

Background: Mechanisms that regulate plasma membrane expression of neuronally toxic DEG/ENaC channels are unclear.
Results: Disruption of ER NRA-2, a Nicalin homolog, enhances C. elegans MEC-10(d) DEG/ENaC neurotoxicity. Immunocytochemistry, TIRF imaging, and electrophysiological assays support that NRA-2 controls relative MEC-10(d) distribution between ER and cell surface to regulate channel activity levels.
Conclusion: NRA-2 regulates surface expression of a mutant DEG/ENaC channel.
Significance: NRA-2 Nicalin can modulate DEG/ENaC pathophysiology.
Keywords: C. elegans; ER Quality Control; Intracellular Trafficking; Ion Channels; Neurodegeneration; C. elegans, Nicalin, Quality Control, Trafficking
Abstract
Hyperactivated DEG/ENaCs induce neuronal death through excessive cation influx and disruption of intracellular calcium homeostasis. Caenorhabditis elegans DEG/ENaC MEC-4 is hyperactivated by the (d) mutation and induces death of touch neurons. The analogous substitution in MEC-10 (MEC-10(d)) co-expressed in the same neurons is only mildly neurotoxic. We exploited the lower toxicity of MEC-10(d) to identify RNAi knockdowns that enhance neuronal death. We report here that knock-out of the C. elegans nicalin homolog NRA-2 enhances MEC-10(d)-induced neuronal death. Cell biological assays in C. elegans neurons show that NRA-2 controls the distribution of MEC-10(d) between the endoplasmic reticulum and the cell surface. Electrophysiological experiments in Xenopus oocytes support this notion and suggest that control of channel distribution by NRA-2 is dependent on the subunit composition. We propose that nicalin/NRA-2 functions in a quality control mechanism to retain mutant channels in the endoplasmic reticulum, influencing the extent of neuronal death. Mammalian nicalin may have a similar role in DEG/ENaC biology, therefore influencing pathological conditions like ischemia.
Introduction
Under conditions of localized oxygen deprivation in the brain, neuronal energy deficits promote an excitotoxic cascade in which ion channels are excessively activated to induce neuronal necrosis (1). Although the primary insult is often localized to a small site, successive rounds of secondary neuronal death ensue as dying neurons release channel-hyperactivating glutamate to lethally excite their neighbors, expanding the region of cell death to markedly exacerbate functional losses. Understanding the molecular mechanisms operative in necrotic neuronal death is critical for the design of neuroprotective strategies, although challenges of in vivo dissection of the death process in higher organisms are substantial.
Genetic dissection of necrosis mechanisms in model organisms has revealed conserved pathways that contribute to necrotic death (1, 2). In C. elegans, specific gain-of-function mutations in DEG/ENaC family channel subunits induce the necrotic death of the neurons in which they are expressed (3–5). MEC-4, a pore-forming subunit of the mechanosensory channel complex in C. elegans touch receptor neurons (TRNs)3 that is required for sensing gentle mechanical stimuli (typically delivered to the animal as a brush with an eyelash hair), can be altered to a neurotoxic form (4). Large side-chain amino acid substitutions for a small amino acid near the pore domain (MEC-4(A713V), referred to here as MEC-4(d)) create a hyperactivated channel that is permeable to Na+ and Ca+2 (6–8). Calcium dysregulation in mec-4(d) neurons is amplified by calreticulin-dependent release of calcium from ER stores (7, 9), which activates cytosolic calpain proteases (10) that ultimately influence deleterious lysosomal activity (11). That the mammalian neuronal DEG/ENaC channel subunit ASIC1 contributes to necrotic death in mouse ischemia models (12–14) supports that conserved mechanisms of neuronal DEG/ENaC hyperactivation toxicity are operative across phyla.
MEC-10 is a second DEG/ENaC channel subunit co-expressed with MEC-4 in C. elegans TRNs (3), and assembling with additional subunits stomatin MEC-2 and paraoxonase MEC-6 to form a touch-transducing complex (8). Introduction of an engineered mec-10 transgene encoding the mec-4(d)-analogous toxic amino acid substitution (namely A693V) to create mec-10(d) induces a low level of necrosis associated with neuronal swelling and featuring a similar toxicity mechanism to mec-4(d) (15). The low level of toxicity associated with the mec-10(d) transgene presents an opportunity to screen for genes that can mutate, or be knocked down with RNAi approaches, to enhance necrosis. Such genes would be anticipated to identify negative regulators of channel biosynthesis and activity or to define factors that normally protect against downstream neurotoxic events.
Here we report that disruption of mammalian nicalin homolog nra-2, an ER protein implicated in C. elegans muscle acetylcholine receptor (AChR) channel maturation (16), enhances MEC-10(d)-induced necrosis via a mechanism that increases surface expression of the MEC-10(d) channel. Our data further underscore the role of excessive channel activity in necrosis induction, identify NRA-2/nicalin as a new player in DEG/ENaC channel biogenesis, and reveal an unexpected focused surface distribution of the MEC-10(d) channel in touch neurons. Because mammalian DEG/ENaC functions modulate cystic fibrosis, renal diseases, hypertension, and ischemia-associated neurotoxicity, insights into regulated surface expression might be exploited in design of interventions that modulate channel activity in human disease.
EXPERIMENTAL PROCEDURES
C. elegans Strains
Nematodes were maintained according to standard procedures (17). Transgenic strains were generated as described (18). RNAi was executed according to, (15) EF hand RNAi collection described in Ref. 47.
Strains/Alleles Used
Strains/alleles used were: N2 (wild type); RB1480: nra-2(ok1731); ZB4038: zdIs5[Pmec-4GFP]; mec-4(u231); ZB4039: zdIs5[Pmec-4GFP]; mec-4(u231); nra-2(ok1731).
Transgenic Strains Used
Transgenic strains used were ZB2057: bzIs111[mec-10(d)]: Pmec-4mec-10(d)::GFP; Pmec-4mCherry; ZB4031: [Pmec-18sid-1; Psng-1YFP]; bzIs111 [Pmec-4mec-10(d)::GFP; Pmec-4mCherry] (generated from strain TU3312 (Pmec-18sid-1;Psng-1YFP) and integrated by X-irradiation, according to standard procedures); ZB4033: Is165[mec-10(d)]; nra-2(ok1731); ZB4034: Is165[mec-10(d); nra-2(ok1731)]; Ex[Pmec-4nra-2; Pttx-3RFP]; SK4005: zdIs5[Pmec-4GFP]; mec-4(u231); ZB4035: bzIs141[Pmec-4mec-4::GFP]; ZB4036: bzIs142 [Pmec-4mec-10::GFP]; ZB2522: bzIs180 Pmec-4mec-10(d)GFP; nre-1(hd20); lin-15B(hd126); ZB4045: bzIs141[Pmec-4mec-4::GFP]; nra-2(ok1731); ZB4046: bzIs142[Pmec-4mec-10::GFP]; nra-2(ok1731); Ex221: [mec-10(d)]; nra-2(ok1731); Ex[Pmec-4nra-2+Pttx-3RFP].
