

Background: Metal-associated β-amyloid (Aβ) aggregates are implicated in the pathogenesis of Alzheimer disease.
Results: Copper bound Aβ(1–42) aggregates, including fibrils, degrade hydrogen peroxide, forming hydroxyl radicals and carbonyls.
Conclusion: Copper-bound Aβ fibrils can retain redox activity.
Significance: Aβ fibrils bound to copper are not inert end points and may be a source of oxidative stress in the Alzheimer brain.
Keywords: Alzheimer Disease, Free Radicals, Hydrogen Peroxide, Oxidative Stress, Reactive Oxygen Species (ROS), β-Amyloid, Copper Ions, Peptide Aggregation, Zinc Ions
Abstract
According to the “amyloid cascade” hypothesis of Alzheimer disease, the formation of Aβ fibrils and senile plaques in the brain initiates a cascade of events leading to the formation of neurofibrillary tangles, neurodegeneration, and the symptom of dementia. Recently, however, emphasis has shifted away from amyloid fibrils as the predominant toxic form of Aβ toward smaller aggregates, referred to as “soluble oligomers.” These oligomers have become one of the prime suspects for involvement in the early oxidative damage that is evident in this disease. This raises the question whether or not Aβ fibrils are actually “inert tombstones” present at the end of the aggregation process. Here we show that, when Aβ(1–42) aggregates, including fibrils, are bound to Cu(II) ions, they retain their redox activity and are able to degrade hydrogen peroxide (H2O2) with the formation of hydroxyl radicals and the consequent oxidation of the peptide (detected by formation of carbonyl groups). We find that this ability increases as the Cu(II):peptide ratio increases and is accompanied by changes in aggregate morphology, as determined by atomic force microscopy. When aggregates are prepared in the copresence of Cu(II) and Zn(II) ions, the ratio of Cu(II):Zn(II) becomes an important factor in the degeneration of H2O2, the formation of carbonyl groups in the peptide, and in aggregate morphology. We believe, therefore, that Aβ fibrils can destroy H2O2 and generate damaging hydroxyl radicals and, so, are not necessarily inert end points.
Introduction
Neuropathological changes in the brain associated with Alzheimer disease (AD)2 include the formation of senile plaques, containing β-amyloid (Aβ) fibrils, and neurofibrillary tangles, composed of phosphorylated Tau protein. According to the “amyloid cascade” hypothesis, the formation of Aβ fibrils and senile plaques in the brain initiates a cascade of events leading to a number of downstream consequences, including the formation of neurofibrillary tangles, neurodegeneration, and the symptom of dementia (1). More recently, however, emphasis has shifted away from amyloid fibrils as the predominant toxic form of Aβ and toward smaller and more soluble aggregates that may include a variety of structures, referred to as “soluble oligomers,” Aβ-derived diffusible ligands (ADDLs) or “protofibrils” (2–8). This change in thinking has been prompted, to some extent, by the finding that the relationship between the numbers of senile plaques and the severity of dementia is poor (9, 10) and by the finding that small, soluble oligomers seem to be more toxic than fibrillar Aβ (11) and, in addition, have potent effects on learning and memory in animals (6, 7, 12, 13).
Damage to the brain in AD also appears to involve other processes such as inflammation and extensive oxidative modification to macromolecules, including proteins, lipids, and nucleic acids (see, for example, Ref. 14 and the references quoted therein). There is evidence for oxidative damage to the brain in a wide variety of neurodegenerative diseases (15, 16), but in AD, it seems to be a particularly early event in the course of the disease (17, 18). During the early stages of AD, the primary Aβ species present are likely to be non-fibrillar, and, therefore, Aβ oligomers have become one of the prime suspects for involvement in this early oxidative damage. This idea has been supported by data suggesting that Aβ has the ability to generate reactive oxygen species (ROS) when in association with certain redox-active transition metal ions. Just over a decade ago, Huang et al. (19, 20) published two major papers highlighting the direct generation of two key ROS, hydrogen peroxide (H2O2) and the hydroxyl radical (•OH), during the incubation of Aβ in vitro, with the latter being especially damaging because of its very high reactivity. Huang et al. (19, 20) also established that Aβ binds strongly to copper, iron, and zinc ions and that, when bound, Aβ not only reduces Cu(II) to Cu(I) and Fe(III) to Fe(II) but also that the resulting peptide-metal ion complex is redox-active. This is important because Cu(I) and Fe(II) ions, when bound to Aβ, are capable of reducing molecular oxygen to H2O2 and the latter to •OH and, hence, can generate these two ROS directly. Moreover, the levels of these metals have been reported to be elevated in the amyloid plaque deposits present in the brains of individuals with AD (21, 22).
