

Abstract
We describe a new rapid and efficient polymerase chain reaction (PCR)-based site-directed mutagenesis method. This procedure is effective with any plasmid and it employs four oligonucleotide primers. One primer contains the desired mutation, the second is oriented in the opposite direction (one of these two primers should be phosphorylated), and the third and fourth should be coding in complementary fashion for a unique restriction site to be introduced in a nonessential region. The method consists of two simultaneous PCR reactions; the PCR products are digested with the enzyme that recognizes the newly introduced unique restriction site and then ligased and used to transform competent bacteria. Additionally, the use of Dpn I facilitates the elimination of template DNA. The newly introduced restriction site is essential for ligation in the correct orientation of the two-PCR products and is further used for mutant screening. Resulting plasmids carry both the new restriction site and the desired mutation. Using this method, more than 20 mutants have already been generated (using two different kinds of templates); all these mutants were sequenced for the desired mutation and transfected into AtT-20 cells and the expressed mutant proteins encoded by the vector were assayed.
INTRODUCTION
Site-directed mutagenesis is an important procedure in studies of gene expression and protein structure-function relationships. Several protocols have been developed to mutate specific sequences in plasmid DNA. All employ oligonucleotide primers containing the desired mutations flanked by bases complementary to target sequences [1]. Primers are extended in vitro, although some procedures have described in vivo extension after transformation of special host bacteria [2].
Polymerase chain reaction (PCR) is a method of amplifying a segment of DNA; the advent of PCR has allowed the introduction of site-specific mutations into DNA by the incorporation of the desired mutations into the oligonucleotide primers. It has become possible to amplify longer sequences (up to 6 kilo base (kb)), and the use of different thermostable DNA polymerases with 3′ → 5′ exonuclease “proofreading” activity allowed to reduce to very few unwanted mutations. Numerous PCR-based methods of site-directed mutagenesis have been developed such as the generation of a mutant fragment, which has to be subcloned in the same vector used as a template [3, 4]. The overlap extension PCR method, that requires a large number of primers [5], and the megaprimer one that uses several rounds of PCR [6, 7] have been reported. These require a well-designed strategy taking into account the length of the mutated fragment, the presence of (two) unique cleavage sites, and the purification of PCR intermediate products used as templates for further PCR reaction rounds (for review see [8]).
In this paper, we describe a new method that allows the rapid generation of desired mutant with a high efficiency once the gene of interest has been cloned in a vector. The method consists of two simultaneous PCR reactions. The resulting products possess a new unique restriction site on one end and one of them is phosphorylated on the other end. We have used this methodology to amplify two halves of the pLNCX2 vector containing human prorenin or prosomatostatin. We have generated more than 20 mutants with different types of mutations: single-point mutations, deletions, or insertions. All the performed mutants were sequenced for the desired mutation in the corresponding insert. Subsequently, they were stably transfected in AtT-20 cells and the expressed mutant proteins encoded by the vector were assayed.
Figure 1.
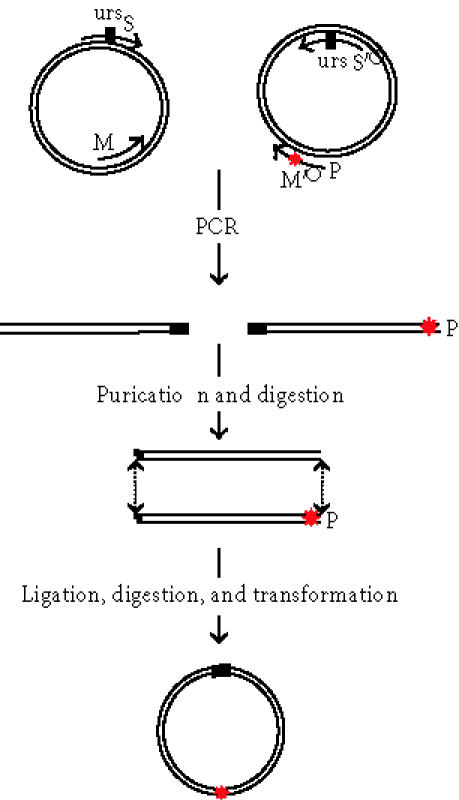
Schematic representation of site-directed mutagenesis strategy: primer M′ contains the desired mutation∗, primer M oriented in the opposite direction is phosphorilated, and the primers S and S′ are complementary and code for newly introduced unique restriction site BamH I (urs). The two-PCR products are digested with BamH I and ligased. After Dpn I digestion, the circular DNA carry the desired mutation.
