

Background: FSP27 depletion increases both basal and stimulated lipolysis.
Results: FSP27 interacts with ATGL via amino acids 120–220 to regulate lipolysis and triglyceride storage in human adipocytes.
Conclusion: FSP27 inhibits ATGL-mediated lipolysis and protects adipocytes against free fatty acid-impaired insulin signaling.
Significance: The novel lipolytic regulation shown here may lead to new treatments for insulin resistance.
Keywords: Adipocyte, Adipose Tissue Metabolism, Adipose Triglyceride Lipase, Diabetes, Diacylglycerol, Fatty Acid, Fatty Acid Metabolism, Insulin Resistance, Obesity
Abstract
In adipocytes, lipolysis is a highly regulated process involving hormonal signals, lipid droplet-associated proteins, and lipases. The discovery of new lipid droplet-associated proteins added complexity to the current model of lipolysis. In this study, we used cultured human adipocytes to demonstrate that fat-specific protein 27 (FSP27), an abundantly expressed protein in adipocytes, regulates both basal and stimulated lipolysis by interacting with adipose triglyceride lipase (ATGL, also called desnutrin or PNPLA2). We identified a core domain of FSP27, amino acids 120–220, that interacts with ATGL to inhibit its lipolytic function and promote triglyceride storage. We also defined the role of FSP27 in free fatty acid-induced insulin resistance in adipocytes. FSP27 depletion in human adipocytes increased lipolysis and inhibited insulin signaling by decreasing AKT phosphorylation. However, reducing lipolysis by either depletion of ATGL or expression of exogenous full-length FSP27 or amino acids 120–220 protected human adipocytes against the adverse effects of free fatty acids on insulin signaling. In embryonic fibroblasts derived from ATGL KO mice, exogenous free fatty acids did not affect insulin sensitivity. Our results demonstrate a crucial role for FSP27-ATGL interactions in regulating lipolysis, triglyceride accumulation, and insulin signaling in human adipocytes.
Introduction
The prevalence of lipotoxicity as a pathogenic mechanism in insulin resistance and type 2 diabetes has sharpened the research focus on lipolysis. Lipolysis is a catabolic branch of triglyceride (TG)3 metabolism that provides fatty acids (FAs) in times of metabolic need. FAs are important substrates for energy production and lipid synthesis. Despite their fundamental physiological importance, an oversupply of FAs is highly detrimental. High concentrations of circulating FAs and triglycerides, observed in both obesity and lipodystrophy, are thought to cause muscle insulin resistance and decreased glucose tolerance (1–5). Therefore, lipolysis plays a very important role in human health and disease (6, 7). Higher organisms store FAs in adipose tissue, a specialized organ that supplies FAs to other high-demand tissues such as liver and muscle. Adipose tissue regulates the balance of FA esterification and lipolysis, thus playing a central role in regulating whole-body metabolism and glucose homeostasis (8).
According to the current model of lipolysis, complete hydrolysis of TGs to FAs and glycerol requires three consecutive steps that involve different enzymes: adipose triglyceride lipase (ATGL, also called desnutrin and PNPLA2), hormone-sensitive lipase, and monoacylglycerol lipase. ATGL is the rate-limiting enzyme for lipolysis in adipocytes, catalyzing the first step of hydrolysis of TG to diacylglycerol and FA (6, 9–12). Although several proteins are known to be involved in the lipolytic machinery, the complex process and the regulation of lipolysis are not fully understood.
Under basal condition in adipocytes, ATGL is mostly localized to endoplasmic reticulum-related membranes in the cytoplasm (13). However, a small fraction of ATGL is complexed with its inhibitor, G0/G1 switch gene 2 (G0S2), on the lipid droplet (LD) (14). Perilipin 1 (PLIN1) prevents the access of a necessary activator, comparative gene identification 58 (CGI-58), to ATGL, thus decreasing lipolysis (6, 15–17). In times of energy demand, β-adrenergic receptor stimulation activates PKA to phosphorylate PLIN1 at multiple sites and release CGI-58. Hormone-sensitive lipase is also phosphorylated and translocates to lipid droplets with PLIN1 acting as a scaffold. Unbound ATGL moves to lipid droplets, where it binds and is stimulated by CGI-58 to acutely increase lipolysis (15, 18). Prolonged β-adrenergic stimulation subsequently down-regulates G0S2 protein expression, thereby releasing more ATGL to sustain lipolysis (14).
Fat-specific protein 27 (FSP27) is a differentiation-regulated protein in adipocytes (19–26). Its expression in various cell types markedly enhances TG deposition and LD size, whereas its depletion causes LD fragmentation and increases TG hydrolysis in adipocytes in vitro and in vivo (25, 27–30). We and others (31, 32) showed that FSP27 interacts with PLIN1 in human adipocytes to promote the formation of large LDs. FSP27 KO mice have a higher mitochondrial oxidative metabolism (25, 27, 30, 33–39), and their white adipocytes show multilocular droplets (25, 30) and increased lipolysis. This increase in energy expenditure protects mice from diet-induced obesity and insulin resistance. Whether this is a direct effect of FSP27 deficiency or a secondary effect of enhanced lipolysis needs to be elucidated. In contrast, a naturally occurring human homozygous nonsense mutation, FSP27 E186X, is associated with partial lipodystrophy, hypertriglyceridemia, and insulin-resistant diabetes (40). We demonstrated that FSP27 expression in omental fat positively correlates with insulin sensitivity in obese humans (26). Together, these results show that FSP27 has opposite effects on insulin sensitivity in mice and humans. Therefore, in this study, we used cultured human primary adipocytes, which provide an excellent model system to study the underlying physiological and biological processes in human adipose biology and their relationship to human metabolic disease.
The objective of this study was to define the role of FSP27 in lipolysis. We hypothesized that FSP27 would regulate ATGL-mediated lipolysis and by doing so it promotes storage of circulating FFAs into adipocytes, thus protecting against FFA-mediated insulin resistance (41–44). We demonstrated that FSP27 directly interacted with ATGL; that FSP27 promoted TG accumulation via its effect on ATGL-mediated lipolysis; that the domain of FSP27, aa 120–220, is necessary and sufficient to interact with ATGL and accumulate TGs; that FSP27 protected human adipocytes from FFA-mediated insulin resistance; and that, as expected, adipocytes derived from ATGL KO mouse embryonic fibroblasts (MEFs) were resistant to exogenous FFA-induced insulin resistance. Taken together, these results support a model in which FSP27 interacts with ATGL to regulate lipolysis and TG accumulation and to protect from FFA-induced insulin resistance in human adipocytes.
EXPERIMENTAL PROCEDURES
Materials
LipidTOX-Deep Red, fetal bovine serum, and culture media were purchased from Invitrogen. Isoproterenol was from Sigma-Aldrich. Human FSP27 siRNA and ATGL siRNA was purchased from Dharmacon (Chicago, IL). p300 cDNA in pCMVβ was purchased from Upstate Biotechnology.
FSP27 Antibodies
FSP27 polyclonal antibodies were raised against full-length recombinant human FSP27 expressed as a TrpE fusion protein. Production of the FSP27-TrpE fusion protein used the Path11 expression system. Following SDS-PAGE, the FSP27-TrpE fusion protein band was excised from the gel and used as an antigen to produce polyclonal antibody in rabbits via contractual arrangement with ProSci Inc. (Poway, CA).
Cell Culture
Human primary preadipocytes, obtained from the Boston Nutrition Obesity Research Center Adipocyte Core, were differentiated into fully mature adipocytes (45). COS-7, 293T, and HeLa cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 50 μg/ml streptomycin, and 50 units/ml penicillin.
Lentivirus Production and Transduction
The day before transfection, ∼10 × 106 293T cells were plated in a 10-cm dish. Recombinant lentiviruses were produced by a five-plasmid transfection procedure as described previously (31, 46). Packaged recombinant lentiviruses were harvested from the cell culture supernatant 48 h after transfection and filtered through 0.45-μm filters. 500 μl of supernatant and 10 μg/ml Polybrene were added to each well of a 12-well plate containing differentiated human adipocytes. After overnight incubation, the medium was changed to a regular maintenance medium (45). Protein expression was observed after 4 days, as we described previously (31).
