-a-ergocryptine methanesulfonate salt, R(−)-propylnorapomorphine hydrochloride, haloperidol, S(−)-raclopride tartrate salt, 5-hydroxytryptamine hydrochloride, d,l-norepinephrine hydrochloride, (R)-(+)-WIN 55,212-2 mesylate salt, R(+)-SCH23390 hydrochloride, pertussis toxin, and cadmium chloride were from Sigma-Aldrich. Okadaic acid and cyclosporin A were obtained from EMB Bioscience (La Jolla, CA). The phosphatase inhibitor cocktails used in this study contain β-glycerophosphate, sodium pyrophosphate, sodium fluoride, and sodium orthovanadate; they were purchased from either Pierce (78426) or Alfa Aesar (J61022). The PP2C inhibitor NSC127153 was obtained from the National Institutes of Health NCI Developmental Therapeutics Program. NSC127153 was dissolved in DMSO (50 mm stock solution) and then diluted in dimethylacetamide/10% d-cyclodextrin/30 mm phosphoric acid before use at indicated concentrations. Cypermethrin was from Santa Cruz Biotechnology (Santa Cruz, CA). Cell culture reagents were from Life Technologies.
Experimental Animals
β-Arrestin 2 KO mice were described previously (20). C57Bl6 D2-KO (B6.129S2-Drd2tm1Low/J) mice were obtained from The Jackson Laboratory (Bar Arbor, ME). For all experiments involving mutant animals, respective WT littermates were used as controls, and all mice were 3–4 months of age. Before experiments, mice were housed 4–5/cage in a humidity-controlled room at 23 °C on a 12-h light-dark cycle with food and water ad libitum. Animal care was approved by the Duke University Institutional Animal Care and Use Committee according to National Institutes of Health guidelines.
Plasmids
HA-tagged human dopamine 1, 2, 3, and 5 receptors were previously described (41) by and obtained from Dr. K. M. Kim (Department of Pharmacology, Chonnam National University, Republic of Korea). Human DRD4, PPM1L, PPM1H, PPM1D, and PHLPP1 cDNAs were purchased from Open Biosystems (Huntsville, AL). Human PPM1K cDNA was obtained from OriGene (Rockville, MD). The human PPM1A, PPM1B, PPM2C, PPM1G, and PPM1F and the 1980 nucleotides encoding the 660 amino-terminal residues of the wild type mouse huntingtin with a stretch of seven glutamines were cloned by total RNA extraction from HEK293T cells and mouse striatum respectively, using ProtoScript first strand cDNA synthesis kit from New England Biolabs. All constructs utilized in this study were made with the proofreading polymerase Phusion from New England Biolabs and subjected to sequencing before use. HTTN660 fragment contains critical sites subjected to post-translational modification, such as the IκB kinase (Ser-13, Ser-16), the Akt (Ser-421) and Cdk5 (Ser-434) phosphorylation sites, and the caspase 2, 3, and 6 cleavage sites (residues 513, 530, 552 and 586) along with cysteine 214 (palmitoylation) to cite a few. Therefore, HTTN660 was used as sensor for assessing D2R-mediated post-translational modification of HTT on Ser-421. Full-length human HTT with 23 or 73 glutamines was a generous gift from Cagla Eroglu, Ph.D. (Cell Biology Department, Duke University, Durham, NC). HTTN660 with 138 glutamines was made by sequential PCR reactions and is composed of the human Huntingtin first exon containing 138 glutamines followed by codons from mouse origin until nucleotide 1980.
Antibodies
The monoclonal anti-huntingtin antibody 2166 was from Chemicon (Temecula, CA). The anti-phospho-HTT Ser-421 was a generous gift of Professor M. R. Hayden (Centre for Molecular Medicine and Therapeutics, University of British Columbia, Vancouver, British Columbia, Canada). See Ref. 17 regarding the validation of the anti-phospho-HTT Ser-421 specificity. The anti-PPM1A polyclonal antibody 76574 was from Abcam (Cambridge, MA). The anti-PPM1B polyclonal antibody AF4396 was from R&D Systems (Minneapolis, MN).
Cell Lysis and Immunoprecipitation Protocols
Extracts Prepared from HEK293T Cells
48 h after transfection, cells were lysed on ice in cold radioimmune precipitation buffer containing protease and phosphatase inhibitor cocktails for 1 h at 4 °C before being subjected to 20 min of centrifugation (14,000 rpm) at 4 °C. The protein concentration of each supernatant was measured using Pierce BCA protein assay. Laemmli buffer containing β-mercaptoethanol was added, and the samples were incubated at 55 degrees for 15–20 min before being subjected to electrophoresis.
Immunoprecipitation of the Huntingtin from Striatal Tissue
A complete mouse striatum was rapidly dissected on an ice-cold surface, and the two parts were mechanically homogenized at 4 °C in 300 μl of lysis buffer (10 mm Tris/HCl, pH 7.4, 2 mm EDTA, 1% Nonidet P-40, 5% glycerol) containing protease and phosphatase inhibitor cocktails. After 30 min on ice, homogenates were cleared by centrifugation (14 000 rpm) for 20 min at 4 °C. After preclearing, immunoprecipitations were conducted overnight in a volume of 600 μl at 4 °C in corresponding lysis buffer using 0.5 mg of supernatant proteins and 2.5 μl of anti-HTT antibody (MAB2166, Chemicon) followed by 2–3 h of incubation with protein A- and G-Sepharose beads (GE Healthcare Biosciences). The beads were then washed four times for 15 min in radioimmune precipitation buffer containing a mixture of protease and phosphatase inhibitors. After 15–20 min of incubation at 55 °C in Laemmli loading buffer (Bio-Rad) containing β-mercaptoethanol, immunoprecipitates were then resolved by SDS-PAGE in 4–12% acrylamide gradient gels (Invitrogen).