In our own work, we used the technique of electron spin resonance spectroscopy in conjunction with spin trapping to confirm that Aβ does, indeed, self-generate H2O2, in our case in the presence of only trace levels of metal ions (23, 24). We also used the same technique to examine the time profile for H2O2 generation during the process of Aβ aggregation (for details, see Ref. 25) and found three characteristic phases, i.e. a brief time lag before any H2O2 was observed, followed by a rapid growth and then a slow decay in concentration. This early “pulse” of H2O2 generation was found to coincide with the presence of structures resembling “oligomers” or protofibrils, and its concentration profile (i.e. the variation with time) was in complete contrast to the aggregation profile (as monitored by thioflavin T and immunoassay methods), which showed the typical sigmoidal growth curve observed previously with Aβ and other amyloids (26, 27). Thus, one of the fundamental molecular mechanisms underlying the pathogenesis of cell damage in AD could be the direct production of ROS by Aβ during the early oligomer phase of its aggregation (23, 28–30). The behavior that we observed for H2O2 is classical of that expected for a reaction intermediate formed during a series of consecutive processes. During the phase where concentration is increasing rapidly, the rate of H2O2 production exceeds the rate of removal. The latter two rates are roughly in balance as the curve flattens off, whereas, during the final “decay” phase, the rate of removal exceeds the rate of formation. The loss of H2O2 during the final phase of this reaction profile could be due to at least two mechanisms. One of these is the direct bimolecular reaction of the peroxide with the peptide. The other is the destruction of the peroxide by the transfer of an electron from any of the redox-active metal ions that might be bound to the peptide. In the case of Fe(II) ions, this is the well known Fenton reaction.
This raises the question whether the β-amyloid fibrils are actually an “inert tombstone” present at the end of the aggregation process, as some researchers believe, or whether they are actively involved in the production and/or removal of ROS. Our published results have suggested that fully formed β-amyloid fibrils do not generate H2O2 (at least within the electron spin resonance detection limit) (25). However, as far as we are aware, there are no studies aimed at determining whether fibrils composed of Aβ can degrade H2O2 in vitro.
The aim of the present investigation was to determine whether Aβ fibrils and other aggregates can degrade H2O2 and whether they could be involved in the generation of •OH with consequent oxidative damage to the peptide. Therefore, we also looked to see whether we could detect the presence of carbonyl groups within the peptide as a marker of this latter damage.
EXPERIMENTAL PROCEEDURES
Peptide Preparation
Recombinant Aβ(1–42) (rPeptide, >97% purity) was kept at −20 °C until prepared for use by a protocol adapted from a personal communication by Manzoni et al. (31). In brief, Aβ was dissolved in 0.01% NH4OH (pH 10.6) to 0.5 mg/ml, vortexed, and then sonicated for 4 × 30 s. The solvent was evaporated using a Thermo Savant SpeedVac concentrator. TFA containing 4.5% thioanisole was subsequently added, vortexed, sonicated for 30 s, and then evaporated under a stream of nitrogen (N2) gas. 1,1,1,3,3,3-hexafluoroisopropanol was then added to give a 0.5 mg/ml solution of peptide, vortexed, and sonicated for 4 × 30 s. The Aβ was then split into working aliquots, and the 1,1,1,3,3,3-hexafluoroisopropanol was removed by evaporation in the SpeedVac.
For aggregation and sampling, 10 mm phosphate buffer (PB) (pH 7.4), with and without test concentrations of metal ions (Cu(II), Fe(II), Fe(III), Mn(II), Zn(II)), was added to Aβ to give a 50 μm solution of the peptide, vortexed, and then sonicated for 4 × 30 s and incubated at 37 °C. After 144 h, samples were taken for thioflavin T (ThT) and atomic force microscopy (AFM) examination. The rest of the sample was spun in a Beckman airfuge for 1 h at 136,000 × g. The supernatant was carefully pipetted off and tested for aggregate content by ThT. The pellets were resuspended and washed in 10 mm PB (pH 7.4) and then spun in the airfuge for a further 1 h. The supernatants were again removed and the pellets resuspended and incubated at 37 °C in 1 mm H2O2 in 10 mm PB for up to 96 h. Samples were taken every 24 h for up to 144 h (Amplex red assay) at 0 and 48 h (coumarin assay) and at 0 and 48, 72, and 144 h (for 2,4-dinitrophenylhydrazine (DNPH) immunoassay). Metal ion concentrations were determined by inductively coupled plasma MS (Thermo Elemental X7) after 200× dilution with 0.1 m HNO3.
Thioflavin T
15 μm ThT was prepared in 50 mm glycine-NaOH buffer (pH 8.5). This was used to prime a Synergy 2 multilabel plate reader (Biotek). Aβ samples were diluted to 25 μm in 10 mm PB, and then 3 × 10-μl samples were aliquoted into a Nunc 96-well black microtiter plate. The samples were then injected with 50 μl of 15 μm ThT, mixed, and read 10 times over 2 min with the plate reader at excitation λ 450 nm and emission λ 482 nm (32). Average fluorescence was calculated over the 2-min time course, together with standard deviations, and plotted as relative fluorescence units.