Adenovirus Transduction
Human ATGL-CFP adenovirus was a gift from Carol Sztalryd and Dawie Gong (University of Maryland), and human FSP27-HA-tagged adenovirus was a gift from Andrew Greenberg (Tufts Medical Center). Virus was added at a multiplicity of infection of 100 to differentiated human adipocytes. Cells were harvested after 24 h, and protein expression was measured.
RNA Isolation and Quantitative Reverse Transcription PCR
Total RNA was isolated from human adipocytes using TRIzol reagent (Invitrogen). cDNA was synthesized according to the instructions of the manufacturer using oligo(dT) primers and avian myeloblastosis virus reverse transcriptase (Roche Diagnostics). Quantitative real-time PCR was performed in a LightCycler using a SYBR Green 1 PCR kit (Roche Diagnostics).
siRNA Transfection of Human Adipocytes
Human adipocytes were transfected with siRNA duplexes using Lipofectamine reagent and PLUS (Invitrogen) on day 9 of differentiation, as described previously (47).
Triglyceride Determination and Lipolysis Measurement
Cells were lysed with TG lysis buffer (0.1% SDS, 1 mm EDTA, and 100 mm Tris-HCl (pH 7.4)), and TG was measured with a TG determination kit (Sigma) according to the instructions of the manufacturer. Human adipocytes were incubated in Krebs-Ringer bicarbonate HEPES buffer supplemented with 4% BSA. The buffer was collected after 2 h to measure glycerol released as a measure of lipolysis. For β-adrenergic stimulation, cells were stimulated with 1 μm isoproterenol.
Immunoprecipitation and Western Blot Analysis
Protein A/G-Sepharose beads (50 μl) (Santa Cruz Biotechnology) were cross-linked to 7 μg of antibody against either ATGL (Thermo Scientific, catalog no. K.751.7), HA (Abcam, catalog no. ab9110, or FLAG (Sigma, catalog no. F1804) using the cross-linker disuccinimidyl suberate (Sigma) for 2 h on a topover rotator at room temperature. Cross-linking was quenched with 50 mm Tris-HCl for 15 min at room temperature. Beads with conjugated ATGL or HA antibodies were washed three times with lysis buffer. Fully differentiated human adipocytes were harvested in 1 ml of lysis buffer containing 20 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1 mm Na2 EDTA, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin (Cell Signaling Technology), phosphatase inhibitor mixture, and protease inhibitor mixture (Roche). Cleared lysates containing 1 mg of extracted protein were incubated with 50 μl of cross-linked ATGL, FLAG, or HA protein A/G-Sepharose beads or with protein A/G-Sepharose beads alone as a control overnight at 4 °C. The beads were rinsed five times with cold lysis buffer, and bound protein was released by boiling in 4× SDS loading buffer for 5 min at 95 °C. Equal volumes of immunoprecipitation sample or 15 μg of cell lysate protein were separated on 10% precast gels (Bio-Rad). Proteins were transferred to PVDF membranes at 50 V and 4 °C for 2 h. Membranes were blocked for 1 h in 5% nonfat milk in Tris-buffered saline-Tween at room temperature. Primary antibody was incubated overnight at 4 °C, and secondary antibody (horseradish peroxidase-coupled goat anti-rabbit (Santa Cruz Biotechnology, catalog no. sc-2030, 1:10,000) or mouse anti-rabbit IgG (Cell Signaling Technology, catalog no. L27A9, 1:6000)) were incubated for 1 h at room temperature. SuperSignal West Dura extended duration substrate (Thermo Scientific, catalog no. 34075) was used for detection. Pictures were taken with an LAS-4000 Fuji Imager.
Insulin Stimulation of Human Adipocytes and Immunoblot Analysis of AKT Phosphorylation
Adipocytes plated in 12-well culture plates were washed three times in prewarmed PBS and then incubated at 37 °C for 3h in Krebs-Ringer HEPES buffer containing 0.01% BSA and 5 mm glucose for insulin and serum starvation. They were incubated for 15 min at 37 °C with Krebs-Ringer HEPES buffer with or without glucose but with 1 nm insulin. Cells were carefully washed three times with ice-cold PBS and harvested in 200 μl of lysis buffer containing phosphatase and protease inhibitor mixtures (Roche). Protein concentration was measured with a Bradford assay (Pierce BCATM protein assay). Equal amounts of protein were separated on 10% polyacrylamide gels and transferred to PVDF membranes. Nonspecific binding was blocked for 1 h at room temperature with 5% nonfat dry milk in Tris-buffered saline-Tween. Membranes were incubated overnight at 4 °C with antibody directed against total AKT (1:1000, Cell Signaling Technology) or phospho-AKT (Ser-473) (1:1000, Cell Signaling Technology). Anti-β-tubulin antibody was used as a loading control (1:10,000, Sigma-Aldrich). Appropriate horseradish peroxidase-conjugated secondary antibodies were used at 1:10,000. Detection was performed as described previously (48).
RESULTS
FSP27 Depletion Increased Both Basal and Stimulated Lipolysis in Human Adipocytes
Studies in our laboratory and by others investigated the role of FSP27 in LD dynamics and lipolysis in murine 3T3-L1 adipocytes (25, 28, 29, 48, 49) and showed that FSP27 knockdown increases both basal and stimulated lipolysis (28, 50). Because a discrepancy exists in the role of FSP27 (also called CIDEC, cell death-inducing DFFA-like effector c) in human compared with murine lipid metabolism (26, 40), we investigated the role of FSP27 in lipolysis in differentiated human adipocytes by FSP27 knockdown and measuring glycerol release in the culture media under both basal (no treatment) and stimulated (1 μm isoproterenol) conditions. Nine days after differentiation, human adipocytes were transiently transfected with siRNA directed against FSP27, and 5 days later (day 14 of differentiation), FSP27 mRNA and protein expression were measured. A 75% decrease in FSP27 mRNA (Fig. 1A) resulted in an 80% decrease in FSP27 protein expression (Fig. 1B). FSP27 depletion increased basal lipolysis by 20% and stimulated lipolysis by 40% (Fig. 1C).
FIGURE 1.

FSP27 depletion increased both basal and stimulated lipolysis in human adipocytes. Nonspecific scrambled (Scr) siRNA was used as a control in all experiments. A, relative mRNA levels in siRNA-transfected human adipocytes. B, immunoblot analysis and quantification of protein expression levels of FSP27 and β-tubulin (loading control) of siRNA-transfected human adipocytes. C, biochemical quantification of basal and stimulated lipolysis on the basis of measurement of glycerol release after 2 h. Data are mean ± S.E. *, p < 0.05; **, p < 0.001, n = 3 (unpaired Student's t test).
FSP27 Interacted with ATGL
ATGL is the rate-limiting hydrolase in human adipocyte lipolysis. Therefore, we investigated whether FSP27 interacts with ATGL to regulate its enzymatic activity. Mature human adipocytes were infected with FSP27-FLAG-HA lentivirus, and anti-FLAG antibodies were used to immunoprecipitate FSP27 protein. Interestingly, FSP27-FLAG-HA coimmunoprecipitated with endogenous ATGL in human adipocytes (Fig. 2A). In a separate experiment, FLAG expressed alone in adipocytes was pulled down specifically with FLAG antibodies. ATGL did not coimmunoprecipitate with FLAG (data not shown). This indicates that ATGL does not bind to the FLAG epitope. To confirm that endogenous proteins coimmunoprecipitate, we showed that ATGL-antibodies immunoprecipitated both ATGL and endogenous FSP27 (Fig. 2B), confirming that endogenous FSP27 and ATGL interact physically in human adipocytes. As a further confirmation, we coexpressed exogenous FSP27-HA and ATGL-CFP in 293T cells and used anti-HA antibody to coimmunoprecipitate FSP27-HA and ATGL-CFP (Fig. 2C). To show that the HA epitope does not pull down ATGL nonspecifically, we expressed ATGL-CFP and p300-HA in 293T cells. p300 (Upstate Biotechnology) is a transcription factor also known as EP300. HA antibody pulled down p300 but not ATGL (supplemental Fig. 1). These results clearly establish a direct physical interaction between FSP27 and ATGL.
FIGURE 2.