Cell Culture and Transfection
HEK293T cells were grown in Eagle's minimum essential medium with Earle's salt supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and a 1:1000 dilution of gentamicin reagent solution (InvitrogenTM). Cells were transiently transfected using a calcium phosphate co-precipitation method. The DNA amounts used for transfection are indicated in the figure legends and are for 10-cm cell dish. STHdhQ7/7 cells obtained from the Coriell Institute for Medical Research (Camden, NJ) were cultivated at 33 °C in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter of glucose, l-glutamine, and sodium pyruvate supplemented with 10% (v/v) fetal bovine serum (FBS) and a 1:100 dilution of antibiotic/antimycotic reagent solution (A5955, Sigma). STHdhQ7/7 cells were transfected according to the manufacturer's recommendations by either MagnetofectionTM (OZ Biosciences, Marseille, France) or GeneCellin (BioCellChallenge, Toulon, France).
Protein-Protein Interaction on Recombinant Proteins
For GST affinity purifications, mouse striatal extracts were incubated with bacterial protein extracts containing recombinant GST-tagged proteins prior to affinity purification as described previously (19).
In Vitro Phosphatase Assay
HTTN660-GFP was purified from HEK293 cells lysates by immunoprecipitation. Recombinant GST-PPM1A and GST-PPM1B were purified from bacteria extracts by GST pulldown. In vitro phosphatase assays were carried out by incubating GST tagged-phosphatases with HTTN660 bound on agarose beads in phosphatase buffer (50 mm Tris/HCl, pH 7.4, 0.1 mg/ml BSA, 0.1% β-mercaptoethanol, 30 μm EDTA, plus or minus 40 mm MgCl2) at 30 °C for 45 min. Reactions were stopped by the addition of Laemmli buffer and boiling at 90 °C for 5 min.
Statistical Analysis
Data are presented as means ± S.E. Data from Western blot were analyzed by t test with a p < 0.05 considered significant: *, p < 0.05; **, p < 0.02; ***, p < 0.001. Regarding the quantification of the phospho-Ser-421 signal in cells expressing the HTTN660-GFP construct, the integrated density of the two main bands migrating at ∼120 and 125 kDa was measured for the total versus phospho-Ser-421 signal using ImageJ software (rsbweb.nih.gov/ij/index.html). Both bands can be detected in total cell lysates, using either an anti-GFP antibody or the anti-HTT MAB2166 antibody. Phosphorylation variations for both fragments were proportionally equivalent despite the experimental conditions. These bands likely reflect two different post-translational modifications of the HTTN660 fragment. Under our experimental conditions, only the 125-kDa fragment retains an interaction with D2R.
RESULTS
D2R Stimulation Promotes HTTN660 Dephosphorylation in HEK Cells
To assess whether the D2 receptor could modulate the Akt-dependent phosphorylation of huntingtin on Ser-421, we transiently expressed D2R along with the first 660 amino-terminal residues of mouse huntingtin (HTTN660) in HEK293T cells. Under these conditions in which HTTN660 is phosphorylated at Ser-421, we observed that apomorphine as well as the D2R-selective agonists bromocriptine and N-n-propylnorapomorphine (NPA) were effective in promoting HTTN660 dephosphorylation at Ser-421 (Fig. 1A). Interestingly, the D2R antagonists, raclopride and haloperidol, increased HTTN660 Ser-421 phosphorylation beyond basal levels (Fig. 1B), suggesting that D2Rs can exert a tonic control over the Akt-dependent phosphorylation site.
FIGURE 1.

D2R activation promotes HTTN660 dephosphorylation on Ser-421 via an okadaic acid-insensitive phosphatase. HA-D2R along with the first 660 amino-terminal residues of the HTT (HTTN660) fused to the GFP were transiently overexpressed in HEK293T (2 μg each). A, Western blots and densitometry analysis of phospho-Ser-421 HTT levels in extracts prepared from cells treated for 45 min with 20 μm apomorphine (Apo), bromocriptine (Bromo), or NPA or untreated controls. B, Western blots and densitometry analysis representing the opposite effect of D2R agonist (apomorphine, 20 μm for 45 min) versus antagonists (haloperidol (Halo) or raclopride (Raclo), 20 μm for 45 min each) on phospho-Ser-421 HTT levels. C and D, cells were incubated with 250 nm OA (C) or 10 μm OA, for 45 min (D) before apomorphine stimulation (20 μm, 45 min). IB, immunoblot. E, cells were pretreated with 10 μm cypermethrin (Cyper) or 2 μm cyclosporin A (CsA), for 30 min before 20 μm apomorphine was applied to the culture medium for 45 min. In all experiments, solvents used to dissolve the different drugs were applied alone as controls. The results presented here are derived from 3–4 sets of independent experiments.
D2R-induced HTTN660 Dephosphorylation Is Mediated by an Okadaic Acid-insensitive Phosphatase
We next investigated which class of phosphatase might be responsible for D2R-mediated HTT dephosphorylation in HEK293T cells. The PP1/PP2A inhibitor okadaic acid (250 nm) not only failed to prevent Ser-421 dephosphorylation downstream of D2R activation but also significantly increased basal phospho-Ser-421 levels (Fig. 1C). Thus, PP1 and PP2A, despite being obvious candidates based on a previous study (21), may not be involved in this event. Even at 10 μm, okadaic acid (OA) was still ineffective at preventing HTT dephosphorylation following apomorphine stimulation (Fig. 1D); at this concentration, OA inhibits PP1 and PP2A but also PP4, PP5, PP6, and PP2B (calcineurin-B, PP3) (22–25). To ascertain that PP2B was not implicated (OA has an IC50 of 4 μm for PP2B (22)), we used two PP2B-specific inhibitors: cypermethrin and cyclosporin A. Both failed to prevent HTT dephosphorylation induced by apomorphine (Fig. 1E), implying that Ser-421 was dephosphorylated by an as yet undefined okadaic acid-insensitive phosphatase activated by D2R.