Cell Death Assay
The percentage of TRN degeneration was scored as described before (15). Briefly, mCherry-labeled TRNs were counted in Is[mec-10(d)] and nra-2(ok1731); Is[mec-10(d)] strains using a Zeiss Imager.D1m microscope and a Zeiss PlanApochromat 20× NA 0.8 objective. The average number of surviving TRNs per animal was calculated as the total number of TRNs counted divided by the total number of animals counted.
C. elegans Cell Culture
C. elegans embryonic cells were cultured following published procedures (19, 20). Briefly, gravid adults were cultured on enriched peptone plates (8P plates) seeded with Escherichia coli strain Na22. Eggs were isolated by lysing animals with a solution containing fresh bleach and 10 n NaOH. Eggs were separated from animal carcasses by treatment with 30% sucrose dissolved in egg buffer solution (118 mm NaCl, 48 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 25 mm Hepes, pH 7.3, 340 mosm). The eggshell was removed by treatment with 2 mg/ml chitinase (Sigma Aldrich, catalogue number C6137), and cells were dissociated by manual pipetting through a 27-gauge needle, resuspended in culture medium (L-15 + 10% FBS + 45 mm sucrose + 100 IU of penicillin and 100 μg/ml streptomycin) and plated on glass coverslips previously covered with peanut lectin (0.5 mg/ml). Cells were seeded at a density of ∼230,000 cells/cm2 and used 4–6 days after seeding. Strains used for all microscopy were Is[mec-10(d)] and nra-2(ok1731); Is[mec-10(d)].
Immunofluorescence Staining and Microscopy
Fluorescent photographs of GFP C. elegans were taken using a Zeiss Axiovert 200m microscope equipped with a GFP filter and a QImaging Rolera e-mc2 camera with MetaMorph software. For MEC-4::GFP and MEC-10:GFP quantification, photographs were taken using the same exposure time (300 ms), and quantifications were performed in ImageJ.
C. elegans primary embryonic cell culture was set up in 35-mm glass bottom cell culture dishes (Bioexpress, catalog number T-2881-16). Primary embryonic cells from C. elegans were fixed in 4% paraformaldehyde for 20 min and permeabilized for 10 min with M9 + 0.1% Tween 20. Cells were blocked in 2% BSA for 1 h at room temperature, incubated with primary antibody for 1 h at room temperature, washed three times with M9 + 0.01% Tween 20, incubated with secondary antibody for 1 h at room temperature, washed three times with M9 + 0.01% Tween 20, and imaged on a Zeiss Axiovert microscope with a 63× NA 1.3 objective. Images were taken at 250-ms exposure, at 1 × 1 binning. Images were deconvolved using Autoquant X3. Measurements were made on thresholded images using MetaMorph software.
Primary Antibodies Used
Primary antibodies used were: mouse polyclonal α-EEA-1 at 1 μg/ml, mouse mAb SQV8 (Developmental Studies Hybridoma Bank) at 1 μg/ml. Secondary antibodies used were: anti-mouse Cy5 (Jackson ImmunoResearch catalog number 115-175-146) at 1:5,000 dilution.
ER Tracker and LysoTracker Staining
C. elegans primary embryonic cell culture was set up as described above. ER tracker Blue White DPX (Life Technologies catalog number MP12353) or LysoTracker Blue DND-22 (Life Technologies catalog number L-7525) were incubated at 1 μm concentration in M9 + 0.01% Tween 20 for 30 min and washed three times with M9 + 0.01% Tween 20 prior to imaging.
TIRF Microscopy
C. elegans primary embryonic cell culture was set up using Is[mec-10(d)] and nra-2(ok1731); Is[mec-10(d)] strains as described above. Cells were imaged live on an Olympus IX70 microscope using an Olympus PlanApo 60× NA 1.45 total internal reflection fluorescence microscopy (TIRFM) objective. Touch neurons in cell culture were identified by mCherry expression using standard wide-field microscopy. TIRF imaging was done using a Melles Griot Ar488 laser (Melles Griot Laser Group, Carlsbad, CA) and an Olympus IX2 Laser Combiner. A micrometer screw was used to change the incident beam angle to 61° for TIRF mode imaging. All images were taken at 500-ms exposure using iVision software (BioVision Technologies Inc., Exeter, PA). Image quantification was done in ImageJ. Images were thresholded, a region of interest was drawn around the cell body, and mean intensity of fluorescent signal was measured. Statistical analysis was performed using GraphPad Prism 6.
Electrophysiology
cRNAs were synthesized using T7 mMESSAGE mMACHINE kit, purified, and run on denaturating agarose gels to check for size and integrity. cRNA quantification was performed spectroscopically. Stage V–VI oocytes were selected among multistaged oocytes dissected by 2-h collagenase treatment (2 mg/ml in Ca2+-free OR2 solution) from Xenopus laevis ovaries. Oocytes were injected with 10 ng/oocyte of each cRNA and incubated in OR2 medium + 500 μm amiloride. OR2 solution consisted of 82.5 mm NaCl, 2.5 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 1 mm Na2HPO4, 0.5 g/liter polyvinyl pyrrolidone, and 5 mm HEPES (pH 7.2), supplemented with penicillin and streptomycin (0.1 mg/ml) and 2 mm sodium pyruvate at 20 °C for 2–3 days before recording. Currents were measured using a two-electrode voltage-clamp amplifier (GeneClamp 500B; Axon Instruments) at room temperature. Electrodes (0.2–0.5 megaohms) were filled with 3 m KCl, and oocytes were perfused with a physiological NaCl solution containing (in mm) 100 NaCl, 2 KCl, 1 CaCl2, 2 MgCl2, and 10 HEPES, pH 7.2. We used the pCLAMP suite of programs (Axon Instruments) for data acquisition and analysis. Currents were filtered at 200 Hz and sampled at 1 kHz. Currents are expressed as control minus 500 μm amiloride (7, 8).