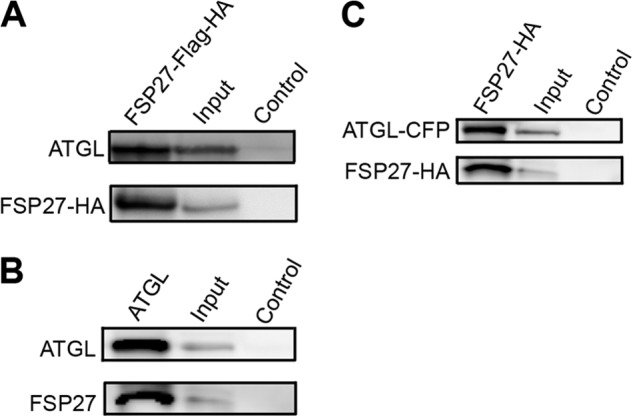
FSP27 coimmunoprecipitated with ATGL. A, in human adipocytes expressing FSP27-FLAG-HA, FSP27 was immunoprecipitated with anti-FLAG antibodies and immunoblotted with ATGL (top panel) or HA (bottom panel) antibody. B, in human adipocytes, ATGL antibodies were used as a bait to pull down endogenous ATGL, followed by immunoblotting with ATGL (top panel) or FSP27 antibody (bottom panel). C, FSP27-HA and ATGL-CFP were expressed in 293T cells using adenovirus. HA antibodies were used to immunoprecipitate FSP27-HA, followed by immunoblotting with ATGL (top panel) or FSP27 (bottom panel) antibodies. Input represents total cell lysates (15 μg), and control represents lysates from human adipocytes infected with control virus and pulled down with FLAG or HA antibody.
FSP27 Decreased ATGL-mediated Lipolysis
Because FSP27 interacts with ATGL and its depletion promotes basal and stimulated lipolysis in human adipocytes, we hypothesized that FSP27 regulates ATGL-mediated lipolysis. To investigate whether FSP27 affected ATGL hydrolase activity, we performed in vitro TG hydrolase assays (10, 51) in which FSP27 was added to cell lysates containing both ATGL and CGI-58. FSP27 did not affect ATGL- or CGI-58-stimulated ATGL hydrolase activity (data not shown). Another recent study complemented these results (50). These findings suggest that an intact cellular machinery might be required for FSP27 to inhibit ATGL-mediated lipolysis. Therefore, we tested whether expression of exogenous FSP27 affected ATGL-mediated lipolysis in cultured human adipocytes. Cells were infected with adenoviral preparations encoding either FSP27 or ATGL or both, and glycerol release in the media was measured. A 2- to 3-fold increase in overall FSP27 expression was observed with adenovirus (supplemental Fig. 2), similar to the effect shown previously with lentivirus (47). As expected, FSP27 expression decreased basal lipolysis by 65% and stimulated lipolysis by 35%, whereas ATGL overexpression increased basal lipolysis by 50% and stimulated lipolysis by 30% (Fig. 3A). Interestingly, coexpression of FSP27 and ATGL significantly suppressed the increase in lipolysis because of ATGL overexpression (Fig. 3A). These results show that FSP27 negatively regulates ATGL-mediated lipolysis in human adipocytes. Because ATGL and FSP27 contained twice as much virus (multiplicity of infection of 200), which could affect the lipolytic rate, we tested the effect of viral load on glycerol released by human adipocytes. After 2.5 h under basal conditions, glycerol release was 0.223 ± 0.04 nmol/μg protein at a multiplicity of infection of 100 and 0.229 ± 0.03 nmol/μg protein at a multiplicity of infection of 200. This indicates that, under our experimental conditions, the viral load did not affect the lipolytic rate in human adipocytes.
FIGURE 3.
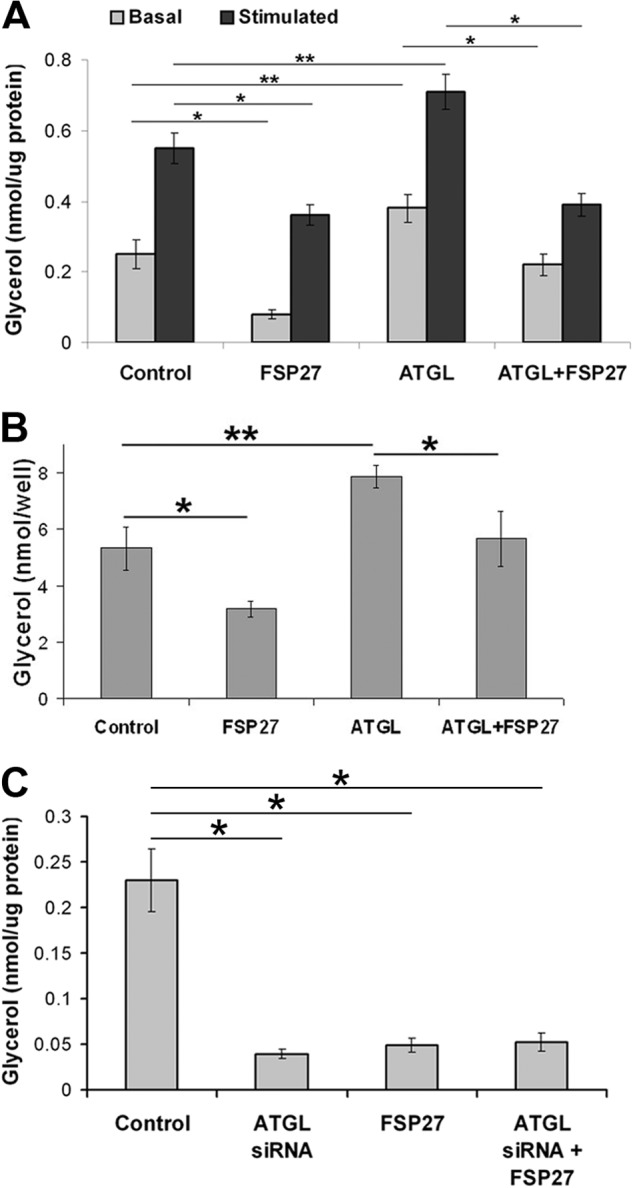
FSP27 expression decreased ATGL-mediated lipolysis. Glycerol released in cell culture media from human adipocytes expressing EGFP, FSP27-HA and/or ATGL (A) and COS-7 cells, measured per well containing an equal number of cells. A and B, control cells were infected with EGFP-containing empty virus. C, human adipocytes after ATGL depletion and/or FSP27-HA expression. A and C, glycerol released in 2.5 h was measured and normalized to total protein. Data are mean ± S.E. *, p < 0.001; **, p < 0.05; n = 3 (unpaired Student's t test).
Next, we asked whether FSP27-mediated suppression of ATGL activity required other factors or proteins present solely in primary human adipocytes. We used COS-7 cells that express very low levels of ATGL and do not express endogenous FSP27. COS-7 cells were infected with either EGFP (control) or FSP27-GFP and/or ATGL-CFP adenovirus. As in human adipocytes (Fig. 3A), lipolysis in COS-7 cells was reduced by FSP27 and induced by ATGL (Fig. 3B). Cells coexpressing FSP27 and ATGL did not show increased lipolysis compared with control cells (Fig. 3B), reaffirming that FSP27 controls ATGL activity to regulate lipolysis independently of adipocyte-specific factors or proteins.
To further confirm that FSP27-mediated reduced lipolysis is due solely to its effect on ATGL activity, the effect of FSP27 was studied on lipolysis in the absence or presence of ATGL. siRNA-mediated ATGL depletion or FSP27 expression in human adipocytes reduced glycerol release in the media by about 80% (Fig. 3C). Expression of FSP27 in ATGL-depleted cells had no additional effect on glycerol release, suggesting that FSP27-mediated decrease in lipolysis is dependent upon its effect on ATGL lipolytic activity. Alternatively, it is possible that glycerol release after ATGL depletion alone is so low that it cannot be reduced further or cannot be detected.