Amplex Red
Stock solutions of 10 mm Amplex red in dimethyl sulfoxide and 1 kilo unit/ml HRP in 10 mm PB (pH 7.4) were prepared and aliquoted into working portions. Each aliquot had 10 s of nitrogen gas introduced into them, and then they were frozen using liquid nitrogen and stored at −80 °C. The Amplex red working solution was prepared at 100 μm Amplex red and 200 mm/ml HRP in 10 mm PB (pH 7.4), aliquoted in 1-ml portions, and kept at −80 °C in the dark. For fluorescence measurements, samples were diluted 1:400 in 10 mm PB, and then three 15-μl samples were pipetted into a Nunc 384-well black plate. 15 μl of Amplex red working solution was then added to each well. The plate was shaken and the fluorescence read on the Victor 2 microplate reader at excitation λ 563 nm and emission λ 587 nm (33). Means ± S.D. were converted to concentration of H2O2 and plotted.
Atomic Force Microscopy
2 μl of 50 μm Aβ(1–42) aggregated with either no added metal ions or test concentrations of CuCl2 and/or ZnCl2 (12.5 or 50 μm) in 10 mm PB (± metal ions) was diluted by 1:10 in MilliQ water and pipetted onto a piece of poly-l-lysine-coated mica. This was allowed to dry and then washed with MilliQ water. This was then imaged using a Digital Instruments multimode scanning probe microscope using tapping mode, taking at least three 10-μm images of each sample, followed by 5-, 2-, and 1-μm scans. The images were then processed using WSxM scanning probe microscopy software (34). Lateral measurements of Aβ aggregates were adjusted to account for the “tip broadening” effect using the equation
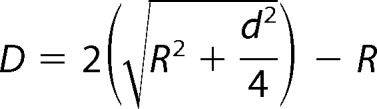 |
where D is the “true” diameter, r is the tip radius, and d is the measured diameter at half vertical height (35).
DNPH Immunoassay for Carbonyl Groups
1 mm H2O2 was added to the aggregated, airfuged, and washed Aβ(1–42) pellets, and 2-μl samples were taken immediately (time zero) and again after 48, 72, or 144 h and added to 6 μl of 10 mm DNPH in 6 m guanidine hydrochloride. These samples were incubated at room temperature for 50 min, giving them a gentle vortex every 10 min. 7 μl of each sample was then added to 1.393 ml of 10 mm PB (pH 7.4), vortexed, and frozen at −80 °C. Analysis of the samples for carbonyl groups was done via immunoassay using methods outlined by Dalle-Donne et al. (36). Briefly, 200-μl samples were plated onto a clear Maxisorb 96-well plate in quadruplicate and incubated overnight at 4 °C. Plates were aspirated and then blocked with 250 μl of 2% milk powder in 10 mm PBS, 0.5% Tween at 37 °C for 1 h with gentle shaking. Plates were then washed four times in 10 mm PBS, and then 200 μl of 1 μg/ml HRP-labeled, anti-DNPH mAb in block was added to each well. The plates were incubated at 37 °C for 1 h with gentle shaking and then washed four times in 10 mm PBS. 200 μl of a 1:500 dilution of europium-labeled streptavidin in 0.05% γ-globulin, 0.5% BSA, 20 μm diethylene triamine pentaacetic acid, 10 mm Tris, and 150 mm NaCl (pH 7.4) was added to each well. The plates were shaken for 10 min, covered in foil, and placed on a rocking table for a further 50 min. After washing four times with 10 mm PBS, 200 μl of enhancer solution (PerkinElmer Life Sciences) was added to each well. Plates were shaken again for 10 min and then read using a Victor 2 microplate reader with the settings for the time-resolved DELFIA system. Means ± S.D. were plotted in relative fluorescence units.
Coumarin Assay for Hydroxyl Radical Formation
Aggregated, airfuged, and washed Aβ(1–42) pellets were resuspended to final concentrations: 1 mm H2O2, 0.1 mm coumarin carboxylic acid in 10 mm PB. Reaction mixtures were then incubated at 37 °C for 48 h. Three 15-μl aliquots were plated onto a black 384-well microtiter plate, and fluorescence was read at excitation λ 390 nm and emission λ 460 nm (37). Means ± S.D. were plotted in relative fluorescence units.
Data Analysis
All experiments were carried out with a minimum of three independent repeats. Representative data are shown. Statistical analyses performed were completed via one-way analysis of variance followed by Tukey's honest significant difference (HSD) post hoc test to correct for multiple pairwise comparisons.
RESULTS
Influence of Cu(II) Ions on the Formation and Decay of H2O2 during the Incubation of Aβ(1–42)
In our previous research, we found that the levels of self-generated H2O2 (as determined by electron spin resonance spectroscopy or Amplex red assay) increased during the incubation of Aβ(1–42) in the absence of any added metal ions, reached a maximum, and then slowly declined as incubation/aggregation continued (16, 25, 38). The self-generation of this H2O2 is believed to proceed via a two-electron transfer from a suitable transition metal ion to O2 (16, 19, 23, 25, 29–30), with its mechanism detailed by others (39, 40). In our case, these metal ions are present at trace levels in the buffers or bound to the peptide during synthesis (24). Some of our results for Aβ(1–42) incubated at 25 μm over a 48-h period and obtained by Amplex red measurements are shown in Fig. 1. When Aβ(1–42) is incubated in the presence of an equimolar concentration of Cu(II), we note an acceleration in the generation of H2O2 over the first few hours. Interestingly, though, there is also a much more rapid deterioration in H2O2 levels at this Cu(II):peptide ratio compared with the curve obtained in the absence of added copper, suggesting that, as incubation proceeds, the later aggregates of Aβ(1–42) bound to Cu(II) actively degrade H2O2. We were intrigued by the increased decay rates in these latter experiments, and, in view of these observations, we examined the ability of Aβ(1–42) fibrils to degrade H2O2, particularly when bound to copper ions. In addition to Cu(II), we also determined the influence of Mn(II), Fe(II), Fe(III), and Zn(II) ions on this process and on carbonyl group formation and of Cu(II) and Zn(II) ions on peptide aggregate morphology (as seen by AFM).