RESULTS
A Targeted RNAi Screen for Calcium-responsive Genes That Modulate Channel-induced Necrosis Identifies ER Chaperone nra-2
To extend molecular understanding of necrosis mechanisms, we sought to identify modulators of genetically induced touch neuron degeneration. Because calcium is a key factor in neuronal necrosis across species (7, 22, 23), we selected a collection of 191 C. elegans genes that encode EF hand calcium-binding proteins for targeted knockdown by feeding RNAi (24). We first constructed a strain with weak necrosis inducer mec-10(d) (3) that co-expressed GFP in TRNs and is sensitive to RNAi in neurons (Pmec-4mec-10(d)::GFP; nre-1(hd20); lin-15B(hd126); screen strategy summarized in Fig. 1A). At 15 °C we observed baseline PLM tail touch neuron degeneration of ∼2%, as described previously (15). We then fed this strain either empty vector control (L4440) or dsRNA EF hand gene clones from the L1 to L4 larval stages and evaluated the extent of neuronal death by counting surviving fluorescent PLM tail TRNs. Of the 191 EF hand genes tested, RNAi directed against clone T05F1.1 (nra-2) exhibited the greatest enhancement of mec-10(d)-induced necrosis: a 3-fold increase as compared with vector control treatment.
FIGURE 1.

nra-2 disruption enhances mec-10(d)-induced necrosis in C. elegans. A, screen strategy for identification of enhancers of mec-10(d)-induced necrosis. Touch receptor neuron channel subunit mec-10(d) encodes an amino acid substitution in the pore that hyperactivates the channel and induces low levels of necrosis in TRNs, as measured by the number of surviving GFP+ cells. Enhancer loci are identified as those that confer increased levels of necrosis after RNAi knockdown, diminishing the number of apparent GFP-expressing neurons. B, RNAi disruption of nra-2 in strains sensitized for neuronal RNAi enhances mec-10(d)-induced necrosis. Shown is the percentage of TRN necrosis in Pmec-18sid-1-expressing nematodes subjected to nra-2(RNAi) or empty vector control L4440. We scored surviving touch neurons by GFP signal; the percentage of degeneration was determined by the formula ((1 − (x/6)) × 100 where x is the number of surviving neurons. Error bars, S.E.; average of three repeat trials (each with at least 35 L4 animals), *, p = 0.0261, Student's t test. C, NRA-2 is an ER-resident chaperone protein. Shown is comparative domain organization of C. elegans NRA-2 and mammalian homolog nicalin. Nicalin, unlike NRA-2, does not contain an EF hand domain. There is 31% identity and 49% similarity between the two proteins. Domain predictions were made using the ScanProsite Tool (available from ExPASy). D, nra-2 deletion enhances mec-10(d)-induced necrosis. We scored the percentage of degeneration as in B. nra-2(Δ) is nra-2(ok1731); rescue transgene is Ex[Pmec-4nra-2; Pttx-3RFP] Error bars, S.E.; average of three repeat trials (each with at least 30 L4 animals), **, p = 0.0033, one-way ANOVA.
Note that nra-2 encodes an ER-resident transmembrane protein homologous to mammalian nicalin (31% identity, 49% similarity). Both proteins contain an N-terminal aminopeptidase domain with poor conservation in the catalytic residues; this domain is not thought to possess peptidase activity. NRA-2 contains an EF hand domain within the aminopeptidase domain that is absent in mammalian nicalin (Fig. 1C).
We confirmed the RNAi phenotype using a second, neuronally sensitized RNAi strain (25) (Pmec-18sid-1; Psng-1YFP in Pmec-4mec-10(d)::GFP; Pmec-4mCherry). We grew this strain at 20 °C on empty vector or nra-2 dsRNA-expressing bacteria from the L1 to L4 larval stages and counted all surviving TRNs at the L4 stage. We found that nra-2 knockdown caused over 4-fold enhancement of mec-10(d)-induced TRN death (Fig. 1B), confirming the enhancer outcome for nra-2(RNAi).
To rule out any extraneous effect of RNAi on death enhancement, we constructed a nra-2(Δ); mec-10(d) double mutant strain. The double mutant exhibited over a 5-fold increase of mec-10(d)-induced TRN degeneration as compared with mec-10(d) alone (Fig. 1D). As further confirmation that nra-2 was the enhancer locus, we expressed the nra-2(+) transgene in TRNs of nra-2(Δ); mec-10(d) animals and observed a 3-fold reduction of TRN degeneration as compared with control nra-2(Δ); mec-10(d) animals, i.e. a restoration of TRN degeneration to mec-10(d) levels (Fig. 1D). This transgene rescue both confirms that the nra-2 mutation confers the death-enhancing phenotype and establishes that nra-2 acts cell autonomously within the touch receptor neuron to modulate necrosis. Because nra-2(Δ) itself does not induce any TRN necrosis (Fig. 1D), our data establish that loss of nra-2 function results in increased necrosis due to mec-10(d)-induced insult.
NRA-2 Does Not Play an Essential Role in the Differential Distribution of MEC-10 and MEC-4 Subunits in Touch Receptor Neurons
NRA-2 encodes a transmembrane ER-resident protein implicated in regulation of acetylcholine receptor (AChR) subunit composition in C. elegans muscle, possibly by allowing only specific subunit combinations of that receptor to exit the ER (16). This role in channel assembly raises the question as to whether NRA-2 might similarly regulate distribution, assembly, or surface expression of the touch-sensing MEC channel. The gentle touch-sensing MEC channel is proposed to be composed of MEC-4 and MEC-10 subunits (3, 26, 27). However, some data suggest that the MEC channel assembled in vivo on neuronal processes is primarily made up of MEC-4 subunits (28) and that the MEC channel at the cell body is heteromeric, containing MEC-4 and MEC-10 subunits. In support of this model, MEC-4::GFP localizes in puncta along the process length (28, 29) (Fig. 2A), but MEC-10::GFP is localized at the cell body (28) (Fig. 2B). Consistent with localized functional roles, mec-4 null mutants are insensitive to touch delivered anywhere along the length of the process, but mec-10 null mutants are insensitive only to touch administered in the vicinity of the soma, and are otherwise sensitive to touch along the process. Thus, MEC-4 homomeric channels might be the primary transducers of touch along the process, but MEC-4/MEC-10 heteromers might function in sensing touch at the cell body (28).
FIGURE 2.

nra-2(Δ) does not induce significant changes in MEC-4 and MEC-10 distribution and is associated with a modest change in overall touch sensation. A, MEC-4::GFP (puncta in processes and dispersed in soma) shows similar patterns in wild type and nra-2(Δ) backgrounds. Images are representative of 30 individual cell observations (scale bar = 5 um). B, MEC-10::GFP (soma restriction) shows similar patterns in wild type and nra-2(Δ) backgrounds. Images are representative of 30 individual cell observations (scale bar = 5 μm). C, MEC-4::GFP intensity in soma in wild type and nra-2(Δ) backgrounds, n = 30 for each strain, error bars, S.E. D, MEC-10::GFP intensity in soma in wild type and nra-2(Δ) backgrounds, n = 30 for each strain, error bars, S.E. E, touch sensitivity was recorded as the fraction of nematodes that respond to the first five touches with an eyelash pick. Error bars, S.E. Data are combined for three trials, 30 animals per experiment), ****, p < 0.0001, one-way analysis of variance. F, percentage of TRN necrosis in mec-4(d) versus nra-2(Δ); mec-4(d) is not significantly different. We scored surviving touch neurons by GFP signal; the percentage of degeneration was determined by the formula ((1 − (x/6)) × 100 where x is the number of surviving neurons. Error bars, S.E.; three trials; n > 35 L4 animals.