FSP27-mediated TG Accumulation Was ATGL-dependent
Given the strong evidence supporting the role of FSP27 in TG storage (23, 25, 26, 28, 29, 40, 48, 49), we hypothesized that FSP27 increases TG levels by decreasing lipolysis via suppression of ATGL activity. We tested this hypothesis by depleting ATGL from cultured human adipocytes with siRNA and expressing FSP27 by adenoviral infection for 48 h. Cells that expressed HA-FSP27 accumulated significantly more TG than control cells expressing EGFP (Fig. 4A). GFP-FSP27 increased TG levels as much as HA-FSP27 did (data not shown). As expected, ATGL knockdown increased cellular TG levels by about 45%. In ATGL-depleted cells, FSP27 increased TG further but only to the same level as in control cells (Fig. 4A). Hence, the effects of ATGL suppression and FSP27 expression on TG accumulation were not additive. These results suggest that FSP27-induced TG accumulation is predominantly ATGL-dependent.
FIGURE 4.
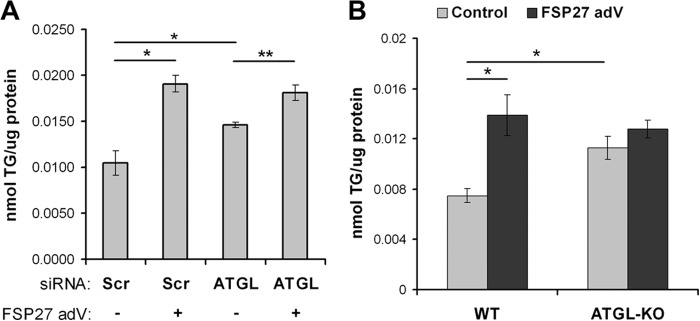
FSP27-mediated TG accumulation was ATGL-dependent. Biochemical quantification of total triglycerides in human adipocytes treated with scrambled siRNA (Scr, control) (A) or siRNA against ATGL in the presence or absence of adenoviral expression of FSP27-HA or EGFP (as a negative control) and adipocytes differentiated from WT or ATGL-KO MEFs (B). FSP27-HA or EGFP (control) were expressed using adenovirus (adV). Total triglyceride concentration was normalized to total protein. Data are mean ± S.E. (paired Student's t test). *p < 0.001, and **p < 0.05, n = 3.
That FSP27 expression still promoted TG accumulation in ATGL-depleted cells may have resulted from incomplete ATGL suppression (only about 80%). Therefore, to test whether FSP27-mediated TG accumulation is completely dependent on ATGL, we compared MEFs from WT and ATGL KO mice. MEFs were cultured and differentiated essentially as described previously (52). As expected, FSP27 expression in adipocytes differentiated from WT MEF increased cellular TG content. However, adipocytes derived from ATGL KO MEFs showed more than 50% higher TG levels than WT cells (Fig. 4B). Interestingly, FSP27 expression had no significant effect on TG levels in ATGL KO-derived adipocytes (Fig. 4B). These data strongly suggest that FSP27-mediated TG accumulation is dependent on inhibiting ATGL-mediated lipolysis.
Identification of the TG Accumulatory Domain of FSP27
Previously, we identified functional domains of FSP27 associated with LD clustering (aa 173–220) and fusion/enlargement (aa 120–210) (48). We analyzed the role of these domains in TG accumulation in COS-7 cells. Full-length FSP27 expression increased total TG content by 50%, but neither the aa 173–220 nor aa 120–210 domains increased TG levels (Fig. 5A). Therefore, to identify the domain of FSP27 associated with TG accumulation, we expressed the GFP-labeled N terminus (aa 1–120) and C terminus (aa 120–239) of FSP27 in COS-7 cells. The aa 1–120 sequence does not localize to LDs (48), and, in this study, it did not affect cellular TG levels (data not shown). However, expression of the aa 120–239 sequence caused TG accumulation equivalent to full-length FSP27 (Fig. 5A). These results show that at least part of the sequence aa 210–239 in the C terminus of FSP27 plays a role in TG accumulation. To identify the smallest domain of FSP27 sufficient to induce TG accumulation, we prepared EGFP-labeled constructs of aa 120–220 and aa 120–230. Their expression in COS-7 cells resulted in TG accumulation similar to that of full-length FSP27 (Fig. 5A), showing that FSP27 (120–220) is sufficient to induce TG accumulation.
FIGURE 5.
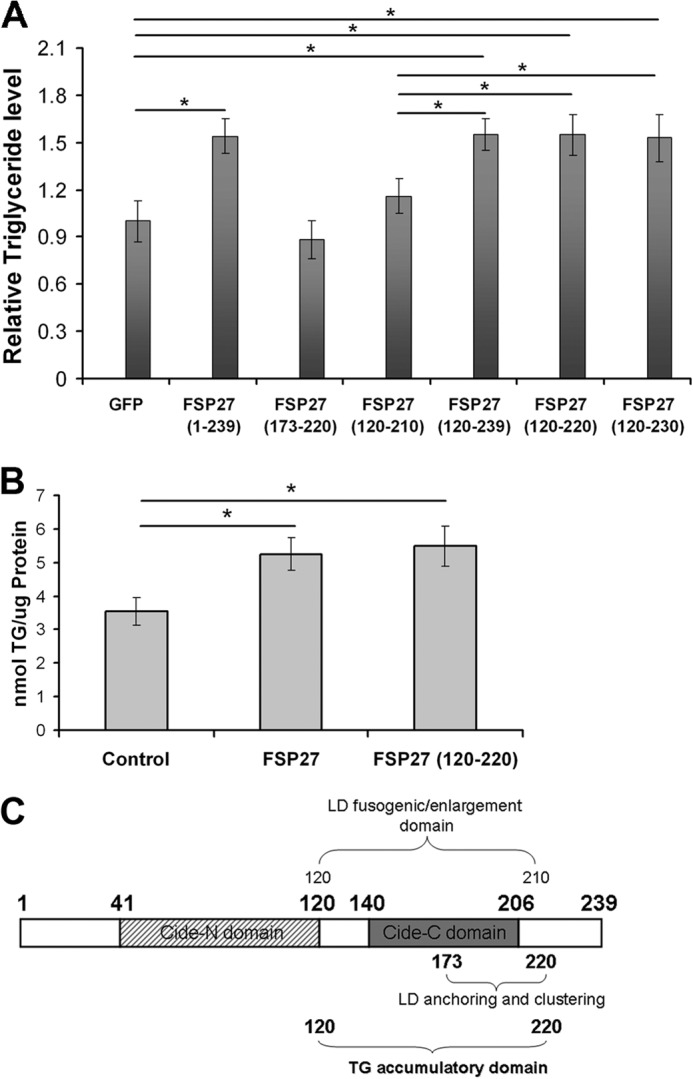
Identification of the TG accumulatory domain of FSP27. A, deletion mutants of FSP27 fused to EGFP were expressed in COS-7 cells to study their role in TG accumulation. *, p < 0.001 (unpaired Student's t test, n = 5). B, FSP27 (120–220) is sufficient to accumulate TGs in human adipocytes. FSP27 and FSP27 (120–220) were expressed in human adipocytes using lentivirus. EGFP containing lentivirus was used as a control. *, p < 0.005; n = 3 (unpaired Student's t test). C, schematic of FSP27 indicating its functional domains.
To confirm the significance of these findings in adipocytes, we repeated these experiments in cultured human adipocytes. Lentivirus-mediated expression of both full-length FSP27 and FSP27 (120–220) induced comparable levels of TG accumulation in adipocytes (Fig. 5B).
The TG Accumulatory Domain of FSP27 Interacted with ATGL and Reduced Lipolysis
We next determined whether FSP27 (120–220) interacts with ATGL and decreases ATGL-mediated lipolysis in human adipocytes. HA-tagged FSP27 (120–220) was expressed in differentiated human adipocytes using lentivirus, as described previously (31), and precipitated with HA antibody. ATGL coimmunoprecipitated with FSP27 (120–220) (Fig. 6A). We then asked whether FSP27 (120–220) affects ATGL-mediated lipolysis. Human adipocytes were infected with FSP27 (120–220) lentivirus and, 3 days later, cells were infected with either ATGL or EGFP control virus. Cells overexpressing ATGL released significantly more glycerol into the medium than cells expressing EGFP, whereas cells overexpressing FSP27 (120–220) alone released less glycerol (Fig. 6B). Interestingly, cells coexpressing FSP27 (120–220) and ATGL underwent less lipolysis than cells overexpressing ATGL alone, suggesting that FSP27 (120–220) suppresses ATGL-mediated lipolysis in human adipocytes.