PPM1A and PPM1B Dephosphorylate HTTN660 Ser-421
Two lines of evidence led us to consider PPM/PP2C family members as potentially responsible for HTT Ser-421 dephosphorylation. PP2Cs are among the few Ser/Thr phosphatases along with PP7 to be OA-insensitive (26, 27). Additionally, sequence similarity between toxofilin, a target of PP2C (28), and the HTT residues surrounding Ser-421 prompted us to consider the PPM/PP2C family as a rational lead, especially considering that PP7 expression is restricted to fetal brain, retina, and lymphocytes (27, 29). We selected 10 different PPM/PP2Cs based on their phylogenetic distances (indicated in black in Fig. 2A). Each phosphatase (i.e. PPM1A, PPM1B, PPM1D, PPM1F, PPM1H, PPM1K, PPM1G, PPM1L, PPM2C, and PHLPP1) was individually co-expressed in HEK293T cells along with D2R and HTTN660 and assessed for their individual ability to affect HTT Ser-421 phosphorylation. Only PPM1A and the closely related PPM1B were consistently able to reduce the extent of HTTN660 Ser-421 phosphorylation (Fig. 2, B, C, D, and F), whereas the eight other members of the PP2C family showed no effects (Fig. 2B and data not shown). In addition, recombinant PPM1A and PPM1B were equally able to dephosphorylate HTTN660 Ser-421 in vitro (Fig. 3A). Collectively, these data point to PPM1A and/or PPM1B as likely candidates for the dephosphorylation of HTT Ser-421.
FIGURE 2.
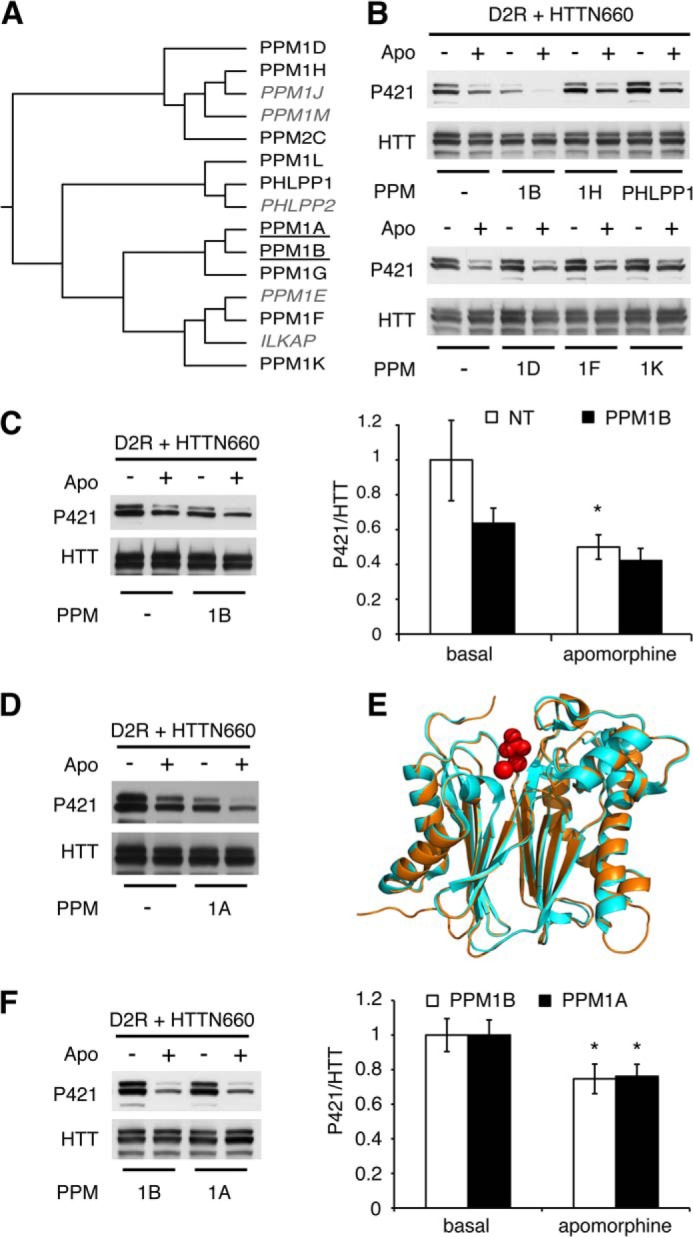
PPM1A and PPM1B modulate HTT Ser-421 phosphorylation status downstream of D2R activation. A, phylogenetic tree of the human PP2C family displaying PPM1A and PPM1B (underlined) as a discrete subfamily. The PP2C proteins tested in this study are indicated in black, and non-tested ones are in gray italics. Sequences were aligned using ClustalW Gonnet matrix. B–D and F, HEK293T cells were transiently transfected with 2 μg of HA-D2R and 2 μg of HTTN660-GFP along with 2 μg of the indicated phosphatase. 48 h after transfection, cells were stimulated by 20 μm apomorphine (Apo) for 45 min. Then cells were lysed, and the phospho-Ser-421-specific signal was detected by Western blot. The results presented in F establish that PPM1A dephosphorylates HTT Ser-421 in an extent identical to what is achieved by PPM1B. E, graphic representation of the structural alignment of the Homo sapiens PPM1A (1a6q, orange) and PPM1B (2p8e, pale blue) obtained by the MatchAlign command in PyMOL (amino acids 3–305). The resulting alignment generated a root mean square deviation of 0.307. In red are the manganese and phosphate, respectively, with which these phosphatases have been co-crystallized. All data presented here are derived from 4–7 sets of independent experiments.
FIGURE 3.