FIGURE 1.
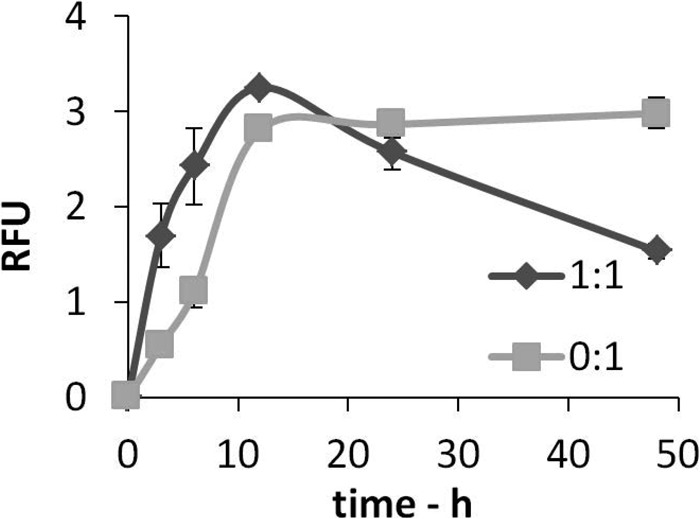
Levels of hydrogen peroxide accumulate rapidly during the early stages of Aβ aggregation in the presence of Cu(II) ions but then decline at longer time points. Aβ(1–42) (25 μm) was incubated in 10 mm PB at 37 °C for up to 48 h in the presence or absence of equimolar Cu(II) ions. Samples were taken at 0, 3, 6, 12, 24, and 48 h and assayed for H2O2 concentration using Amplex red. Data are mean ± S.D. (n = 3). RFU, relative fluorescence unit.
Aβ(1–42) Aggregation in the Presence of Different Metal Ions and the Effect of the Resulting Aggregates on Hydrogen Peroxide Decay and Carbonyl Group Formation
We first studied the aggregation of Aβ(1–42) over 144 h in the presence or absence of five different metal ions at a metal ion:Aβ(1–42) molar ratio of 1:1 (Fig. 2A) using the ThT method to detect aggregation. We note a decrease in the final level of fluorescence in the presence of some of the metal ions. This decrease could be due to less fibril formation, although the quenching of the fluorescence in the presence of individual metal ions could also be involved. This equimolar metal ion:peptide ratio was selected following a series of preliminary experiments that suggested that a clear difference between the effects of different treatments on H2O2 degradation and carbonyl group formation was readily observed at this ratio. Aβ(1–42) aggregation proceeded in the presence of Mn(II), Fe(II), Fe(III), Zn(II), and Cu(II) ions, or in their absence, to generate ThT-positive aggregates. When these aggregates were centrifuged at high speed in a Beckman airfuge and washed, the supernatants were always ThT-negative, indicating that the ThT-positive peptide aggregates had all been retained in the pellets. In subsequent experiments, this pelleted material was resuspended and incubated with 1 mm H2O2 to determine the effects of mature aggregates on the degradation of H2O2.
FIGURE 2.

Aggregates formed by incubation of Aβ(1–42) in the presence of Cu(II) ions degrade H2O2 and contain carbonyl groups. Aβ(1–42) (50 μm) was incubated in 10 mm PB at 37 °C for 144 h in the presence or absence of an equimolar concentration of a range of different metal ions, as indicated, prior to centrifugation for 1 h in a Beckman airfuge. The supernatant was removed for analysis, and the pellet containing aggregated Aβ was resuspended in 10 mm PB and then centrifuged for a further 1 h before removal of the second supernatant (Wash). The final pellet was resuspended in 1 mm H2O2 to an Aβ concentration of 25 μm. This final suspension was incubated at 37 °C for up to 72 h and assayed for H2O2 concentration every 24 h and for carbonyl group formation at 0 and 48 h. A, ThT fluorescence data (mean ± S.D., n = 3) for the Aβ samples prior to pelleting, the first supernatant, and the subsequent wash. RFU, relative fluorescence unit. B, metal ion concentrations (average of two independent inductively coupled plasma MS analyses) for the Aβ samples prior to pelleting, the first supernatant and subsequent wash. C, Amplex red data (mean ± S.D., n = 3) for the final suspension incubated with H2O2. D, carbonyl group formation data (mean ± S.D., n = 4) for the final suspension incubated with H2O2. *, p ≤ 0.001; Tukey's HSD.