FIGURE 6.
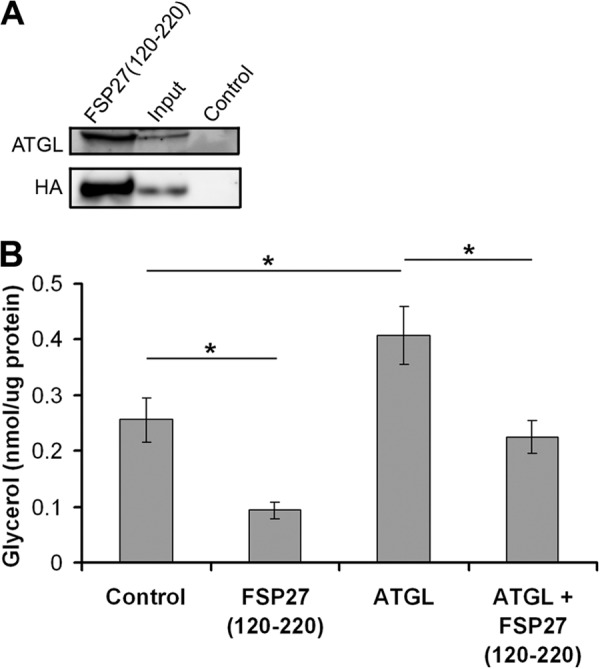
FSP27 (120–220) coimmunoprecipitated ATGL and reduced lipolysis. A, in human adipocytes, adenovirally expressed FSP27(120–220)-HA was immunoprecipitated using anti-HA antibody and immunoblotted with ATGL (top panel) or HA (bottom panel) antibody. B, human adipocytes were infected with EGFP (control), FSP27(120–220)-HA, or ATGL using lentivirus or adenovirus, respectively, and glycerol release in the media was measured. *, p < 0.005; n = 3 (unpaired Student's t test).
FSP27 Protected Human Adipocytes against FFA-induced Impairment of Insulin Signaling
FFAs impair insulin signaling and promote insulin resistance in adipocytes (5, 44, 53–55). Given our finding that FSP27 depletion in human adipocytes increased lipolysis and, hence, increased FFA levels, we tested whether FSP27 depletion affects insulin signaling. Cultured human adipocytes were transfected with either scrambled or FSP27 siRNA, as in Fig. 1, and then starved and subsequently stimulated with 1 nm insulin. siRNA-mediated FSP27 knockdown decreased insulin-mediated stimulation of AKT phosphorylation (Fig. 7A).
FIGURE 7.
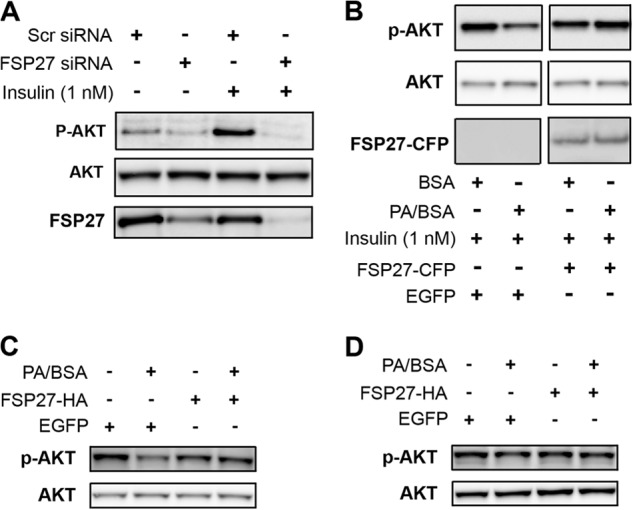
FSP27 protected human adipocytes against FFA-induced insulin resistance. A, insulin-stimulated AKT phosphorylation in human adipocytes after siRNA-mediated FSP27 knockdown. Scr, scrambled; P-AKT, phospho-AKT. B, insulin-stimulated AKT phosphorylation in human adipocytes after overnight treatment with 100 μm PA/BSA in the presence or absence of FSP27-CFP or EGFP (control). C, FSP27-HA expression protects adipocytes differentiated from WT MEFs against 100 μm PA/BSA-mediated inhibition of insulin-stimulated AKT phosphorylation. D, 100 μm PA/BSA or FSP27-HA expression had no effect on insulin-stimulated AKT activation in adipocytes differentiated from ATGL KO MEFs.
We next tested whether FSP27 overexpression protected adipocytes against FFA-induced insulin resistance. Palmitic acid (PA) induces insulin resistance in various insulin-responsive cell types (42, 53, 56). Therefore, we treated cells overnight with 100 μm PA complexed to BSA, which, in preliminary experiments, significantly decreased insulin-stimulated AKT phosphorylation in human adipocytes. BSA alone was used as a control. FSP27-CFP or EGFP (as a control) was overexpressed in cultured human adipocytes that were then treated overnight with PA/BSA. PA/BSA decreased insulin-stimulated AKT phosphorylation in cells expressing EGFP, whereas expression of FSP27-CFP protected the adipocytes from PA-induced insulin resistance (Fig. 7B). Of note, FSP27-HA also protected cells from PA-induced insulin resistance (data not shown), confirming that this effect was not an artifact of the CFP tag. In fact, we showed previously that HA, Myc, GFP, and FLAG tags do not affect FSP27 localization and function (28, 48).
Adipocytes Derived from ATGL KO MEFs Were Resistant to FFA-induced Insulin Resistance
Compared with WT mice, ATGL KO mice are more sensitive to insulin (57) and less susceptible to high-fat diet-induced insulin resistance (58). Our finding that FSP27 interacted with ATGL and induced TG accumulation in an ATGL-dependent manner (Figs. 2 and 4) suggested that FSP27 protects against FFA-induced insulin resistance via its effect on ATGL activity. To test this possibility, we compared MEFs derived from WT and ATGL KO mice. Overnight treatment of WT MEFs with 100 μm PA/BSA significantly decreased insulin-stimulated AKT phosphorylation, and this decrease was prevented in cells expressing FSP27-HA (Fig. 7C). Interestingly, PA/BSA did not affect AKT activation in ATGL KO MEFs, nor did FSP27-HA affect AKT phosphorylation (Fig. 7D). These results may suggest that the FSP27 protective effect on FFA-induced insulin resistance may act via suppression of ATGL activity. They do not prove that the effect of FSP27 on AKT phosphorylation may also be independent of its inhibitory role on ATGL activity. However, they do suggest that exogenous FFAs must go through a process of TG accumulation followed by ATGL-mediated lipolysis to affect insulin signaling.
FSP27 (120–220) Protected Human Adipocytes against FFA-induced Insulin Resistance
Given our finding that the aa 120–220 domain of FSP27 was associated with TG accumulation, we next tested whether it was comparable with full-length FSP27 in protecting against FFA-impaired insulin signaling. FSP27 (120–220) or EGFP (as a control) was expressed in differentiated human adipocytes, which were then challenged with 100 μm PA/BSA. PA/BSA inhibited insulin-stimulated AKT phosphorylation in control cells (Fig. 8, second and fourth lanes) but not in cells expressing FSP27 (120–220) (Fig. 8, sixth and eighth lanes).
FIGURE 8.
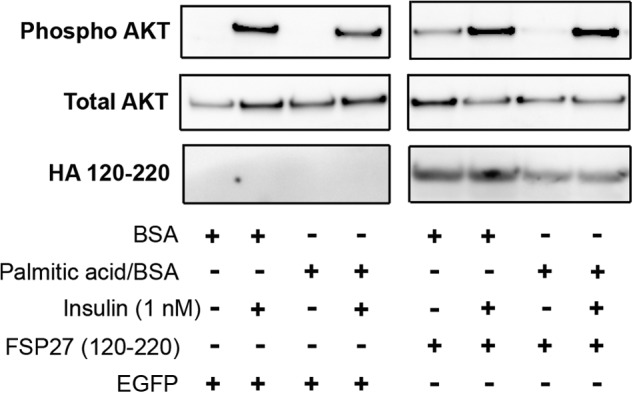
FSP27 (120–220) protected against FFA-induced insulin resistance in human adipocytes. Shown is insulin-stimulated AKT phosphorylation in human adipocytes. The core FSP27domain associated with TG accumulation, aa 120–220, was expressed using lentivirus, with EGFP as a control. The cells were treated overnight with 100 μm PA/BSA. The blots show AKT phosphorylation in basal and insulin-stimulated conditions. FSP27 (120–220) protected human adipocytes from inhibition of AKT phosphorylation by exogenous PA.