If NRA-2 plays a role in restricting MEC-4 and MEC-10 to different regions within the touch neuron, the distributions of MEC-4::GFP and/or MEC-10::GFP might be altered in the nra-2 mutant background. We constructed lines that were MEC-4::GFP; nra-2(Δ) and MEC-10::GFP; nra-2(Δ) and compared basic subunit distribution ± nra-2. (Note that both translational fusion constructs have C-terminal tags and can functionally rescue cognate mutations (28, 30).) Our comparison of distribution of tagged MEC-4 and MEC-10 proteins did not reveal any striking distribution changes ± nra-2 (Fig. 2, A and B). General level of channel expression as measured by fluorescence intensity also appeared similar ± nra-2 (Fig. 2, C and D). Thus analysis of tagged reporters does not support a major role for NRA-2 in changing the distinctive distribution of MEC-4::GFP and MEC-10::GFP channel subunits or in regulating overall GFP-tagged subunit stabilities.
To address whether native subunits at physiological expression levels might be differentially affected by nra-2 deficiency, we scored touch sensitivity over the process length ± nra-2 (primarily a score of MEC-4 function). We observed that the nra-2(Δ) mutants were only modestly touch-insensitive (Fig. 2E). Moreover, when we touched in the vicinity of the cell body ± nra-2, we noted only a slight change from 2 to 10% touch-insensitive in nra-2(Δ) (n = 100, not significant by paired two-tailed t test). Taken together, our data support that most mechanotransducing complexes are distributed and function even when NRA-2 is lacking. These data suggest that NRA-2 might act preferentially on toxic rather than wild type DEG/ENaC subunits.
nra-2(Δ) Does Not Enhance mec-4(d)-induced Neuronal Death
We identified nra-2(RNAi) as an enhancer of mec-10(d)-induced necrosis, but potential effects on the stronger necrosis inducer mec-4(d) were unknown. To test whether nra-2(Δ) can enhance mec-4(d)-induced neuronal death, we constructed nra-2(Δ); mec-4(d) double mutants and measured the extent of TRN death. As reported previously, mec-4(d) induces a high level of TRN degeneration (∼85% of cells die), in striking contrast to mec-10(d) (3, 4). We found that the absence of nra-2 does not enhance neuronal death further than occurs in mec-4(d); nra-2(+) (Fig. 2F). These data, together with localization and touch sensitivity data, are also consistent with a model in which NRA-2 does not play a major role in MEC-4 biology, although we cannot be certain of a “ceiling effect” for mec-4(d)-induced necrosis in which higher enhanced levels of neuronal death are not possible. Our findings raise the possibility that NRA-2 might interact specifically with only the MEC-10 or MEC-10(d) subunits in vivo.
MEC-10(d) Concentration in the ER Compartment Is Diminished When nra-2 Is Absent
Toxicity of the MEC-4(d) channel subunit is associated with enhanced channel activity (6–8, 31), and thus enhanced MEC-10(d) channel activity is a plausible mechanism for increased cell death. Given that NRA-2 is an ER protein involved in AChR channel maturation, we wondered whether NRA-2 acts in the ER to limit surface expression of the MEC-10(d) channel. To address this hypothesis, we first monitored subcellular distribution of MEC-10(d)::GFP in various intracellular compartments, ± nra-2.
We elected to work with cultured C. elegans touch neurons (20, 32), which allowed us greater freedom to use immunohistochemical and imaging techniques that are impractical in vivo. We measured colocalization of MEC-10(d)::GFP with fluorophore-labeled antibodies for endosome (EEA-1) and Golgi body (mAbSQV8), and we dye-labeled lysosomes and ER in cultured TRNs of nra-2(Δ);mec-10(d)::GFP and control mec-10(d)::GFP animals (Fig. 3, A–H). Mean MEC-10(d)::GFP colocalization with endosomes, lysosomes, or Golgi bodies did not significantly differ between nra-2(Δ) and control animals. Mean ER colocalization of the MEC-10(d)::GFP and ER, however, was ∼ 25% for nra-2 mutants as compared with 50% for control mec-10(d) animals (Fig. 3I). We compared total ER-associated immunofluorescence for control and mutant strains and found no significant difference in levels, suggesting that the decrease in MEC-10(d)::GFP fluorescence in nra-2 mutants is not due to a general decrease in the volume of the ER (Fig. 3J). Our data suggest that at the intracellular level, MEC-10(d)::GFP is localized primarily to the ER, and that the absence of nra-2 reduces ER-associated MEC-10(d)::GFP.
FIGURE 3.

nra-2(Δ) lowers the proportion of MEC-10(d) present in the ER. A–H, representative examples of distribution of MEC-10d::GFP relative to compartment reporters for endosomes (EEA-1 antibody (Ab) staining (WT (A) and nra-2(Δ) (B)), SQV-8 mAb staining (WT (C) and nra-2(Δ) (D), LysoTracker staining (WT E and nra-2(Δ) (F), and ER Tracker staining (WT G and nra-2(Δ) (H). Thresholded images were measured for colocalization of red organelle marker with green MEC-10(D)::GFP. Scale bar: 3 μm. I, quantification of MEC-10(d)::GFP accumulation in subcellular compartments. Deconvolved thresholded images were measured for colocalization using MetaMorph. Error bars, S.E. n > 20. *, p < 0.05, Student's t test. J, quantification of ER area/cell. Deconvolved thresholded stacks were measured for total ER-associated fluorescence using MetaMorph. Error bars, S.E. n > 25. p = 0.633 by unpaired two-tailed t test.
NRA-2 Regulates Surface Expression of MEC-10(d)
Low levels of MEC-10(d)::GFP in the ER might reflect enhanced degradation or enhanced trafficking out of the ER to the cell surface. Given the necrosis-enhancing phenotype of nra-2 deletion and the overall similar fluorescence levels for MEC-10(d)::GFP ± nra-2, the latter model seemed more plausible. To test the hypothesis that low MEC-10(d)::GFP in the ER might result from altered trafficking from ER to the surface, we quantified surface distribution of MEC-10(d)::GFP in cultured TRNs of nra-2(Δ);mec-10(d)::GFP and control mec-10(d)::GFP animals by TIRFM. TIRFM uses an angled incident laser beam to illuminate surface fluorescent probes by inducing total internal reflection of the incident beam at the interface of coverslip and culture medium (33). The resultant low intensity evanescent wavelets penetrate ∼150 nm into the medium and thereby excite fluorescent proteins at or near the plasma membrane. TIRFM thus allows live imaging of plasma membrane-associated proteins with high X-Y resolution and very low noise.