The first supernatant and the subsequent wash were also examined by inductively coupled plasma MS to determine the metal ion content in these samples. The results are summarized in the table in Fig. 2B and clearly show that, in the case of Cu(II) and Zn(II), the supernatant and wash contained only very low concentrations of these metal ions, indicating that the great majority of copper and zinc ions are bound to Aβ. This observation is not surprising in view of the latest data for the dissociation constant (Kd) for Cu(II) bound to Aβ(1–42), which lies in the range 10 pm to 100 nm (41). Our failure to detect free Cu(II) ions is in agreement with data observed by others (employing two unrelated techniques), who also found the absence of any free Cu(II) ions, thus supporting a very high affinity of Aβ(1–42) for Cu(II) (20). The Kd for Zn(II) is believed to be somewhat lower, at 1–20 μm (41). Notably, in our experiments, the other metal ions (Mn(II), Fe(II), and Fe(III)) did not show clear evidence for binding to Aβ.
We next examined the ability of resuspended pellets of Aβ(1–42) aggregates formed in the presence of different metal ions (Mn(II), Fe(II), Fe(III), Zn(II), and Cu(II)) to destroy 1 mm H2O2, employing Amplex red to monitor H2O2 levels over further incubation periods of up to 72 h. This concentration of H2O2 was selected following a series of trial experiments aimed at establishing the best conditions for our experiments. It is notable (Fig. 2C) that all of the treatments, apart from Cu(II), showed only a small deterioration in H2O2 levels with time, but when Aβ(1–42) was bound to Cu(II) ions at a 1:1 incubation ratio, more than 50% of the H2O2 was lost over a 72-h incubation period.
We then examined the ability of Aβ(1–42) aggregates formed in the presence or absence of the different metal ions for evidence of carbonyl group formation. Carbonyl formation is well established and used widely as a measure of oxidative damage via reaction with ROS (36, 42). In these latter experiments, resuspended pellets of the Aβ(1–42) aggregates were incubated at 37 °C with H2O2 for a further 48 h. Significant formation of carbonyl groups was only evident when the peptide was incubated with Cu(II) ions (analysis of variance, F(6, 21) = 174.0, p < 0.001, Tukey's HSD, p < 0.001) (Fig. 2D).
Influence of the Cu(II):Aβ(1–42) Ratio on H2O2 Levels, Aggregate Morphology, and Carbonyl Group Formation
Measurements were taken of H2O2 levels (by Amplex red assay) during incubation (at 37 °C) of 1 mm H2O2 with Aβ(1–42) preaggregated for 144 h in the presence of Cu(II) ions. Here the molar ratio of Cu(II):Aβ(1–42) was varied from zero added Cu(II) ions to 1:1 with the peptide. The results of these experiments are shown in Fig. 3A. Not only do they clearly indicate a decline in the H2O2 level with incubation, but they also show that this decline became greater as the Cu(II) ion concentration was increased with respect to the peptide and was most pronounced when the Cu(II):Aβ(1–42) ratio reached 1:1.
FIGURE 3.

Aβ(1–42) aggregates formed in the presence of Cu(II) ions degrade H2O2 in a concentration-dependent manner and generate hydroxyl radicals. Aβ(1–42) (50 μm) was incubated for 144 h, as before, at the indicated molar ratios of Cu(II):Aβ. Samples were taken for AFM imaging, and the Aβ aggregates were then pelleted and washed as before. The final pellets were resuspended in 1 mm H2O2 to an Aβ concentration of 25 μm and incubated at 37 °C for up to 144 h. A, Amplex red results (data are mean ± S.D., n = 3) for samples of the final suspension assayed every 24 h. B, amount of carbonyl groups formed (data are mean ± S.D., n = 4) for samples of the final suspension taken at 0, 72, and 144 h. RFU, relative fluorescence unit. C, representative AFM images of the initial aggregates formed (2-μm scans). D, coumarin fluorescence assay for hydroxyl radicals for samples of the final suspension taken at 0 and 48 h. Data are mean ± S.D., n = 3. *, p ≤ 0.001; Tukey's HSD.
These results indicate a substantial loss of H2O2 with incubation. Consequently, to investigate possible oxidative damage to Aβ(1–42), we next investigated the formation of carbonyl groups in the peptide (via DNPH immunoassay) at various Cu(II):Aβ(1–42) molar ratios. As can be seen in Fig. 3B, the formation of carbonyl groups in the peptide was enhanced when Cu(II) ions were present (maximizing with Cu(II):Aβ(1–42) at a molar ratio between 1:5 and 1:2).