In vitro dephosphorylation of huntingtin and impact of PPM1A/B inhibition on D2R-mediated huntingtin dephosphorylation. A, recombinant PPM1A and PPM1B dephosphorylate HTT Ser-421 in vitro. 40.106 HEK293 cells were transfected with 20 μg of HTTN660-GFP. 48 h after transfection, HTTN660-GFP was immunoprecipitated and used as substrate for in vitro phosphatase assay in the presence of indicated concentrations of GST-PPM1A or GST-PPM1B. Exclusion of PPM1A/B or magnesium chloride served as negative controls. Phospho-Ser-421 and total HTT levels were then assessed by immunoblotting. Data were collected from 2 sets of independent experiments for each phosphatase. B and C, HEK293T cells transfected with 2 μg of HA-D2R and 2 μg of HTTN660-GFP in the absence (B) or in the presence of 4 μg of PPM1B (C) were subjected to 50 μm 1272153 or the corresponding solution of dimethylacetamide/DMSO (DMA/DMSO) used to dissolve this compound (see “Experimental Procedures”) for 60 min prior to apomorphine stimulation (20 μm, 45 min).
PPM1A/B Pharmacological Inhibition and PPM1B Knockdown Impairs D2R-mediated HTTN660 Dephosphorylation
Considering that PPM1A or PPM1B overexpression enhanced HTTN660 dephosphorylation and that PPM1A inhibitor 127153 (30) blocked D2R-induced HTT dephosphorylation in cells (Fig. 3B), we sought to establish which of these two phosphatases was engaged downstream of D2R stimulation. Here we provide evidence that compound 127153 also inhibits PPM1B because this inhibitor blocked D2R-mediated phospho-Ser-421 dephosphorylation even when PPM1B is overexpressed (Fig. 3C). This result could have been anticipated considering the high sequence homology shared by the two phosphatases (their catalytic domains present 78% identity), the conservation of the residues engaged in inhibitor binding (data not shown), as well as the almost perfect structure alignment of PPM1A with PPM1B (Fig. 2E). Thus, this pharmacological inhibitor was not suitable to establish the selectivity of action between these two closely related phosphatases, which have been reported to share common substrates (31, 32). Therefore, we resolved to individually knocking them down in HEK cells. Although a stable cell line with a specific PPM1A knockdown of 48 ± 2% failed to affect D2R-driven HTTN660 dephosphorylation (Fig. 4A), a PPM1B knockdown of 37 ± 7% significantly reduced the D2R-mediated HTTN660 dephosphorylation (Fig. 4B) when compared with control cells expressing an shRNA deprived of target in human cells (i.e. shRNA 936). These data suggested that even a modest decrease in PPM1B expression could significantly impair HTTN660 dephosphorylation in response to D2R activation, however, without totally ruling out a potential involvement of PPM1A in this process.
FIGURE 4.

Functional consequences of PPM1A/B knockdown on huntingtin dephosphorylation downstream of D2R activation. A and B, HEK293T stable cell lines expressing an shRNA deprived of target in human cells (sh 936) or targeted against PPM1A (sh 494) (A) or expressing an shRNA against PPM1B (sh 1182) (B) were transiently transfected with 2 μg of HA-D2R and 2 μg of HTTN660-GFP. 48 h after transfection, cells were stimulated by 20 μm apomorphine (Apo) for 45 min. Then the cells were lysed, and the phospho-Ser-421 and HTT signals, as well as PPM1A/B knockdown relative to actin, were detected by Western blot. All data presented here are derived from 3–4 sets of independent experiments, and up to 6 sets in B.
D2R-induced HTTN660 Dephosphorylation Is Gαi-dependent
We sought to determine whether D2R-driven HTT dephosphorylation was a G protein- or β-arrestin-dependent event. D1 class receptors (D1 and D5) couple to Gαs and D2 class receptors (D2, D3, and D4) couple to Gαi/o. In HEK293T cells, D1 class receptor stimulation did not result in any significant changes in HTTN660 phosphorylation (Fig. 5A). In contrast, D2 and D4 receptor activation promoted HTTN660 dephosphorylation (Fig. 5A). These results along with the observation that the β2-adrenergic receptor (Gαs-coupled) and the 5HT2cR (Gαq-coupled) failed to dephosphorylate HTTN660 (Fig. 5C), might suggest that any Gαi/o-coupled receptors could trigger HTT dephosphorylation. However, this does not appear to be the case as cannabinoid 1 receptor activation did not promote HTTN660 dephosphorylation, whereas α2a-adrenergic receptor activation actually had the opposite effect with a marked increase in Ser-421 phosphorylation (Fig. 5C). Nevertheless Gαi/o activation by D2R is necessary because pertussis toxin treatment abolished D2R-mediated HTTN660 dephosphorylation in HEK293 cells (Fig. 5D). Accordingly, we observed that a mutated D2R, deficient in β-arrestin binding while retaining full G protein activation capabilities (33), promoted HTTN660 dephosphorylation as the wild type receptor did (Fig. 5E), supporting that D2R-driven HTT dephosphorylation is β-arrestin 2-independent. This observation was further supported by the kinetic of this dephosphorylation event, which was evident 5 min after D2R stimulation (Fig. 5B) and therefore consistent with a G protein-dependent mechanism. Collectively, these data suggest that D2R-induced HTT phospho-Ser-421 dephosphorylation is Gαi-dependent, but also likely to rely upon the ability of the receptor to bring together the relevant partners.
FIGURE 5.

D2R-induced HTT dephosphorylation is Gαi-mediated. A, D1 class receptors (left panels) and D2 class receptors (right panels) were individually expressed in HEK293T cells along with HTTN660. Dopamine receptors expressing cells were stimulated by 20 μm apomorphine (Apo) for 45 min prior to cell lysis. B, HEK293T cells transiently expressing D2R and HTTN660 (2 μg each) were exposed or not exposed to apomorphine (20 μm) for the indicated time prior to cell lysis. C, β2-adrenergic receptor (β2-AR), serotoninergic 2c receptor (5HT2cR, Val-Gly-Val isoform), α2a-adrenergic receptor (α2a-AR), and the cannabinoid 1 receptor (CB1R) were individually expressed in HEK293T cells (2 μg each) along with HTTN660-GFP (2 μg). Receptor-expressing cells were stimulated by 20 μm isoproterenol (iso), 5-hydroxytryptamine (5-HT), norepinephrine (NE), or WIN55212 (WIN), respectively, for 45 min prior to cell lysis and Western blot analysis of the phospho-Ser-421 signal. D, HEK cells were incubated or not incubated in the presence of pertussis toxin (PTX, 350 ng/ml, overnight) before stimulation by apomorphine (20 μm, 45 min). E, HEK293T cells expressing the wild type D2R (WT D2R) or a D2R deficient in β-arrestin binding (mutant D2R) along with HTTN660 (2 μg each) were stimulated or not stimulated by 20 μm apomorphine for 45 min prior to cell lysis. For all results, data were collected from at least 3 sets of independent experiments.