DISCUSSION
This study demonstrates the interaction of the LD protein FSP27 with ATGL, the rate-limiting enzyme for lipolysis in adipocytes. This interaction leads to reduced lipolysis and enhanced lipid storage capacity in adipocytes. FSP27-mediated TG accumulation was reduced significantly in the absence of ATGL, indicating that the protective effect of FSP27 on cellular TG content of adipocytes depends largely on ATGL-mediated lipolysis. We identified aa 120–220 as the TG accumulatory domain of FSP27 and showed that this domain also interacts with ATGL and inhibits ATGL-mediated lipolysis. FSP27 knockdown increased lipolysis and impaired insulin-stimulated AKT activation in mature human adipocytes. Interestingly, overexpression of FSP27 or ATGL deficiency protected human adipocytes from FFA-induced impairment of AKT activation. The TG-accumulatory FSP27 (120–220) domain was necessary and sufficient to protect human adipocytes from FFA-induced impairment of AKT activation. The discovery of the FSP27-ATGL interaction and its role in lipolysis, TG storage, and insulin sensitivity in human adipocytes provides a new potential avenue for targeting insulin resistance in humans.
We and others found previously that FSP27 depletion caused LD fragmentation (25, 28, 29) and increased basal and stimulated lipolysis in murine 3T3-L1 adipocytes and human primary adipocytes (28, 29). A recent study proposed that FSP27 might limit the presence of ATGL on LDs (50). The observation that multilocular LDs form in the absence of FSP27 suggests that increasing total LD surface area (a consequence of LD fragmentation) increases the access of ATGL to the LD surface and, subsequently, increases lipolysis. This study shows that FSP27 interacts directly with ATGL. This suggests that, besides regulating LD morphology, there is an additional level of regulation of lipolysis by FSP27. Gain-of-function effects were elucidated by overexpressing ATGL and/or FSP27 in COS-7 cells and mature human adipocytes. Interestingly, FSP27 expression suppressed ATGL-mediated lipolysis in both cell types, indicating a direct role of FSP27 in regulating ATGL-mediated lipolysis. This study, however, did not address whether FSP27 interacts with ATGL only on the LD surface. Because FSP27 and ATGL are present in other compartments, such as the endoplasmic reticulum, it is plausible that FSP27 interacts with ATGL in these compartments as well. Nonetheless, our study clearly demonstrates a role for FSP27 in regulating ATGL-mediated lipolysis in human adipocytes.
Adipocyte lipolysis is stimulated when catecholamines bind to β-adrenergic receptors on the cell surface (6, 59, 60). Through the action of a stimulatory G protein, adenylate cyclase is activated, leading to increased intracellular cAMP and activation of cAMP-dependent protein kinase (PKA). PKA phosphorylates both PLIN1 and hormone-sensitive lipase (6, 59–62). PKA-dependent phosphorylation of PLIN1 causes the release of CGI-58 and initiation of lipolysis by activating ATGL (15, 17, 18). In the current model of lipolysis, PLIN1 binds CGI-58 in the basal state, limiting its access to ATGL (15, 17). It is not yet clear what restrains ATGL from interacting with CGI-58. We propose that the scaffold protein PLIN1 interacts with FSP27, which then binds ATGL at the LD surface and limits access of ATGL to CGI-58 (Fig. 9). Upon lipolytic stimulation, CGI-58 is released from phosphorylated PLIN1, allowing it to bind directly to ATGL and activate it. It is not known whether FSP27 and CGI-58 share the same binding domain on ATGL.
FIGURE 9.

A hypothetical model of FSP27 regulation of lipolysis that is supported by the results of this study. The hypothesis is that PLIN1 scaffolds FSP27 at the lipid droplet surface where FSP27 interacts with ATGL and decreases lipolysis. A, under basal conditions, FSP27 decreases the access of ATGL to its coactivator CGI-58 (CGI), thereby diminishing lipolysis (dashed downward arrow). HSL, hormone-sensitive lipase. B, when FSP27 is absent in basal conditions, ATGL is free to interact with CGI-58, leading to increased lipolysis (solid downward arrow). C, upon β-adrenergic stimulation in the presence of FSP27, PKA activation results in phosphorylation of PLIN1 and hormone-sensitive lipase, causing the release of CGI-58, which binds to and stimulates ATGL (15, 18). Unbound ATGL is translocated to lipid droplets, and G0S2 is down-regulated to increase ATGL-mediated lipolysis (downward arrow). D, upon β-adrenergic stimulation in the absence of FSP27, the otherwise FSP27-sequestered ATGL is now available for CGI-58 binding, resulting in even higher levels of lipolysis (downward arrow). TAG, triacyl glycerol; and P, phosphorylation.
This model is supported by our finding that FSP27 depletion increases lipolysis in human adipocytes under both basal and stimulated conditions. We speculate that ATGL, when not bound to FSP27, might gain access to CGI-58 under both basal and stimulated conditions (Fig. 9, B and D). Alternatively, FSP27 regulation of LD morphology might also affect the process of ATGL-regulated lipolysis, a possibility that remains to be tested. Moreover, G0S2 inhibits ATGL activity (14). Prolonged β-adrenergic stimulation down-regulates G0S2 expression, thereby releasing more ATGL for sustained lipolysis. Future studies are required to demonstrate whether FSP27 is involved in ATGL-G0S2 and ATGL-CGI-58 interactions.
The mechanism of action of FSP27-mediated TG accumulation in adipocytes is not fully understood. We hypothesized that FSP27 reduces lipolysis of stored TGs and, thereby, increases TG content. This conclusion is supported by the observation that FSP27-mediated TG accumulation in human adipocytes was impaired upon ATGL depletion. Also, overexpression of FSP27 in ATGL-silenced adipocytes had no further inhibitory effect on lipolysis, suggesting that FSP27-mediated regulation of lipolysis and TG accumulation were specifically ATGL-dependent. However, it is possible that FSP27 might regulate lipolysis by other mechanisms in addition to ATGL catalytic activity. For example, interaction of FSP27 with ATGL at the LD surface may facilitate interactions with other intrinsic factors that may, in turn, maximize FSP27 regulation of lipolysis and increase TG accumulation. This possibility might explain why TG levels were lower in ATGL-silenced cells without FSP27 overexpression than with it (Fig. 4A). However, these results do not exclude the possibility that FSP27 provides additional protective shielding effects for LD-associated TGs, similar to that shown for PAT family proteins (59, 60). Furthermore, the effects of ATGL phosphorylation on FSP27-ATGL interaction remain to be determined.
We showed previously that FSP27 regulates LD morphology by clustering and fusion of the droplets via its aa 120–210 domain (48). This study demonstrated that the aa 120–220 domain, but not the previously defined aa 120–210 domain, induced TG accumulation at levels comparable with full-length FSP27. The aa 120–220 domain spans the LD targeting, clustering, and enlargement regions of FSP27 (48), suggesting that the regulation of LD morphology is essential for FSP27-mediated TG accumulation. Furthermore, our results show that aa 210–220 are necessary, but not sufficient, for TG accumulation. For example, a peptide sequence corresponding aa 210–220 did not cause TG accumulation (data not shown), and the truncated FSP27 variant aa 173–220 did not affect cellular TG levels (Fig. 5). Overall, our domain deletion studies show that aa 120–220 of FSP27 is the core domain required for the FSP27-mediated decrease in lipolysis and TG storage in adipocytes.