MATERIALS AND METHODS
This method consists in amplifying by PCR the two halves of a plasmid using four primers, of which one or two contain the desired mutation(s) and two are complementary coding for an additional unique restriction site (Figure 1). The gene of interest (either prosomatostatin or prorenin) was cloned into pLNCX2 vector in Xho I site (Clontech, Basel, Switzerland) according to standard procedures. Then, two pairs of primers (Mx/S versus M′x/S′) were designed in order to amplify either the “right” or the “left” half of the plasmid.
M1: 5′P-GGTGCCATAGCCGGGTT3′,
M2: 5′P-GGGAGCGCCGGTGACACAGCC3′,
M3: 5′P-TCTCTGCAGCTCAAGCCTCAT3′,
M′1: 5′CCGAGAAAAGAAAGCTGGCTGCAAG3′,
M′2: 5′GCTGCTGCCGCGGGGAAGCAG3′,
M′3: 5′AGGCCTCCAGGTTTTTCACCTTTCCGATGATCTGCTAACTCAAACCC3′
S: 5′GTCTTCAAGGATCCATACCAG3′
S′: 5′CTGGTATGGATCCTTGAAGAC3′.
A mutagenic phosophorylated primer M′, complementary to the insert, carries the desired mutation and is coupled to S′. A primer “back-to-back” M of the mutagenic primer is coupled to the second selection primer S; this one could carry an additional mutation if desired. The 5′ ends of the two mutagenic primers meet, but do not overlap.
The “selection” primers called S and S′ are designed to change a nonessential restriction site EcoR I of the plasmid to BamH I, a restriction site that does not exist neither in the vector nor in the insert. PCR reactions are then performed with a proofreading thermostable DNA polymerase to avoid unwanted mutations. DNA polymerase (Pfu Turbo or Pfu) was purchased from Stratagene (Europe) or from Promega (Wallisellen, Switzerland). The primers M1/M′1, M2/M′2, and M3/M′3 were used to perform single mutation, deletion, and insertion, respectively.
The reaction mixture (100 μL) contains 100 ng of the native plasmid, 20 pmol of each primer, 10 nM of each dNTP (Roche, Rotkreuz, Switzerland), and 3 U of the Pfu polymerase (Stratagene, Europe or Promega, Wallisellen, Switzerland) in 1x Pfu buffer. This mixture is then subjected to a thermal cycling on a thermalcycler (Tgradient from Biometra, Switzerland) at 95°C for 30 second for denaturation, followed by 30 second for annealing (the temperature depends on the pair of primers) and then for 6 minutes at 72°C for extension. A total of 12 cycles was used and the procedure was ended by 10 minutes extension at 72°C. The reaction products are analyzed directly by electrophoresis in a 1% agarose gel. Complementary PCR fragments were copurified on Microcon PCR (Amicon®, Millipore, Bedford, Mass, USA) and eluted in 20 μL of ultrapure H2O. Ten μL are then digested for 2 hours at 37°C by BamH I; this reaction is purified by a passage through Microcon-PCR centrifugal filter device and eluted into 20 μL of ultrapure H2O. An aliquot of 4 μL is incubated with T4 DNA ligase (2.5 μL of 10x ligase buffer, 2.5 μL (7.5 U) of enzyme, and 16 μL of H2O) over night at room temperature (2 hour incubation at 37°C could be sufficient but for practical reasons over night incubation was used). The ligation reaction is then digested for 2 hours at 37°C with Dpn I (Promega) (to the 25 μL of ligase reaction, we added 8 μL of 10x buffer, 45 μL of H2O, and 2 μL (20 U) of enzyme) in order to eliminate the wild-type template. Ten μL of this mixture is used to transform XL1-blue E. Coli bacteria. For transformation assay, competent cells (100 μL) were incubated with 10 μL of reaction mixture for 30 minutes at 4°C, the cells were heat shocked for 45 second at 42°C. The transformation mixture was diluted four-fold with SOC medium and grown for one hour at 37°C. The transformed bacteria are plated on LB 80 μg/mL ampicillin-containing plates and grown over night at 37°C.