Our TIRFM imaging revealed a striking concentration of surface MEC-10(d)::GFP at a site near the nucleus, either with or without nra-2. The biological reason for this highly restricted distribution of MEC-10(d)::GFP is unclear, but might underlie its role in touch perception near the cell body (see “Discussion”). Importantly, our TIRFM studies showed that mean fluorescence intensity of surface-localized MEC-10(d)::GFP was significantly increased in the nra-2(Δ) background (p < 0.0001, ∼1200 units control, ∼2400 units nra-2(Δ); Fig. 4, A and B). Furthermore, we normalized the mean intensity of surface fluorescence to the mean surface area of the cell body for each strain and observed a significant increase in normalized surface fluorescence in the nra-2(Δ) background (Fig. 4C). These data suggest that loss of nra-2 function results in an increase of surface-associated MEC-10(d)::GFP. Coupled with data supporting decreased ER-associated MEC-10(d)::GFP in nra-2(Δ), our data suggest a role for NRA-2 in ER retention of MEC-10(d). The increased MEC-10(d) surface expression in the nra-2(Δ) background, accompanied by the expected increased mutant channel ion influx, could be the mechanistic basis for the enhanced neurotoxicity.
FIGURE 4.

TIRF imaging supports that proportionally more MEC-10(d) is situated at the cell surface in nra-2(Δ) mutants. A, representative images of nra-2(+) and nra-2(Δ) neurons that express mec-10(d)::GFP in culture. MEC-10(d)::GFP fluorescence is visualized under TIRF conditions, and the cell boundary is traced in a red outline. All images were taken using a 60× NA 1.45 Zeiss objective. Scale bar: 3 μm. B, quantification of mean surface channel expression measured by TIRF microscopy. Exposure time for all images was 500 ms. Error bars, S.E. n = 50 cells. ****, p < 0.0001, Student's t test. C, quantification of mean intensity of MEC-10(d)::GFP relative to area of cell body. Error bars, S.E. n > 30. *, p < 0.03 by unpaired two-tailed t test.
NRA-2 limits MEC Channel Currents in an Oocyte Expression System
Co-expression of MEC-4(d), MEC-2, and MEC-6 in Xenopus oocytes generates an amiloride-sensitive channel with a large inward Na+ current as well as a Ca2+-activated Cl− current that reveals inward Ca2+ conductance (6–8). MEC-4(d) is essential for the generation of both currents and, interestingly, the addition of MEC-10(d) diminishes these currents (6–8, 34). Our in vivo and cell culture data suggest that enhanced necrosis in TRNs of nra-2 mutants may be attributed to increased ion influx due to increased surface expression of MEC-10(d) channels. We tested this model by measuring Na+ current in oocytes expressing MEC-4, MEC-10(d), MEC-2, and MEC-6 (henceforth described as the MEC-10(d) complex), with or without NRA-2 (Fig. 5, A and B). We found that MEC-10(d) complex-expressing oocytes showed a mean inward current of 1.5 μA. The addition of NRA-2 to the injected MEC-10(d) subunits reduced mean inward current to 0.15 μA. That co-expression of NRA-2 with the MEC-10(d) complex suppressed Na+ current by ∼90% is consistent with a negative role for NRA-2 in MEC-10(d) currents (Fig. 5C).
FIGURE 5.

NRA-2 suppresses MEC-10(d)-induced currents in Xenopus oocytes. A and B, representative traces of Na+ current in Xenopus oocytes induced by expression of MEC channel subunits containing MEC-10(d) without NRA-2 (A) and with co-injection of NRA-2 (B). Voltage protocol was from −160 mV to + 100 mV from a holding potential of −30 mV. C, current amplitude histogram of Na+ current recorded at −100 mV in oocytes, with and without co-injection of NRA-2 (n = 9). **, p < 0.01, Student's t test. D, the percentage of current suppression of MEC-4(d) and/or MEC-10(d)-induced Na+ current by NRA-2 co-injection in oocytes. p values indicate significance of suppression over controls not injected with NRA-2, by Student's t test.
We wondered whether the Na+ current suppression was due to a MEC-10(d)-specific interaction with NRA-2, as suggested by genetic studies. Surprisingly, however, we found that the addition of NRA-2 could also suppress currents in oocytes expressing only MEC-4 and MEC-4(d) (p < 0.05; Fig. 5D), indicating that the MEC-10(d) subunit is not essential for NRA-2 impact. Notably, combinations that included MEC-4(d) without native MEC-4(+) were not susceptible to NRA-2-induced current suppression, with either MEC-10(+) or MEC-10(d) co-expressed (Fig. 5D). These data support the in vivo scoring of neuronal death ± nra-2 in mec-4(d) mutants (Fig. 2F) and suggest that a MEC-4(+) subunit might be needed for NRA-2 impact on channel surface expression or that a channel composed of a specific number of mutant versus wild type subunits is impacted by NRA-2. Note that a limitation of our assay is that it is not possible to address whether M4/M10 channels or channels composed of M10d only could be suppressed because no current is detectable with these combinations (6, 8).
In sum, from our electrophysiological studies in the oocyte expression system, we underscore three observations: (i) NRA-2 limits currents for the co-expressed MEC-4, MEC-10(d), MEC-6, MEC-2 channel; (ii) the presence of at least one MEC-4 appears essential to allow NRA-2 current suppression in a channel complex containing either MEC-4(d) or MEC-10(d); and (iii) NRA-2 current suppression does not require the presence of either MEC-10 or MEC-10(d). These results suggest that the mechanism of NRA-2 action may not be MEC-10(d)-specific, but rather might involve a more complex capacity of NRA-2 to sense the correct conformation of an assembled, or partially assembled, channel complex that involves more than one MEC subunit.
A Model for NRA-2 Influence on MEC-10(d) Neurotoxicity
Our data suggest that NRA-2 acts in the ER to mediate surface expression of properly folded/assembled MEC-4/MEC-10 channels (summary in Fig. 6). Correctly folded heteromers traffic to the plasma membrane. When the aberrantly folded MEC-10(d) or MEC-4(d) subunit is present, the specific three-dimensional conformation of the assembling channel (which includes MEC-4) is sensed as being abnormal, and NRA-2 preferentially retains those channels in the ER. In the absence of NRA-2, more channels that contain aberrant subunits escape quality control in the ER, which creates more leaky channels at the cell membrane, leading to enhanced neurotoxicity.
FIGURE 6.