Because research by others (43) has indicated that the morphology of the Aβ aggregate formed depends on the Cu(II):peptide ratio, we examined the morphology of our aggregates, prepared at 0, 1:4, and 1:1 molar ratios, by AFM. The resulting images are illustrated in Fig. 3C and clearly indicate that the aggregate morphology was affected by the Cu(II):Aβ(1–42) ratio. In the absence of any added Cu(II) ions, the aggregates formed showed typical amyloid fibril morphology. Two populations of fibrils were observed, one with an approximate height of 3.71 ± 0.78 nm and a width of 9.19 ± 0.12 nm (n = 19) and the other of approximately double that size with a height of 6.88 ± 0.56 and a width of 18.45 ± 0.50 nm (n = 13), the latter being consistent with two smaller fibrils entwined together. As the ratio of Cu(II):Aβ(1–42) was increased to 1:4, some small, amorphous, non-fibrillar “granular” aggregates were seen in addition to the fibrillar material. This fibrillar material has similar proportions to the fibrils generated with no copper added, with an approximate height of 3.96 ± 1.05 nm and a width of 7.91 ± 0.18 nm (n = 14). As carbonyl formation maximized at this Cu(II):Aβ(1–42) molar ratio, AFM was also performed to check for carbonyl-induced changes to fibril morphology following incubation with H2O2. The images obtained were similar to those presented in Fig. 3C (at the 1:4 ratio) and showed that fibrillar Aβ remained the predominant form present. At a Cu(II):Aβ(1–42) ratio of 1:1, no fibrillar aggregates were present, and only small, non-fibrillar, amorphous aggregates were observed. These aggregates have an approximate height of 1.49 ± 0.41 nm (n = 19). Differences in morphology are clear, particularly when the Cu(II):peptide ratio is 1:1.
Finally, in this part of our research, we sought to obtain information on the redox activity of the peptide bound to Cu(II) ions by the investigation of the possible conversion of H2O2 into •OH (via coumarin fluorescence assay). The results of this part of our investigation are illustrated in Fig. 3D. As can be seen in this Fig. 3D, the level of •OH detected increased dramatically as the peptide to Cu(II) ion ratio increased, thus indicating the redox activity of the copper-peptide complex. Also illustrated in Fig. 3D are the results of our control experiments, which indicate that neither H2O2 itself nor the buffer (PB) facilitated the formation of any •OH in the absence of the peptide or Cu(II) ions.
We designed an additional set of experiments to investigate the influence of copper ions on fibrils preformed in their absence. A sample of Aβ(1–42) was preaggregated for 144 h in the absence of any deliberately added Cu(II) ions, incubated for a further 24 h in the presence of Cu(II) ions, and washed. Then, the redox activity toward H2O2 was determined. These experiments were undertaken at three different Cu(II):peptide molar ratios (i.e. no added Cu(II) ions, a ratio of 1:4, and a ratio of 1:1), with the results showing a great similarity in the ability to reduce H2O2 levels at each Cu(II):peptide ratio regardless of whether the Cu(II) ions were present during aggregation or added to preformed fibrils (see Fig. 4).
FIGURE 4.
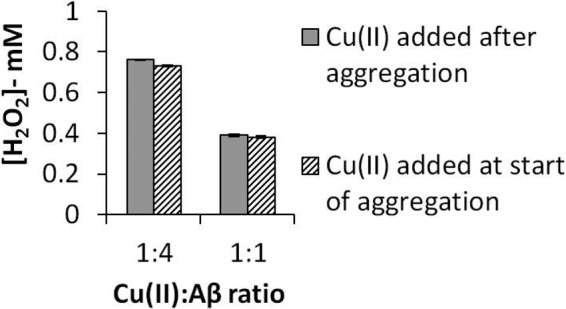
Aβ(1–42) aggregates formed in the presence of Cu(II) ions, or with Cu(II) ions added after aggregation, show comparable abilities to degrade H2O2. Aβ(1–42) (50 μm) was incubated for 144 h, as before, with Cu(II) ions present at the indicated molar ratios or with no added Cu(II) ions. The fibrils formed from Aβ incubated with no added Cu(II) were pelleted, washed, and incubated with Cu(II) ions at these same molar ratios for 24 h. They were then pelleted and washed as before. The final pellets were resuspended in 1 mm H2O2 to an Aβ concentration of 25 μm and incubated at 37 °C for 72 h. Amplex red data for the final levels of H2O2 (mean ± S.D., n = 3) are shown.
Influence of the Cu(II):Zn(II) Ratio on H2O2 Levels and Aggregate Morphology
As noted above, high levels of both copper and zinc are present in Aβ plaques, and, as such, these and some other metal ions have been implicated by some researchers in the pathogenesis of AD. However, much of this research has concentrated on the influence of “single” metal ions rather than on aggregates formed in the presence of “multiple” ions. Consequently, in this part of our research, we considered the effect of competitive binding between or, potentially, binding of both Cu(II) and Zn(II) to Aβ(1–42) and examined how this might affect H2O2 degradation and aggregate morphology.
Our results for the effect of the Cu(II):Zn(II) molar ratio on H2O2 concentration with incubation at 37 °C for up to 72 h are shown in Fig. 5A. Experiments at 50 μm Cu(II) and Zn(II):Cu(II) ratios of 0:1, 1:4, and 1:1 are shown in the three left columns of Fig. 5A. These results show that the H2O2 concentration not only declined with the length of incubation but also that this decline was less at higher Zn(II) levels. The three center columns of Fig. 5A show our results at 12.5 μm Cu(II) ion concentration but with Zn(II):Cu(II) ratios of 0:1, 1:1, and 4:1. This set of experiments shows the same trend with incubation as observed at 50 μm Cu(II), but the decline in H2O2 concentration over time was less at a Cu(II) ion concentration of 12.5 μm than that observed at 50 μm. Finally, the three right columns in Fig. 5A show the results obtained with zero added Cu(II) ions but with Zn(II) concentrations of 0, 12.5, and 50 μm. With both zero Cu(II) and Zn(II) ion concentrations, a very small decline in H2O2 levels with incubation was noted, but when Zn(II) ions were present, there was virtually no change in these levels.