RESULTS
The four PCR primers S, S′, M, and M′ (in Figure 1) were used in one round of two independent PCR. Primer M′ had the desired mutation. The segment α was amplified in one PCR reaction using primers S (containing the unique restriction site) and M in the presence of template DNA pLNCX2 containing prosomatostatin or prorenin. In order to avoid possible PCR-induced errors, only 12 cycles of PCR are performed using rather large amounts of template DNA (100 ng). Primers S′ and M′ were used to amplify the fragment β. The PCR products were analyzed on agarose gel and yielded the expected DNA molecular weight, 3 kb for fragment α and 3.5 kb for fragment β of the pLNCX2-prosomatostatin vector (Figure 2, lanes 1 and 2). The fragments α and β exhibit the same unique restriction site (BamH I), at the 5′ end for α and at the 3′ end for β, which in addition is phosphorylated at its 5′ end. These two introduced modifications after digestion with BamH I allowed a correct segment orientation for the vector construction carrying the desired mutation with the newly introduced restriction site. Figure 2 (lane 3) demonstrates an efficient ligation with almost all PCR-generated DNA conversion into larger molecular weight DNA. The remaining template DNA was eliminated by Dpn I digestion (Figure 2, Lane 4).
Figure 2.

PCR products generated from pLNCX2-SOM and their ligation followed with Dpn I treatment. Lane 1: fragment α, lane 2: fragment β, lane 3: fragment α and β ligased, lane 4: ligation and Dpn I treatment, and lane 5: marker X (Roche diagnostics, Mannheim, Germany). DNA samples were fractionated in 1% agarose gel and stained with ethidium bromide.
After DNA bacterial transformation, ampicillin-resistant individual colonies were grown over night at 37°C in LB broth containing (80 μg/mL) ampicillin. Plasmid DNA was prepared by alkaline lysis using conventional methods and treated with BamH I or EcoR I for mutants screening (Figure 3). Because the BamH I site replaced one of the two pLNCX2 EcoR I sites, digestion by BamH I or EcoR I linearized the obtained mutants (instead of generation of two bands in the wild type). BamH I-resistant colonies represent residual DNA template after Dpn I treatment, which is confirmed by EcoR I treatment (Figure 3).
Figure 3.

Minipreps analysis for mutant screening: clone 4 generates a 1.6-kb DNA fragment with EcoR I and is resistant to BamH I digestion. It may represent residual DNA after Dpn I digestion. Lane5: marker X (Roche diagnostics, Mannheim, Germany). DNA samples were fractionated in 1% agarose gel, stained with ethidium bromide.
Larger amount of DNA of one of these mutants was prepared by Midipreps (Wizard plus, Promega, Wallisellen, Switzerland), the obtained DNA was sequenced to confirm the presence of desired mutations and to exclude the possible unwanted mutations. The DNA was then transfected into AtT20 cells. With this protocol, we have generated 20 mutations on two different inserts cloned into pLNCX2 (Clontech). Within a few weeks, 10 mutations were successfully produced on prosomatostatin (7 single-point mutations, 1 insertion, and 2 deletions); 7 mutations used the prosomatostatin wild type as a template and 3 used one of the first mutants as a template. For prosomatostatin primers, M1 and M′1 were used to perform a single mutation (Arg → Lys), M2 and M′2 to delete 13 amino acids in proregion, and M3 and M′3 allowed the insertion of bradykinin sequence at C-terminus region to substitute somatostatin-28 coding sequence (Figures 4 and 5). Within the same time frame, another 12 mutations on prorenin (11 were single-point mutations and one deletion) were carried out; 7 used prorenin wild type as a template and 5 used again these first mutants as templates. For the second set of mutants, the unique restriction site Mlu I was used.
Figure 4.
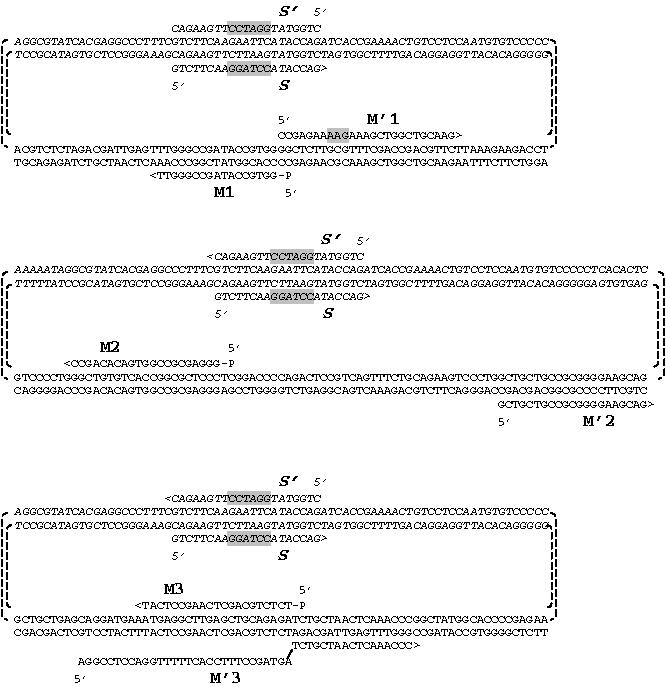
Schematic representation of the used vector and primers design to perform the three different kinds of mutations. M1/M′1 for single mutation, M2/M′2 for deletion, M3/M′3 for insertion, and S1 and S′1 are the mutagenesis common primers.