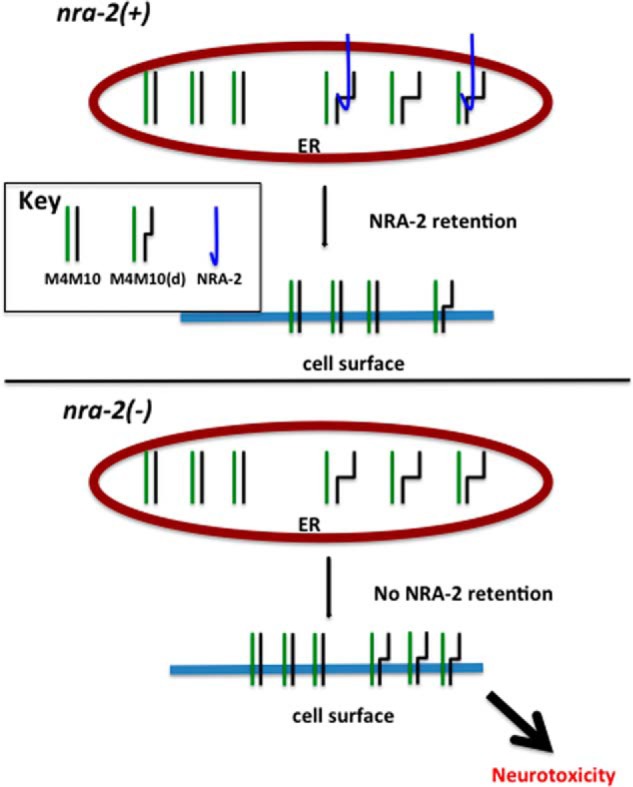
A model of NRA-2 function in C. elegans TRNs. A working model of NRA-2 interaction with a mechanosensory channel composed of wild type and MEC-10(d) subunits is shown. Top, in the ER, NRA-2 functions to evaluate assembling MEC channel complexes, and mutant MEC-10(d)-containing channels are preferentially retained in the ER. Bottom, in an nra-2(Δ) background, there is relaxed retention of mutant MEC-10(d)-containing channels in the ER. Consequently, more MEC-10(D)-containing channels are trafficked to the surface, and the resulting accumulation and function of toxic channels lead to enhanced neurotoxicity.
DISCUSSION
Our studies document the role of ER-resident chaperone protein NRA-2 in modulation of MEC-10(d) channel-induced necrosis in C. elegans. Combining genetic approaches, live imaging, immunohistochemistry, and electrophysiological assays in Xenopus oocytes, we show that loss of nra-2 function results in an increase in the surface expression of toxic MEC-10(d) channels to enhance TRN necrosis. Conversely, NRA-2 limits channel activity in heterologous expression assays. We propose that NRA-2 acts in the ER to distinguish specific conformations of assembling channel complexes, restricting exit of immature or malformed channels. Our work underscores the theme of enhanced channel activity in necrosis induction, extends understanding of NRA-2 function in channel biogenesis, and suggests a potential in vivo molecular strategy for increasing/decreasing surface expression of DEG/ENaC subunits that might be of therapeutic value in some human disease states.
Increased MEC-10(d) Channel Surface Expression Is Associated with Enhanced Neurotoxicity
Previous work has supported that increased activity of mutant DEG/ENaC channels and their associated Na+ and Ca+2 influx initiates neuronal necrosis (4, 7, 31, 35). Here we have shown that nra-2(RNAi) and nra-2 deletion enhance mec-10(d)-dependent neuronal loss via a cell-autonomous mechanism that increases MEC-10(d) surface expression. In the absence of NRA-2, more MEC-10(d) channels reach the neuronal surface, where they collectively conduct higher cation influx than normal, enhancing neurotoxicity. Thus our data reinforce the idea that increased channel activity, either induced via subunit abnormality or induced by increased numbers of plasma membrane channels, is a necrosis-initiating insult.
An Unusual Subcellular Localization of MEC-10(d)::GFP Subunits
Our TIRFM studies highlighted a striking and unexpected concentration of surface-expressed MEC-10(d)::GFP to a highly localized spot at the periphery of the TRN soma. The MEC-10(d)::GFP is not concentrated near lysosomal compartments, but rather is more prevalently partitioned between the ER and the cell surface domain. Neurotoxic outcomes support that MEC-10(d) is a component of an active channel. The concentrated MEC-10::GFP distribution contrasts strongly with the punctate distribution of MEC-4::GFP subunits in the process thought to correspond to gentle touch-transducing touch complexes (29). The dramatic difference in distribution is consistent with previous studies indicating that MEC-4 is needed for response to touch stimulation all along the neuronal process, whereas MEC-10 is needed for responses to touch stimuli only near the soma (28). Restriction of DEG/ENaCs to specific raft domains is known for both invertebrate and mammalian members of this channel class (29, 36–38). Interestingly, studies of βNaC distribution in mouse C-fiber small diameter neurons suggest a soma rather than process distribution (surface expression not assayed) (39), and neuronal ASIC1a has been observed to be largely ER-concentrated, with regulated surface expression under conditions in which ASIC2a is not similarly distributed (40), suggesting differential trafficking and localization as a feature of the channel class shared across phyla. Still, why a distinct MEC-10 channel type might be needed in a focused cellular domain in the soma is not clear. One possibility is that the MEC-10-enriched domain corresponds to a specialized mechanosensory site. Alternatively, this cellular domain might constitute a holding site for end-stage MEC channel surface assembly prior to transport down the process. MEC-10, which can negatively regulate channel activity (8, 34), might be held and added to the MEC complex only under specific physiological conditions, or might even provide a chaperone function. The function of this MEC-10-concentrating cellular domain clearly remains to be elucidated. Because MEC-10 is plays a minor role in gentle touch, a novel activity could be anticipated.
Expanding Understanding of NRA-2 Function
NRA-2 was originally identified as a protein that co-immunoprecipitated with a muscle-specific C. elegans acetylcholine receptor (41). Later studies revealed a role for nra-2 in AChR subunit selection and/or surface expression in muscle (16). More specifically, this study showed that specific AChR subunits co-expressed in muscle were selected against being included into drug-sensitive AChR complexes when NRA-2 was active (16). Similarly, our work shows that NRA-2 functions in ER retention of channels containing mutant MEC-10 subunits. The mild effect of nra-2(Δ) on mec-4-based touch sensitivity could be consistent with an increased (but low level) inclusion of the channel-inhibiting MEC-10 subunit in the MEC-4 channels or more inappropriately folded complexes assembling, although more broad effects on other channels and neuron physiology cannot be ruled out. An effect linked to NRA-2 Ca2+ binding capability could also be operative. Interestingly, however, NRA-2 does not impact mec-4(d)-induced neurodegeneration, suggesting that NRA-2 might recognize only specific channel compositions. Importantly, the oocyte experiments support that NRA-2 does not need the presence of MEC-10 in the channel complex to exert its effects, but it can also act on channel complexes made of wild type and mutant MEC-4 (but not made of MEC-4 mutant only). Taken together, our data suggest that NRA-2 recognizes and selects protein complex to exit the ER, likely based on the integrity of their tertiary/quaternary structure. Because ER calcium contributes to folding efficacy and conformational evaluation of forming complexes, the implication of calcium-binding EF hand protein NRA-2 in MEC complex quality control raises the possibility that the EF hand domain contributes to efficiency of assembly.