FIGURE 5.
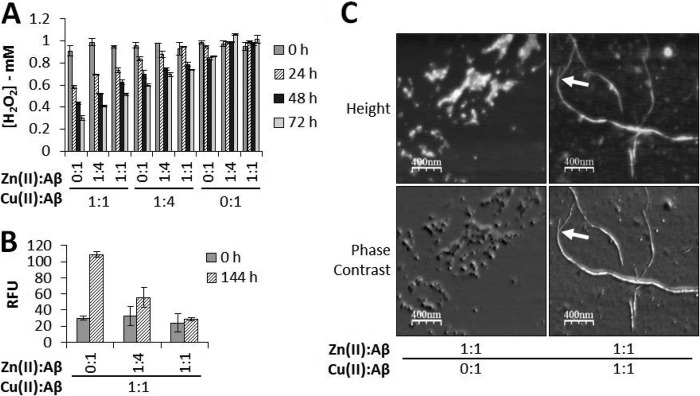
Zn(II) ions prevent the Aβ(1–42)/copper-mediated degradation of H2O2. Aβ(1–42) (50 μm) was incubated for 144 h in PB in the presence of Cu(II) and/or Zn(II) ions at the indicated molar ratios. Samples were taken for AFM imaging, and the aggregates were pelleted and washed as before. The final pellet was resuspended in 1 mm H2O2 to an Aβ concentration of 25 μm and then incubated at 37 °C for up to 144 h. Samples were removed for Amplex red analysis of H2O2 concentration by assay every 24 h and for DNPH carbonyl detection at 0 and 144 h. A, Amplex red results (data are mean ± S.D., n = 3). B, carbonyl groups detected (data are mean ± S.D., n = 4). RFU, relative fluorescence unit. C, representative AFM images of the aggregates formed at the indicated molar ratios of Cu(II) and/or Zn(II) ions (2-μm scans). The arrows indicate where two fibers entwine.
Carbonyl group formation (Fig. 5B) revealed some supporting information. Fig. 5B shows that, as the ratio of Zn(II):Cu(II) was increased from 0:1 to 1:1, the level of carbonyl group formation in the aggregated peptide declined steadily. As zinc increased relative to copper, fewer carbonyl groups were detected, consistent with our results given in Fig. 5A, which shows a reduced loss in H2O2 levels as zinc concentration increases. The observation that, at the highest concentration of Zn(II) to Cu(II), H2O2 levels declined but we detected no obvious increase in carbonyl groups formed may be linked to the sensitivity of this assay or morphological changes that occur at these concentrations of Cu(II) and Zn(II). In either case, Cu(II) bound to the aggregates remains at a sufficient level for bound Cu(II) ions to convert H2O2 to •OH.
Again, we removed some samples to examine their AFM images, and these are illustrated in Fig. 5C. These latter images reveal some important information. When only Zn(II) is present, small, amorphous, non-fibrillar (fairly spherical) aggregates were seen and have an approximate height of 5.97 ± 1.40 nm and a width of 27.50 ± 2.73 nm (n = 16). However, Fig. 5C shows that, at a Zn(II):Cu(II) ratio of 1:1, both some non-fibrillar and some fibrillar materials were clearly visible. Two populations of fibers were measured, one with an approximate height of 2.01 ± 0.54 nm and a width 8.00 ± 0.58 nm (n = 11) and the other with a height of 4.62 ± 0.86 nm and a width 14.68 ± 0.90 nm (n = 11), clearly formed via the association of two smaller fibers. One such example is indicated in Fig. 5C (arrows).
DISCUSSION
In an earlier publication, we presented the results of our study of the formation and decay of H2O2 during the incubation of both Aβ(1–40) and the oxidized form of the ABri peptide associated with familial British dementia (25). In both cases, we were able to show that the self-generation of H2O2 occurred as a short “burst” during the very early stages of aggregation when oligomers were the main species present. After this short burst of activity, H2O2 levels reached a maximum before declining as incubation continued. We speculated that that the decline was either due to a direct reaction of H2O2 with the peptide or due to conversion of H2O2 into •OH via redox activity that then attacked the peptide.
A closer examination of these earlier results indicates that this decline in H2O2 levels continues when fibrillar aggregates are the majority species present. This decline could be due to the conversion of the some of the peroxide into •OH via the redox activity of any metal ion bound to the peptide. Consequently, in this work, we focused our research on investigating the possibility that, when Aβ(1–42) fibrils are bound to a redox-active metal ion such as Cu(II), the metal ion retains its redox activity and, therefore, is capable of degrading H2O2 and releasing the highly reactive •OH radical. This highly reactive radical, if formed in the vicinity of the peptide, could inflict substantial oxidative damage on the peptide via a range of reactions.