Optimal fat storage is essential to maintain a healthy metabolic phenotype in adipocytes and, ultimately, the whole body (5, 44, 53–55). Previous studies established that FFAs inhibit AKT phosphorylation and, hence, promote insulin resistance, which, in turn, causes metabolic syndrome. To define the role of FSP27 in insulin signaling in human adipocytes, we demonstrated that insulin-stimulated AKT activation was inhibited by siRNA-mediated FSP27 silencing. FSP27 overexpression protected the adipocytes from FFA-induced insulin resistance. We also found that exogenous FFAs had no effect on insulin-stimulated AKT activation in ATGL-deficient MEFs (Fig. 7 D). These results suggest that FSP27 might protect adipocytes from the deleterious effects of FFAs via suppression of ATGL-mediated lipolysis. It further suggests that exogenous FFAs do not directly affect insulin signaling but, rather, undergo a process of esterification and then lipolysis to impair insulin signaling. These results complement a previous finding that ATGL KO mice are protected from diet-induced insulin resistance (58). This could explain the higher rate of lipolysis and insulin resistance observed in obese humans compared with lean individuals. Indeed, our previous study showed that FSP27 expression is higher in white adipose tissue of obese insulin-sensitive people than in obese insulin-resistant people (26). Those observations support our present findings that higher FSP27 expression decreases ATGL-mediated lipolysis and, hence, improves storage of FFAs as TGs in human adipocytes.
Supplementary Material
Acknowledgments
We thank Dr. Keith Tornheim for critical reading of the manuscript.
This work was supported, in whole or in part, by NIDDK, National Institutes of Health Grants R56DK094815, P30DK046200, and 8KL2TR000158 (to V. P.) from Boston University Parent Clinical and Translational Science Institute Grant UL1-TR000157. This work was also supported by Grant SFB Lipotox F30, Wittgenstein Award Z136 (to R. Z.), and Grant P25193 (to A. L.), which are funded by the Austrian Science Fund (FWF) and Grant 12CVD04 from the Foundation Leducq (to R. Z.).

This article contains supplemental Figs. 1 and 2.
- TG
- triglyceride
- FA
- fatty acid
- ATGL
- adipose triglyceride lipase
- LD
- lipid droplet
- FFA
- free fatty acid
- aa
- amino acid(s)
- MEF
- mouse embryonic fibroblast
- CFP
- cyan fluorescent protein
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Unger R. H., Clark G. O., Scherer P. E., Orci L. (2010) Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 1801, 209–214 [DOI] [PubMed] [Google Scholar]
- 2. Boden G. (1997) Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 46, 3–10 [PubMed] [Google Scholar]
- 3. Boden G., Chen X., Ruiz J., White J. V., Rossetti L. (1994) Mechanisms of fatty acid-induced inhibition of glucose uptake. J. Clin. Invest. 93, 2438–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dresner A., Laurent D., Marcucci M., Griffin M. E., Dufour S., Cline G. W., Slezak L. A., Andersen D. K., Hundal R. S., Rothman D. L., Petersen K. F., Shulman G. I. (1999) Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 103, 253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferrannini E., Barrett E. J., Bevilacqua S., DeFronzo R. A. (1983) Effect of fatty acids on glucose production and utilization in man. J. Clin. Invest. 72, 1737–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zechner R., Zimmermann R., Eichmann T. O., Kohlwein S. D., Haemmerle G., Lass A., Madeo F. (2012) Fat Signals: lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15, 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lass A., Zimmermann R., Oberer M., Zechner R. (2011) Lipolysis: a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 50, 14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajala M. W., Scherer P. E. (2003) Minireview: the adipocyte: at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144, 3765–3773 [DOI] [PubMed] [Google Scholar]
- 9. Villena J. A., Roy S., Sarkadi-Nagy E., Kim K. H., Sul H. S. (2004) Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 279, 47066–47075 [DOI] [PubMed] [Google Scholar]
- 10. Zimmermann R., Strauss J. G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., Zechner R. (2004) Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386 [DOI] [PubMed] [Google Scholar]
- 11. Ahmadian M., Wang Y., Sul H. S. (2010) Lipolysis in adipocytes. Int. J. Biochem. Cell Biol. 42, 555–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jenkins C. M., Mancuso D. J., Yan W., Sims H. F., Gibson B., Gross R. W. (2004) Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 279, 48968–48975 [DOI] [PubMed] [Google Scholar]
- 13. Soni K. G., Mardones G. A., Sougrat R., Smirnova E., Jackson C. L., Bonifacino J. S. (2009) Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 122, 1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X., Lu X., Lombès M., Rha G. B., Chi Y. I., Guerin T. M., Smart E. J., Liu J. (2010) The G0/G1 switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 11, 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Granneman J. G., Moore H. P., Krishnamoorthy R., Rathod M. (2009) Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (ABHD5) and adipose triglyceride lipase (ATGL). J. Biol. Chem. 284, 34538–34544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., Kienesberger P., Strauss J. G., Gorkiewicz G., Zechner R. (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 3, 309–319 [DOI] [PubMed] [Google Scholar]
- 17. Subramanian V., Rothenberg A., Gomez C., Cohen A. W., Garcia A., Bhattacharyya S., Shapiro L., Dolios G., Wang R., Lisanti M. P., Brasaemle D. L. (2004) Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 279, 42062–42071 [DOI] [PubMed] [Google Scholar]
- 18. Miyoshi H., Perfield J. W., 2nd, Souza S. C., Shen W. J., Zhang H. H., Stancheva Z. S., Kraemer F. B., Obin M. S., Greenberg A. S. (2007) Control of adipose triglyceride lipase action by serine 517 of perilipin A globally regulates protein kinase A-stimulated lipolysis in adipocytes. J. Biol. Chem. 282, 996–1002 [DOI] [PubMed] [Google Scholar]
- 19. Danesch U., Hoeck W., Ringold G. M. (1992) Cloning and transcriptional regulation of a novel adipocyte-specific gene, FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like proteins interact with sequences required for differentiation-dependent expression. J. Biol. Chem. 267, 7185–7193 [PubMed] [Google Scholar]
- 20. Su A. I., Cooke M. P., Ching K. A., Hakak Y., Walker J. R., Wiltshire T., Orth A. P., Vega R. G., Sapinoso L. M., Moqrich A., Patapoutian A., Hampton G. M., Schultz P. G., Hogenesch J. B. (2002) Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 99, 4465–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu S., Matsusue K., Kashireddy P., Cao W. Q., Yeldandi V., Yeldandi A. V., Rao M. S., Gonzalez F. J., Reddy J. K. (2003) Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARγ1) overexpression. J. Biol. Chem. 278, 498–505 [DOI] [PubMed] [Google Scholar]
- 22. Williams P. M., Chang D. J., Danesch U., Ringold G. M., Heller R. A. (1992) CCAAT/enhancer binding protein expression is rapidly extinguished in TA1 adipocyte cells treated with tumor necrosis factor. Mol. Endocrinol. 6, 1135–1141 [DOI] [PubMed] [Google Scholar]
- 23. Kim J. Y., Liu K., Zhou S., Tillison K., Wu Y., Smas C. M. (2008) Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am. J. Physiol. Endocrinol. Metab. 294, E654–E667 [DOI] [PubMed] [Google Scholar]
- 24. Liu K., Zhou S., Kim J. Y., Tillison K., Majors D., Rearick D., Lee J. H., Fernandez-Boyanapalli R. F., Barricklow K., Houston M. S., Smas C. M. (2009) Functional analysis of FSP27 protein regions for lipid droplet localization, caspase-dependent apoptosis, and dimerization with CIDEA. Am. J. Physiol. Endocrinol. Metab. 297, E1395–E1413 [DOI] [PubMed] [Google Scholar]
- 25. Nishino N., Tamori Y., Tateya S., Kawaguchi T., Shibakusa T., Mizunoya W., Inoue K., Kitazawa R., Kitazawa S., Matsuki Y., Hiramatsu R., Masubuchi S., Omachi A., Kimura K., Saito M., Amo T., Ohta S., Yamaguchi T., Osumi T., Cheng J., Fujimoto T., Nakao H., Nakao K., Aiba A., Okamura H., Fushiki T., Kasuga M. (2008) FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Invest. 118, 2808–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Puri V., Ranjit S., Konda S., Nicoloro S. M., Straubhaar J., Chawla A., Chouinard M., Lin C., Burkart A., Corvera S., Perugini R. A., Czech M. P. (2008) Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 7833–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Puri V., Czech M. P. (2008) Lipid droplets: FSP27 knockout enhances their sizzle. J. Clin. Invest. 118, 2693–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puri V., Konda S., Ranjit S., Aouadi M., Chawla A., Chouinard M., Chakladar A., Czech M. P. (2007) Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 282, 34213–34218 [DOI] [PubMed] [Google Scholar]