An NRA-2-related Complex Might Influence Channel Biogenesis across Species
It is noteworthy that reported regulated surface expression of mammalian channel ASIC1a shares striking features with the MEC-10(d) data we observe: concentration in ER at a site surrounding the nucleus and differential trafficking of the ASIC1a subunit (for ASIC1a, under serum or insulin deprivation, but co-expressed ASIC2a is not impacted) to enhance channel activity (40). Regulated ER retention may thus underlie differential DEG/ENaC subunit expression across species.
NRA-2 is a resident ER protein that is most homologous to human nicalin and more distantly related to the nicastrin subunit of the mammalian γ-secretase complex. Nicastrin can promote maturation and trafficking of other proteins in the γ-secretase complex and has been suggested to act as a nucleation center to regulate inclusion of specific components of the complex (42). Mammalian nicalin works in a complex with NOMO (43) and TMEM147 (44) and can modulate Nodal/TGFβ growth factor signaling in Zebrafish (43) via an unclear mechanism that does not appear to involve nicalin protease activity.
Human nicalin can partially rescue nra-2 defects in C. elegans muscle, and NOMO-homologous NRA-4 co-immunoprecipitations along with NRA-2 when AChR is targeted (16). Moreover, nra-4 mutants have similar defects to nra-2 mutants and to nra-2; nra-4 double mutants in C. elegans muscle, supporting roles in the same process and activity in the same complex. Considering structural and functional similarities along with our clear demonstration of NRA-2 influence on MEC-10(d) surface expression, we speculate that the mammalian Nicalin/NOMO complex might also influence DEG/ENaC surface expression (and possibly other channels). If so, this step might be considered for therapeutic modulation of DEG/ENaC channel activity in cystic fibrosis (45), renal disease (46), pain management, and possibly ischemic stroke (21).
Acknowledgments
We thank Dr. Donald Winkelmann for help with TIRF microscopy and instrument sharing, Jian Xue and Helen Ushakov for generating the Ex221 transgenic nra-2 rescue strain, and Dr. Barth Grant and Dr. Anne Norris for help with immunofluorescence experiments. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by National Institutes of Health Office of Research Infrastructure Programs (Grant P40 OD010440).
This work was supported by National Institutes of Health Grant R01 NS034435 and the New Jersey Commission on Spinal Cord Injury Research (to M. D.) and by American Cancer Society Grant RGS-09-043-01-DDC and National Institutes of Health Grant RO1 NS070969 (to L. B.).
- TRN
- touch receptor neuron
- ER
- endoplasmic reticulum
- DEG/ENaC
- degenerin/epithelial Na+ channel
- TIRF
- total internal reflection fluorescence
- TIRFM
- TIRF microscopy
- AChR
- acetylcholine receptor.
REFERENCES
- 1. McCall K. (2010) Genetic control of necrosis – another type of programmed cell death. Curr. Opin. Cell Biol. 22, 882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Driscoll M., Gerstbrein B. (2003) Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat. Rev. Genet. 4, 181–194 [DOI] [PubMed] [Google Scholar]
- 3. Huang M., Chalfie M. (1994) Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature 367, 467–470 [DOI] [PubMed] [Google Scholar]
- 4. Driscoll M., Chalfie M. (1991) The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349, 588–593 [DOI] [PubMed] [Google Scholar]
- 5. Shreffler W., Magardino T., Shekdar K., Wolinsky E. (1995) The unc-8 and sup-40 genes regulate ion channel function in Caenorhabditis elegans motorneurons. Genetics 139, 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chelur D. S., Ernstrom G. G., Goodman M. B., Yao C. A., Chen L., O'Hagan R., Chalfie M. (2002) The mechanosensory protein MEC-6 is a subunit of the C. elegans touch-cell degenerin channel. Nature 420, 669–673 [DOI] [PubMed] [Google Scholar]
- 7. Bianchi L., Gerstbrein B., Frøkjaer-Jensen C., Royal D. C., Mukherjee G., Royal M. A., Xue J., Schafer W. R., Driscoll M. (2004) The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat. Neurosci. 7, 1337–1344 [DOI] [PubMed] [Google Scholar]
- 8. Goodman M. B., Ernstrom G. G., Chelur D. S., O'Hagan R., Yao C. A., Chalfie M. (2002) MEC-2 regulates C. elegans DEG/ENaC channels needed for mechanosensation. Nature 415, 1039–1042 [DOI] [PubMed] [Google Scholar]
- 9. Xu K., Tavernarakis N., Driscoll M. (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca2+ release from the endoplasmic reticulum. Neuron 31, 957–971 [DOI] [PubMed] [Google Scholar]
- 10. Syntichaki P., Xu K., Driscoll M., Tavernarakis N. (2002) Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419, 939–944 [DOI] [PubMed] [Google Scholar]
- 11. Luke C. J., Pak S. C., Askew Y. S., Naviglia T. L., Askew D. J., Nobar S. M., Vetica A. C., Long O. S., Watkins S. C., Stolz D. B., Barstead R. J., Moulder G. L., Brömme D., Silverman G. A. (2007) An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 130, 1108–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chu X. P., Xiong Z. G. (2013) Acid-sensing ion channels in pathological conditions. Adv. Exp. Med. Biol. 961, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yermolaieva O., Leonard A. S., Schnizler M. K., Abboud F. M., Welsh M. J. (2004) Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U.S.A. 101, 6752–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiong Z. G., Zhu X. M., Chu X. P., Minami M., Hey J., Wei W. L., MacDonald J. F., Wemmie J. A., Price M. P., Welsh M. J., Simon R. P. (2004) Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698 [DOI] [PubMed] [Google Scholar]
- 15. Zhang W., Bianchi L., Lee W. H., Wang Y., Israel S., Driscoll M. (2008) Intersubunit interactions between mutant DEG/ENaCs induce synthetic neurotoxicity. Cell Death Differ. 15, 1794–1803 [DOI] [PubMed] [Google Scholar]
- 16. Almedom R. B., Liewald J. F., Hernando G., Schultheis C., Rayes D., Pan J., Schedletzky T., Hutter H., Bouzat C., Gottschalk A. (2009) An ER-resident membrane protein complex regulates nicotinic acetylcholine receptor subunit composition at the synapse. EMBO J. 28, 2636–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fire A. (1986) Integrative transformation of Caenorhabditis elegans. EMBO J. 5, 2673–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Christensen M., Estevez A., Yin X., Fox R., Morrison R., McDonnell M., Gleason C., Miller D. M., 3rd, Strange K. (2002) A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503–514 [DOI] [PubMed] [Google Scholar]