Determination of Interacting Partners
In contrast to other phosphatases, PPM/PP2Cs are monomeric and therefore do not rely upon targeting subunits to interact with and dephosphorylate their substrates (34). Therefore, it was reasonable to assume that PPM1A and/or PPM1B should be able to interact or at least get in proximity to the HTT protein. Thus, we evaluated whether molecular interactions might exist between D2R, HTT, and PPM1A/B to decipher the interacting partners. In HEK293T cells, the endogenous full-length HTT protein was co-immunoprecipitated along with D2R but not D1R (Fig. 6A) nor D3 and D4 (Fig. 6B), illustrating the selectivity of D2R-Huntingtin interaction. This interaction was not affected by the presence of 73 glutamines within the full-length HTT (Fig. 6C) nor the addition of 138 glutamines within the HTTN660 construct (Fig. 6D). Despite a lack of agonist effect, PPM1B was co-immunoprecipitated with D2R in HEK293T cells (Fig. 6E). Additionally, D2R co-immunoprecipitated PPM1B, but not PPM1A, when these phosphatases were overexpressed in HEK293T cells (Fig. 6F), whereas GST pulldown experiments using PPM1A or PPM1B as bait showed that they were equally able to pull down the HTT protein from striatal extracts (Fig. 6G). Finally, PPM1A interacts with the full-length HTT protein in mouse striatum because they can be co-immunoprecipitated together (Fig. 6I, left panel). Despite both PPM1A and PPM1B being expressed in striatal neurons (Fig. 6H), only PPM1A was able to co-immunoprecipitate the full-length HTT protein expressed by these neurons (Fig. 6I, right panel). These observations suggest that both PPM1A and PPM1B have the ability to get in close proximity to HTT.
FIGURE 6.

Molecular interactions existing between D2R, HTT, and PPM1A/B. A and B, HEK293T cells transfected or not transfected (NT) with 1 μg of HA-D1R or 2 μg of HA-D2R (A) or 2 μg of HA-D2R, HA-D3R, or HA-D4R (B) were lysed 48 h after transfection, and HA-tagged receptors were immunoprecipitated (IP). The endogenous full-length HTT protein co-immunoprecipitated with the receptor was detected by Western blot. C, HEK293T cells were transfected or not transfected with 2 μg of the mouse 3HA-D2R along with 2 μg of the full-length human HTT containing either 23 (wild type) or 73 (mutated) glutamines (73Q or 23Q). 48 h after transfection, cells were lysed, and HA-tagged receptor was immunoprecipitated. The full-length HTT protein was specifically co-immunoprecipitated with D2R regardless of the glutamine stretch length or the presence of D2R agonist (apomorphine (Apo), 20 μm, 45 min). D, HEK293T cells were transfected with 2 μg of HA-D2R and with or without (NT) 2 μg of HTTN660-GFP containing either 7 glutamines (wild type) or 138 glutamines (mutated). 48 h after transfection, the cells were stimulated or not stimulated with 20 μm apomorphine for 45 min, and then D2R was immunoprecipitated. The amount of HTTN660 co-immunoprecipitated along with the receptor was not influenced neither by the number of glutamine repeats nor by the phosphorylation state of the HTT fragments. E and F, HEK293T cells transfected by 2 μg of HA-D2R and 2 μg of PPM1B (E), or 2 μg of 3HA-PPM1A or 3HA-PPM1B (F) were stimulated or not stimulated by apomorphine (20 μm, 45 min) before being lysed 48 h after transfection, and then D2R was immunoprecipitated. G, GST pulldown experiments using mouse striatal extracts indicate that both PPM1A and PPM1B were equally able to retain a specific interaction with the striatal HTT. Experiments were repeated twice using separate sets of striatal extracts and recombinant GST fusion proteins. H, striatal cells STHdhQ7/7 in culture were lysed, and the endogenous expression of PPM1A and PPM1B was confirmed by Western blot. I, striatal full-length HTT (left panel) was co-immunoprecipitated along with PPM1A antibody. The same experiment was conducted using striatal cells STHdhQ7/7 in culture (right panel). No interaction was retained in the absence of specific antibodies, indicating that PPM1A interacts with HTT in mouse striatum and that this interaction occurs in striatal cells. Except for G, data were collected from 3 sets of independent experiments.