- 29. Keller P., Petrie J. T., De Rose P., Gerin I., Wright W. S., Chiang S. H., Nielsen A. R., Fischer C. P., Pedersen B. K., MacDougald O. A. (2008) Fat-specific protein 27 regulates storage of triacylglycerol. J. Biol. Chem. 283, 14355–14365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Toh S. Y., Gong J., Du G., Li J. Z., Yang S., Ye J., Yao H., Zhang Y., Xue B., Li Q., Yang H., Wen Z., Li P. (2008) Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS ONE 3, e2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grahn T. H., Zhang Y., Lee M. J., Sommer A. G., Mostoslavsky G., Fried S. K., Greenberg A. S., Puri V. (2013) FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem. Biophys. Res. Commun. 432, 296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun Z., Gong J., Wu H., Xu W., Wu L., Xu D., Gao J., Wu J. W., Yang H., Yang M., Li P. (2013) Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 4, 1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez-Botas J., Anderson J. B., Tessier D., Lapillonne A., Chang B. H., Quast M. J., Gorenstein D., Chen K. H., Chan L. (2000) Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat. Genet. 26, 474–479 [DOI] [PubMed] [Google Scholar]
- 34. Tansey J. T., Sztalryd C., Gruia-Gray J., Roush D. L., Zee J. V., Gavrilova O., Reitman M. L., Deng C. X., Li C., Kimmel A. R., Londos C. (2001) Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 98, 6494–6499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zimmermann R., Lass A., Haemmerle G., Zechner R. (2009) Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim. Biophys. Acta 1791, 494–500 [DOI] [PubMed] [Google Scholar]
- 36. Castro-Chavez F., Yechoor V. K., Saha P. K., Martinez-Botas J., Wooten E. C., Sharma S., O'Connell P., Taegtmeyer H., Chan L. (2003) Coordinated upregulation of oxidative pathways and downregulation of lipid biosynthesis underlie obesity resistance in perilipin knockout mice: a microarray gene expression profile. Diabetes 52, 2666–2674 [DOI] [PubMed] [Google Scholar]
- 37. Zhou Z., Yon Toh S., Chen Z., Guo K., Ng C. P., Ponniah S., Lin S. C., Hong W., Li P. (2003) Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat. Genet. 35, 49–56 [DOI] [PubMed] [Google Scholar]
- 38. Dobrzyn P., Dobrzyn A., Miyazaki M., Cohen P., Asilmaz E., Hardie D. G., Friedman J. M., Ntambi J. M. (2004) Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc. Natl. Acad. Sci. U.S.A. 101, 6409–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ntambi J. M., Miyazaki M., Stoehr J. P., Lan H., Kendziorski C. M., Yandell B. S., Song Y., Cohen P., Friedman J. M., Attie A. D. (2002) Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. U.S.A. 99, 11482–11486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rubio-Cabezas O., Puri V., Murano I., Saudek V., Semple R. K., Dash S., Hyden C. S., Bottomley W., Vigouroux C., Magré J., Raymond-Barker P., Murgatroyd P. R., Chawla A., Skepper J. N., Chatterjee V. K., Suliman S., Patch A. M., Agarwal A. K., Garg A., Barroso I., Cinti S., Czech M. P., Argente J., O'Rahilly S., Savage D. B., and LD Screening Consortium (2009) Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 1, 280–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Itani S. I., Ruderman N. B., Schmieder F., Boden G. (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 51, 2005–2011 [DOI] [PubMed] [Google Scholar]
- 42. Nguyen M. T., Satoh H., Favelyukis S., Babendure J. L., Imamura T., Sbodio J. I., Zalevsky J., Dahiyat B. I., Chi N. W., Olefsky J. M. (2005) JNK and tumor necrosis factor-α mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 280, 35361–35371 [DOI] [PubMed] [Google Scholar]
- 43. Yu C., Chen Y., Cline G. W., Zhang D., Zong H., Wang Y., Bergeron R., Kim J. K., Cushman S. W., Cooney G. J., Atcheson B., White M. F., Kraegen E. W., Shulman G. I. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236 [DOI] [PubMed] [Google Scholar]
- 44. Guilherme A., Virbasius J. V., Puri V., Czech M. P. (2008) Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee M. J., Wu Y., Fried S. K. (2012) A modified protocol to maximize differentiation of human preadipocytes and improve metabolic phenotypes. Obesity 20, 2334–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mostoslavsky G., Kotton D. N., Fabian A. J., Gray J. T., Lee J. S., Mulligan R. C. (2005) Efficiency of transduction of highly purified murine hematopoietic stem cells by lentiviral and oncoretroviral vectors under conditions of minimal in vitro manipulation. Mol. Ther. 11, 932–940 [DOI] [PubMed] [Google Scholar]
- 47. Lee M. J., Pickering R. T., Puri V. (2013) Prolonged efficiency of siRNA-mediated gene silencing in primary cultures of human preadipocytes and adipocytes. Obesity, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jambunathan S., Yin J., Khan W., Tamori Y., Puri V. (2011) FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation. PLoS ONE 6, e28614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gong J., Sun Z., Wu L., Xu W., Schieber N., Xu D., Shui G., Yang H., Parton R. G., Li P. (2011) Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 195, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang X., Heckmann B. L., Zhang X., Smas C. M., Liu J. (2013) Distinct mechanisms regulate ATGL-mediated adipocyte lipolysis by lipid droplet coat proteins. Mol. Endocrinol. 27, 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gruber A., Cornaciu I., Lass A., Schweiger M., Poeschl M., Eder C., Kumari M., Schoiswohl G., Wolinski H., Kohlwein S. D., Zechner R., Zimmermann R., Oberer M. (2010) The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J. Biol. Chem. 285, 12289–12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soukas A., Socci N. D., Saatkamp B. D., Novelli S., Friedman J. M. (2001) Distinct transcriptional profiles of adipogenesis in vivo and in vitro. J. Biol. Chem. 276, 34167–34174 [DOI] [PubMed] [Google Scholar]
- 53. Bays H., Mandarino L., DeFronzo R. A. (2004) Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab. 89, 463–478 [DOI] [PubMed] [Google Scholar]
- 54. Boden G., Shulman G. I. (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and β-cell dysfunction. Eur. J. Clin. Invest. 32, 14–23 [DOI] [PubMed] [Google Scholar]
- 55. Lewis G. F., Carpentier A., Adeli K., Giacca A. (2002) Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 23, 201–229 [DOI] [PubMed] [Google Scholar]
- 56. Van Epps-Fung M., Williford J., Wells A., Hardy R. W. (1997) Fatty acid-induced insulin resistance in adipocytes. Endocrinology 138, 4338–4345 [DOI] [PubMed] [Google Scholar]
- 57. Haemmerle G., Moustafa T., Woelkart G., Büttner S., Schmidt A., van de Weijer T., Hesselink M., Jaeger D., Kienesberger P. C., Zierler K., Schreiber R., Eichmann T., Kolb D., Kotzbeck P., Schweiger M., Kumari M., Eder S., Schoiswohl G., Wongsiriroj N., Pollak N. M., Radner F. P., Preiss-Landl K., Kolbe T., Rülicke T., Pieske B., Trauner M., Lass A., Zimmermann R., Hoefler G., Cinti S., Kershaw E. E., Schrauwen P., Madeo F., Mayer B., Zechner R. (2011) ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoy A. J., Bruce C. R., Turpin S. M., Morris A. J., Febbraio M. A., Watt M. J. (2011) Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology 152, 48–58 [DOI] [PubMed] [Google Scholar]
- 59. Brasaemle D. L. (2007) Thematic review series: adipocyte biology: the perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559 [DOI] [PubMed] [Google Scholar]
- 60. Bickel P. E., Tansey J. T., Welte M. A. (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 1791, 419–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sztalryd C., Xu G., Dorward H., Tansey J. T., Contreras J. A., Kimmel A. R., Londos C. (2003) Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 161, 1093–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tansey J. T., Huml A. M., Vogt R., Davis K. E., Jones J. M., Fraser K. A., Brasaemle D. L., Kimmel A. R., Londos C. (2003) Functional studies on native and mutated forms of perilipins: a role in protein kinase A-mediated lipolysis of triacylglycerols. J. Biol. Chem. 278, 8401–8406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.