- 20. Sangaletti R., Bianchi L. (2013) A method for culturing embryonic C. elegans cells. J. Vis. Exp. 79, e50649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kweon H. J., Suh B. C. (2013) Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep. 46, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamashima T. (2004) Ca2+-dependent proteases in ischemic neuronal death: a conserved 'calpain-cathepsin cascade' from nematodes to primates. Cell Calcium 36, 285–293 [DOI] [PubMed] [Google Scholar]
- 23. Sendrowski K., Rusak M., Sobaniec P., Iłendo E., Dąbrowska M., Boćkowski L., Koput A., Sobaniec W. (2013) Study of the protective effect of calcium channel blockers against neuronal damage induced by glutamate in cultured hippocampal neurons. Pharmacol. Rep. 65, 730–736 [DOI] [PubMed] [Google Scholar]
- 24. Kamath R. S., Martinez-Campos M., Zipperlen P., Fraser A. G., Ahringer J. (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2, RESEARCH0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Calixto A., Chelur D., Topalidou I., Chen X., Chalfie M. (2010) Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gessmann R., Kourtis N., Petratos K., Tavernarakis N. (2010) Molecular modeling of mechanosensory ion channel structural and functional features. PLoS One 5, e12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Hagan R., Chalfie M., Goodman M. B. (2005) The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat. Neurosci. 8, 43–50 [DOI] [PubMed] [Google Scholar]
- 28. Chatzigeorgiou M., Grundy L., Kindt K. S., Lee W. H., Driscoll M., Schafer W. R. (2010) Spatial asymmetry in the mechanosensory phenotypes of the C. elegans DEG/ENaC gene mec-10. J. Neurophysiol. 104, 3334–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang S., Arnadottir J., Keller C., Caldwell G. A., Yao C. A., Chalfie M. (2004) MEC-2 is recruited to the putative mechanosensory complex in C. elegans touch receptor neurons through its stomatin-like domain. Curr. Biol. 14, 1888–1896 [DOI] [PubMed] [Google Scholar]
- 30. Hong K., Mano I., Driscoll M. (2000) In vivo structure-function analyses of Caenorhabditis elegans MEC-4, a candidate mechanosensory ion channel subunit. J. Neurosci. 20, 2575–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brown A. L., Fernandez-Illescas S. M., Liao Z., Goodman M. B. (2007) Gain-of-function mutations in the MEC-4 DEG/ENaC sensory mechanotransduction channel alter gating and drug blockade. J. Gen. Physiol. 129, 161–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bianchi L., Driscoll M. (2006) Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses. WormBook 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin-Fernandez M. L., Tynan C. J., Webb S. E. (2013) A 'pocket guide' to total internal reflection fluorescence. J. Microsc. 252, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown A. L., Liao Z., Goodman M. B. (2008) MEC-2 and MEC-6 in the Caenorhabditis elegans sensory mechanotransduction complex: auxiliary subunits that enable channel activity. J. Gen. Physiol. 131, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y., Matthewman C., Han L., Miller T., Miller D. M., 3rd, Bianchi L. (2013) Neurotoxic unc-8 mutants encode constitutively active DEG/ENaC channels that are blocked by divalent cations. J. Gen. Physiol. 142, 157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sedensky M. M., Siefker J. M., Koh J. Y., Miller D. M., 3rd, Morgan P. G. (2004) A stomatin and a degenerin interact in lipid rafts of the nervous system of Caenorhabditis elegans. Am. J. Physiol. Cell Physiol. 287, C468–C474 [DOI] [PubMed] [Google Scholar]
- 37. Eshcol J. O., Harding A. M., Hattori T., Costa V., Welsh M. J., Benson C. J. (2008) Acid-sensing ion channel 3 (ASIC3) cell surface expression is modulated by PSD-95 within lipid rafts. Am. J. Physiol. Cell Physiol. 295, C732–C739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huber T. B., Schermer B., Müller R. U., Höhne M., Bartram M., Calixto A., Hagmann H., Reinhardt C., Koos F., Kunzelmann K., Shirokova E., Krautwurst D., Harteneck C., Simons M., Pavenstädt H., Kerjaschki D., Thiele C., Walz G., Chalfie M., Benzing T. (2006) Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc. Natl. Acad. Sci. U.S.A. 103, 17079–17086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. García-Añoveros J., Samad T. A., Zuvela-Jelaska L., Woolf C. J., Corey D. P. (2001) Transport and localization of the DEG/ENaC ion channel BNaC1α to peripheral mechanosensory terminals of dorsal root ganglia neurons. J. Neurosci. 21, 2678–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chai S., Li M., Branigan D., Xiong Z. G., Simon R. P. (2010) Activation of acid-sensing ion channel 1a (ASIC1a) by surface trafficking. J. Biol. Chem. 285, 13002–13011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gottschalk A., Almedom R. B., Schedletzky T., Anderson S. D., Yates J. R., 3rd, Schafer W. R. (2005) Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis elegans. EMBO J. 24, 2566–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shah S., Lee S. F., Tabuchi K., Hao Y. H., Yu C., LaPlant Q., Ball H., Dann C. E., 3rd, Südhof T., Yu G. (2005) Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447 [DOI] [PubMed] [Google Scholar]
- 43. Haffner C., Frauli M., Topp S., Irmler M., Hofmann K., Regula J. T., Bally-Cuif L., Haass C. (2004) Nicalin and its binding partner Nomo are novel Nodal signaling antagonists. EMBO J. 23, 3041–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dettmer U., Kuhn P. H., Abou-Ajram C., Lichtenthaler S. F., Krüger M., Kremmer E., Haass C., Haffner C. (2010) Transmembrane protein 147 (TMEM147) is a novel component of the Nicalin-NOMO protein complex. J. Biol. Chem. 285, 26174–26181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hobbs C. A., Da Tan C., Tarran R. (2013) Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J. Physiol. 591, 4377–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alvarez de la Rosa D., Navarro-González J. F., Giraldez T. (2013) ENaC modulators and renal disease. Curr. Mol. Pharmacol. 6, 35–43 [DOI] [PubMed] [Google Scholar]
- 47. Zhang W. (2008) Molecular and Genetic Dissection of Neuronal Necrotic-like death in Caenorhabditis elegans. Ph.D. thesis, Rutgers University, Piscataway, NJ [Google Scholar]