D2R Modulates HTT Phosphorylation Status in Vivo
To assess whether D2R-mediated HTTN660 dephosphorylation observed in HEK cells might have physiological relevance, we first measured the Ser-421 phosphorylation levels of the endogenous HTT following apomorphine stimulation of D2R-transfected striatal cells (STHdhQ7/7) (35). These immortalized cells do not express measurable D1R or D2R as determined by binding experiments (data not shown). In line with the results obtained with HTTN660, the endogenous HTT was dephosphorylated on Ser-421 in response to D2R activation following transfection of the receptor into those cells (Fig. 7A, middle panel). In addition, cadmium chloride, a recently described potent and selective inhibitor of the PPM/PP2C family (36), fully prevented D2R-driven dephosphorylation of the endogenous full-length HTT in STHdhQ7/7 cells (Fig. 7A, right panel). These results support the involvement of PPM/PP2C members downstream of D2R activation in striatal cells. As shown in Fig. 7B, HTT Ser-421 phosphorylation measured in striatal extracts from mice treated with NPA (3 mg/kg, intraperitoneal, 15 min), a highly potent and selective D2R agonist, with respect to other dopaminergic receptors, showed an ∼30% decrease in the phospho-Ser-421 signal when compared with saline-injected wild type mice. Conversely, mice injected with raclopride (a D2R-selective antagonist: 2 mg/kg, intraperitoneal, 30 min) exhibited a 2-fold increase in phospho-Ser-421 signal when compared with saline-injected controls (Fig. 7C). Consistent with the pharmacological profiles observed above, the NPA-induced decrease in HTT phosphorylation was lost in D2R knock-out animals when compared with wild type littermates (Fig. 7D), implying that this effect is mediated through D2R in the striatum. Finally, HTT Ser-421 phosphorylation levels were not affected in β-arrestin 2 knock-out animals when compared with wild type littermates (Fig. 7E), supporting our suggestion that Akt inhibition does not account for D2R-driven HTT dephosphorylation because D2R-mediated inhibition of Akt in mouse striatum is β-arrestin 2-dependent (19).
FIGURE 7.

D2R activation promotes HTT Ser-421 dephosphorylation in vivo. A, STHdhQ7/7 cells were transfected or not transfected (NT) with HA-D2R. 48 h after transfection, the cells were serum-starved 6 h prior to agonist stimulation. In 7 sets of independent experiments, the cells were pretreated or not pretreated with cadmium chloride (5 μm, 6 h in serum-deprived DMEM) before apomorphine (Apo) stimulation (20 μm apomorphine, 35 min). Then the full-length HTT was immunoprecipitated. Western blots and densitometry analysis of phospho-Ser-421 HTT relative levels are shown. B and C, wild type mice were injected with a saline solution or NPA (3 mg/kg, intraperitoneal, 15/20 min) (B) or raclopride (Raclo, 2 mg/kg, intraperitoneal, 30 min) (C). Then the full-length striatal HTT was immunoprecipitated, and the phospho-Ser-421 was assessed. D, D2R knock-out mice were injected with a saline solution or NPA (3 mg/kg, intraperitoneal, 15/20 min), and then the full-length striatal HTT was immunoprecipitated, and the phospho-Ser-421 was assessed. E, analysis of the phospho-Ser-421 HTT relative levels in the striatum of β-arrestin 2 knock-out (BARR2 KO) mice versus wild type littermates under basal condition was conducted as stated above.
DISCUSSION
Huntingtin Ser-421 phosphorylation by Akt directs the anterograde vesicular transport function of the wild type HTT and decreases the toxicity of the mutated HTT. Because in the striatum D2R activation has been shown to promote Akt inactivation through a β-arrestin 2-PP2A complex, we investigated the propensity of D2R to modulate the phosphorylation status of HTT Ser-421. Here we show that D2R promotes HTT dephosphorylation of Ser-421 not only in cells but also in the mouse striatum under non-pathological conditions. Our data indicate that this dephosphorylation event may depend upon a previously unappreciated ability of D2R to engage PPM/PP2C family members rather than a D2R-mediated inhibition of Akt by PP2A, or direct dephosphorylation of HTT by PP2A. Despite PPM1B having been shown to interact with mGluR3 (37) and β-arrestin 2 (38), PPM/PP2Cs have not previously been functionally associated with any GPCR signaling pathways.
In an effort to decipher whether this new target of D2R signaling relied upon G protein- or β-arrestin-dependent signaling, we used a pharmacological approach (pertussis toxin treatment) to demonstrate that Gαi activation is necessary to promote HTT Ser-421 dephosphorylation in vitro. However, other Gαi-coupled GPCRs (i.e. α2a-AR and CB1R) failed to promote HTT Ser-421 dephosphorylation, illustrating the versatility of signaling pathways originating from different GPCRs seemingly coupled to the same G protein class. Conversely a mutated D2R deficient in β-arrestin binding retains full capacity to dephosphorylate HTT. These findings are consistent with the in vivo observation that β-arrestin 2 knock-out mice display normal HTT Ser-421 levels when compared with wild type. Therefore, the D2R-driven HTT Ser-421 dephosphorylation that we report here is not a consequence of the D2R-mediated Akt inactivation by PP2A, which requires β-arrestin 2 (19). Hence the HTT Ser-421 dephosphorylation we observe in response to D2R activation appears to be due exclusively to the action of a phosphatase. Pharmacological experiments conducted in HEK293 cells demonstrated that PP1, PP2A, and PP2B were not involved in HTT Ser-421 dephosphorylation downstream of D2R activation. These results, along with a sequence similarity screen of HTT Ser-421 surrounding residues, prompted us to turn our attention toward the members of the PPM/PP2C family, which are insensitive to common phosphatase inhibitors (25). Expression of various members of the PPM/PP2C family and the use of a pharmacological inhibitor (127153) along with shRNA experiments point to PPM1A and/or PPM1B as strong candidates for the D2R-mediated dephosphorylation of HTT Ser-421. In contrast to the PPP family, PPM/PP2Cs are presumably monomeric functional entities. Consequently, their activities toward specific substrates should require their scaffolding in close proximity to the protein target. Accordingly, we demonstrate that PPM1A interacts with HTT in striatal cells and in vivo, whereas PPM1B interacts with D2R in HEK293 cells (this interaction was not validated in vivo, due to a lack of satisfactory antibodies). The PPM1A/B inhibitor (compound 127153), despite its effectiveness at preventing D2R-induced HTT dephosphorylation in vivo (data not shown), is not suited to evaluate the functional implication of these phosphatases in vivo because 127153 also inhibits PP1, PP2A, and PP2B (30), which can all dephosphorylate HTT Ser-421 (18, 21, 39, 40). However, given the recent report that cadmium chloride is a selective inhibitor of the PPM/PP2C family over other phosphatase families that target phospho-serines (36), we used this agent to demonstrate that the selective inhibition of the PPM/PP2C family fully prevents D2R-mediated HTT dephosphorylation in striatal neurons. However, ultimately, despite our demonstration that both PPM1A and PPM1B are expressed in striatal neurons, a definitive proof for their biological contribution in D2R-mediated HTT dephosphorylation will require conditional knock-out of these phosphatases in D2R-expressing neurons and their functional assessment in animal models.