Figure 5.


Example of successful deletion and insertion. A: 13 residues were deleted from the N-terminus of human somatostatin cDNA. B: 9 residues coding for bradykinin and a stop codon (30 nucleotides) were introduced at the C-terminus of somatostatin precursor.
DISCUSSION
This new mutagenesis method allows performing mutations more efficiently in less time than with traditional methods. Moreover, it is possible to achieve many types of mutation, for example, insertions, single-point mutations, and deletions, depending on the design of the primers. Figure 5 shows an example of deletion and insertion created in human prosomatostatin cDNA. In addition, the first generated mutant can be used as a template for a second round of mutations as long as it is possible to introduce an additional unique restriction site that is required for a correct vector reconstruction and mutant screening. Indeed, this can even be taken further by using such a second mutant as a template for a third round of mutation, and in that case, the first primers (S and S′) with the unique restriction site BamH I could be reused.
The method is rapid and can generate an abundance of mutated segments; once the PCR is carried out, the following steps are straightforward manipulations. This method is as efficient as the method that uses a single inverted PCR with two primers to generate a full-length mutant plasmid [5, 6]. In addition, the present method has several other advantages. It allows the introduction of multiple-distant mutations (up to 55 bp, depending on the maximum length of the primers), as long as the 5′ ends of the two mutagenic primers (M and M′) meet and as one or both primers carry the desired mutations. Alternatively, since the 5′ end of the M′ and the 3′ end of the complementary sequence of M (Figures 1 and 4) are designed to meet, any length of nucleotide sequence in between is deleted. The size of the deletion region depends exclusively on the design of the primers used on the vector DNA sequence Figure 4. Furthermore, insertions can also be made, theoretically, without limit in the size; the M′ primer has to be designed that its 5′ end codes for the sequence to be inserted and its 3′ end is complementary to the successive sequence. We managed to insert 30 nucleotides with this method (Figure 5B), but we did not use any larger sequence.
Another important potential advantage of this method is the rapid construction of chimera proteins. The corresponding cDNA of the two different proteins should be cloned in the same vector (or two very similar vectors). One PCR performed on the first plasmid will amplify the 5′ end of the first cDNA, and the second on the second plasmid will amplify the 3′ end of the other cDNA. As for other types of mutation, the primers should be designed in order to keep the right reading frame. It is also possible to combine the different mutatgenic reactions and carry out mutated chimeric proteins.
A straightforward success of this method requires that the PCR fragment that contains the bacterial antibiotic resistance and plasmid replication region (fragment β) should not be phosphorylated. Otherwise, residual PCR products that are not completely digested (by BamH I in our case) can close up during the ligation reaction and transform bacteria, resulting in the presence of some clones on the LB Agar Petri, which contain a short vector constituted only by a half of the plasmid.
In conclusion, we have developed a new, highly efficient PCR-based site-directed mutagenesis method, which allows for single mutation as well as double or multiple combined mutations including single-point mutation, deletion, and insertion.
References
- Smith M. In vitro mutagenesis. Annu Rev Genet. 1985;19:423–462. doi: 10.1146/annurev.ge.19.120185.002231. [DOI] [PubMed] [Google Scholar]
- Kunkel T A, Roberts J D, Zakour R A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki R K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16(15):7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Ge L, Rudolph P. Simultaneous introduction of multiple mutations using overlap extension PCR. Biotechniques. 1997;22(1):28–30. doi: 10.2144/97221bm03. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Sommer S S. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8(4):404–407. [PubMed] [Google Scholar]
- Angelaccio S, Bonaccorsi di Patti M C. Site-directed mutagenesis by the megaprimer PCR method: variations on a theme for simultaneous introduction of multiple mutations. Anal Biochem. 2002;306(2):346–349. doi: 10.1006/abio.2002.5689. [DOI] [PubMed] [Google Scholar]
- Ling M M, Robinson B H. Approaches to DNA mutagenesis: an overview. Anal Biochem. 1997;254(2):157–178. doi: 10.1006/abio.1997.2428. [DOI] [PubMed] [Google Scholar]