During the assessment of the molecular interactions existing between D2R, HTT, and PPM1A/B, we observed that D2R, but no other dopaminergic receptors, interacts with the full-length HTT protein. In view of the profound perturbations induced by the presence of a polyglutamine stretch in the mutated HTT, the D2R/HTT interaction that we report here might have drastic consequences on D2R trafficking and/or signaling in a pathological context. Therefore, although it was not directly relevant to our study, we felt compelled to evaluate whether D2R would retain the ability to interact with the mutated HTT. In Fig. 6, C and D, we provide some evidence showing that the presence of a polyglutamine stretch does not affect D2R/HTT interaction nor mHTT Ser-421 dephosphorylation following D2R stimulation. Considering the previously reported function of phospho-Ser-421 in protecting against mHTT-induced toxicity, D2R-driven mHTT dephosphorylation may participate in the enhanced sensitivity of medium spiny neurons toward mHTT-induced toxicity. Accordingly, some of the beneficial effects of D2R antagonism observed in Huntington disease models may depend upon the inhibition of this dephosphorylation event.
The data presented in this study provide both in vitro and in vivo compelling evidence for the selective regulation of HTT Ser-421 phosphorylation by D2R and the involvement of PPM/PP2C family members in this process. However, the complete chain of molecular events leading to the observed D2R-mediated activation of PPM1A and/or PPM1B requires further investigations. Nonetheless, this study sheds light on how GPCRs, which are known to regulate a plethora of substrate phosphorylation in cells, can also regulate dephosphorylation of substrates by a previously unappreciated family of phosphatases. Identification of other substrates that are dephosphorylated by PPM1A/B in response to D2R activation might lead to a better appreciation of dopaminergic physiological functions in health and disease.
Acknowledgments
We are grateful to Professor Michael Hayden for providing us with the phospho HTT Ser-421 antibody, to Drs. Xueyan Duan and Xin-Hua Feng for shRNAs against PPM1A/1B, and to Dr. David Rubinsztein for the HTTN588-138Q plasmid.
This work was supported, in whole or in part, by National Institutes of Health Grants NS-19576 and MH073853 (to M. G. C.) and a Natural Sciences and Engineering Research Council of Canada (NSERC) discovery grant (to J. M. B.).
We refer in this study to the HTT Ser-421 to avoid confusion with previous studies describing HTT phosphorylation by Akt. Human HTT Ser-421 corresponds to Ser-398 of the mouse HTT (containing 7 glutamines) examined in this study.
- mHTT
- mutated huntingtin protein
- HTT
- huntingtin protein
- HD
- Huntington disease
- D2R
- dopamine D2 receptor
- PPM/PP2C
- protein phosphatase metallo-dependent family
- PPP
- protein phosphatases
- PP1
- protein phosphatase 1
- PP2A
- protein phosphatase 2A
- OA
- okadaic acid
- NPA
- N-n-propylnorapomorphine
- GPCR
- G protein-coupled receptor
- AR
- adrenergic receptor
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 2. Cyr M., Sotnikova T. D., Gainetdinov R. R., Caron M. G. (2006) Dopamine enhances motor and neuropathological consequences of polyglutamine expanded huntingtin. FASEB J. 20, 2541–2543 [DOI] [PubMed] [Google Scholar]
- 3. Cyr M., Beaulieu J.-M., Laakso A., Sotnikova T. D., Yao W.-D., Bohn L. M., Gainetdinov R. R., Caron M. G. (2003) Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 11035–11040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charvin D., Vanhoutte P., Pagès C., Borrelli E., Caboche J. (2005) Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc. Natl. Acad. Sci. U.S.A. 102, 12218–12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charvin D., Roze E., Perrin V., Deyts C., Betuing S., Pagès C., Régulier E., Luthi-Carter R., Brouillet E., Déglon N., Caboche J. (2008) Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol. Dis. 29, 22–29 [DOI] [PubMed] [Google Scholar]
- 6. Jankovic J., Beach J. (1997) Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology 48, 358–362 [DOI] [PubMed] [Google Scholar]
- 7. Zheng G., Dwoskin L. P., Crooks P. A. (2006) Vesicular monoamine transporter 2: role as a novel target for drug development. AAPS J. 8, E682–E692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huntington Study Group (2006) Tetrabenazine as antichorea therapy in Huntington disease: a randomized controlled trial. Neurology 66, 366–372 [DOI] [PubMed] [Google Scholar]
- 9. Luo S., Vacher C., Davies J. E., Rubinsztein D. C. (2005) Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aiken C. T., Steffan J. S., Guerrero C. M., Khashwji H., Lukacsovich T., Simmons D., Purcell J. M., Menhaji K., Zhu Y.-Z., Green K., Laferla F., Huang L., Thompson L. M., Marsh J. L. (2009) Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284, 29427–29436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu X., Greiner E. R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J. S., Thompson L. M., Wetzel R., Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Atwal R. S., Desmond C. R., Caron N., Maiuri T., Xia J., Sipione S., Truant R. (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460 [DOI] [PubMed] [Google Scholar]
- 13. Havel L. S., Wang C.-E., Wade B., Huang B., Li S., Li X.-J. (2011) Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum. Mol. Genet. 20, 1424–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sacco F., Perfetto L., Castagnoli L., Cesareni G. (2012) The human phosphatase interactome: An intricate family portrait. FEBS Lett. 586, 2732–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Colin E., Zala D., Liot G., Rangone H., Borrell-Pagès M., Li X. J., Saudou F., Humbert S. (2008) Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 27, 2124–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Humbert S., Bryson E. A., Cordelières F. P., Connors N. C., Datta S. R., Finkbeiner S., Greenberg M. E., Saudou F. (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 [DOI] [PubMed] [Google Scholar]
- 17. Warby S. C., Chan E. Y., Metzler M., Gan L., Singaraja R. R., Crocker S. F., Robertson H. A., Hayden M. R. (2005) Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 14, 1569–1577 [DOI] [PubMed] [Google Scholar]
- 18. Pardo R., Colin E., Régulier E., Aebischer P., Déglon N., Humbert S., Saudou F. (2006) Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 26, 1635–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beaulieu J. M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 20. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 21. Metzler M., Gan L., Mazarei G., Graham R. K., Liu L., Bissada N., Lu G., Leavitt B. R., Hayden M. R. (2010) Phosphorylation of Huntingtin at S421 in YAC128 Neurons Is Associated with Protection of YAC128 Neurons from NMDA-Mediated Excitotoxicity and Is Modulated by PP1 and PP2A. J. Neurosci. 30, 14318–14329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bialojan C., Takai A. (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Biochem. J. 256, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brewis N. D., Street A. J., Prescott A. R., Cohen P. T. (1993) PPX, a novel protein serine/threonine phosphatase localized to centrosomes. EMBO J. 12, 987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen M. X., McPartlin A. E., Brown L., Chen Y. H., Barker H. M., Cohen P. T. (1994) A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 13, 4278–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiser D. C., Shenolikar S. (2003) Use of protein phosphatase inhibitors. in Current Protocols in Protein Science, Chapter 13, Unit 13.10, John Wiley & Sons, Inc., Hoboken, NJ: [DOI] [PubMed] [Google Scholar]
- 26. Cohen P., Cohen P. T. (1989) Protein phosphatases come of age. J. Biol. Chem. 264, 21435–21438 [PubMed] [Google Scholar]
- 27. Huang X., Honkanen R. E. (1998) Molecular cloning, expression, and characterization of a novel human serine/threonine protein phosphatase, PP7, that is homologous to Drosophila retinal degeneration C gene product (rdgC). J. Biol. Chem. 273, 1462–1468 [DOI] [PubMed] [Google Scholar]
- 28. Jan G., Delorme V., David V., Revenu C., Rebollo A., Cayla X., Tardieux I. (2007) The toxofilin-actin-PP2C complex of Toxoplasma: identification of interacting domains. Biochem. J. 401, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Montini E., Rugarli E. I., Van de Vosse E., Andolfi G., Mariani M., Puca A. A., Consalez G. G., den Dunnen J. T., Ballabio A., Franco B. (1997) A novel human serine-threonine phosphatase related to the Drosophila retinal degeneration C (rdgC) gene is selectively expressed in sensory neurons of neural crest origin. Hum. Mol. Genet. 6, 1137–1145 [DOI] [PubMed] [Google Scholar]
- 30. Rogers J. P., Beuscher A. E., 4th, Flajolet M., McAvoy T., Nairn A. C., Olson A. J., Greengard P. (2006) Discovery of protein phosphatase 2C inhibitors by virtual screening. J. Med. Chem. 49, 1658–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hishiya A., Ohnishi M., Tamura S., Nakamura F. (1999) Protein phosphatase 2C inactivates F-actin binding of human platelet moesin. J. Biol. Chem. 274, 26705–26712 [DOI] [PubMed] [Google Scholar]
- 32. Sun W., Yu Y., Dotti G., Shen T., Tan X., Savoldo B., Pass A. K., Chu M., Zhang D., Lu X., Fu S., Lin X., Yang J. (2009) PPM1A and PPM1B act as IKKβ phosphatases to terminate TNFα-induced IKKβ-NF-κB activation. Cell. Signal. 21, 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lan H., Liu Y., Bell M. I., Gurevich V. V., Neve K. A. (2009) A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol. Pharmacol. 75, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi Y. (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 35. Trettel F., Rigamonti D., Hilditch-Maguire P., Wheeler V. C., Sharp A. H., Persichetti F., Cattaneo E., MacDonald M. E. (2000) Dominant phenotypes produced by HD mutation in STHdhQ111 striatal cells. Hum. Mol. Genet. 9, 2799–2809 [DOI] [PubMed] [Google Scholar]
- 36. Pan C., Liu H.-D., Gong Z., Yu X., Hou X.-B., Xie D.-D., Zhu X.-B., Li H.-W., Tang J.-Y., Xu Y.-F., Yu J.-Q., Zhang L.-Y., Fang H., Xiao K.-H., Chen Y.-G., Wang J.-Y., Pang Q., Chen W., Sun J.-P. (2013) Cadmium is a potent inhibitor of PPM phosphatases and targets the M1 binding site. Sci. Rep. 3, 2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flajolet M., Rakhilin S., Wang H., Starkova N., Nuangchamnong N., Nairn A. C., Greengard P. (2003) Protein phosphatase 2C binds selectively to and dephosphorylates metabotropic glutamate receptor 3. Proc. Natl. Acad. Sci. U.S.A. 100, 16006–16011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiao K., McClatchy D. B., Shukla A. K., Zhao Y., Chen M., Shenoy S. K., Yates J. R., 3rd, Lefkowitz R. J. (2007) Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pineda J. R., Pardo R., Zala D., Yu H., Humbert S., Saudou F. (2009) Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington's disease. Mol. Brain 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosenstock T. R., de Brito O. M., Lombardi V., Louros S., Ribeiro M., Almeida S., Ferreira I. L., Oliveira C. R., Rego A. C. (2011) FK506 ameliorates cell death features in Huntington's disease striatal cell models. Neurochem. Int. 59, 600–609 [DOI] [PubMed] [Google Scholar]
- 41. Cho D. I., Beom S., Van Tol H. H., Caron M. G., Kim K. M. (2006) Characterization of the desensitization properties of five dopamine receptor subtypes and alternatively spliced variants of dopamine D2 and D4 receptors. Biochem. Biophys. Res. Commun. 350, 634–640 [DOI] [PubMed] [Google Scholar]