As a preliminary to our main experiments, we tested the ability of preaggregated Aβ(1–42), formed by incubating the peptide with various metal ions, to destroy H2O2. Our results are summarized in Fig. 2C. A very small decrease in H2O2 levels over 72 h of incubation can be detected when the aggregate is made in the presence of Mn(II), Fe(II), or Fe(III) and also in the absence of any added metal ion, but the most dramatic decline by far is noted when Aβ is incubated with Cu(II). This latter observation is strongly supported by the results of our detection of the formation of carbonyl groups within Aβ(1–42) when Cu(II) was bound to the peptide (see Fig. 2D). Consequently, we conclude that oxidation is clearly evident when the aggregates are bound to Cu(II), which must, therefore, remain redox-active in the aggregated peptide.
To examine the retention of redox activity in these preformed aggregates, we extended the incubation period to 144 h and examined the influence of the Cu(II):Aβ(1–42) molar ratio on H2O2 levels (see Fig. 3A). We noted that there was a small decline in the peroxide level when only sample preparation levels of metal ions were present (i.e. in the absence of deliberately added Cu(II)). However, a considerably greater decline was noted when copper ions were incubated with, and bound to, the peptide. The rate of degradation of H2O2 increased as more Cu(II) was included to a maximum at a Cu(II):Aβ(1–42) ratio of 1:1 (Fig. 3A). The level of •OH generated by the Aβ(1–42) aggregates formed at different molar ratios of Cu(II) supported these observations (Fig. 3D). Increasing Cu(II) generated more •OH, with a maximum at 1:1, further indicating that loss of H2O2 was due to conversion to •OH.
We also examined AFM images of the various samples (Fig. 3C) because we are aware that the morphology of the aggregates made in the presence of Cu(II) changes with the Cu(II):Aβ(1–42) molar ratio. In agreement with previous results (for example, see Ref. 43), we observed that, toward a 1:1 ratio, the aggregate becomes amorphous and non-fibrillar. At a 1:4 ratio, fibrils are the predominant aggregate observed. When no added copper ions are present, the fibrils tend to be more intertwined, with both single and multiple fibers (with dimensions approximately double those of the single fibers) being seen. These observations compare very favorably with those published previously by others (43). Our main conclusion from this part of research is that the aggregates formed at different Cu(II):Aβ(1–42) molar ratios up to 1:1 retain their redox activity irrespective of their morphology and are capable of converting H2O2 into •OH. However, of particular relevance to AD, the fibrils formed at lower concentrations of Cu(II) also have this redox activity. In further support of the fact that copper-bound Aβ fibrils within senile plaques may also have this capacity, we showed that Cu(II) ions bound to the peptide are redox-active whether they are present during aggregation or are added to preformed fibrils.
We also found evidence for oxidative damage to the peptide via the formation of carbonyl groups (Fig. 3B). The formation of carbonyl groups is well established as an indicator of the attack on proteins by ROS (36, 42). This chemical modification can alter protein secondary and tertiary structure, which can be detrimental to protein functionality. Many carbonylated proteins are targeted for digestion by the 20 S proteasome, and they can inhibit proteasome activity (44). There is also some evidence that they are particularly prone to aggregation (45). Protein carbonylation could, therefore, contribute to neuronal degeneration and dysfunction in neurodegenerative disorders through these mechanisms. However, the role of carbonylation of Aβ in the pathogenesis of AD is unclear.
In this study, carbonyl adducts were not evident in Aβ aggregates prior to the addition of H2O2 independently of their metal ion content and, therefore, of their morphology. This suggests that carbonylation is not a major driving force for Aβ amorphous aggregation or fibrillization under these experimental conditions. Upon addition of H2O2, the formation of carbonyl adducts maximized at a Cu(II):Aβ(1–42) molar ratio of between 1:2 and 1:5, when fibrils are the predominant type of aggregate. This may indicate that the morphology of the aggregates is important in determining whether the •OH formed then oxidize the peptide to form carbonyls, with fibrillar aggregates being more easily oxidized. However, there are two other alternative explanations for this. First, the conformational arrangement of the protein in amorphous aggregates could conceal carbonyl groups that are formed to a greater extent than in the fibrillar aggregates and, therefore, inhibit the reaction between DNPH and the anti-DNPH mAb. The second possibility is that genuine fibrillar aggregates formed in the presence of subequimolar concentrations of Cu(II) adhere more tightly to the microtiter plates than the amorphous aggregates formed with equimolar Aβ(1–42) and Cu(II) and, therefore, give a higher signal for carbonyl groups. Regardless, the oxidative damage detected here, in the form of carbonyl group formation, confirms our observation that redox activity is retained in Aβ(1–42) aggregates produced in the presence of up to an equimolar concentration of Cu(II), with substantial conversion of H2O2 to •OH.
It is known that both copper and zinc ion concentrations are high in the brains of AD patients (for example, see Refs. 46, 47 and the references quoted therein), and the study of Aβ in the presence of these ions has received considerable attention. Most of this research has concentrated on the influence of either Cu(II) or Zn(II) on the aggregation and redox activity of Aβ. In comparison, little attention has been focused on the aggregates formed when Aβ(1–42) is coincubated in the presence of both of these ions (48). Therefore, we investigated preformed Aβ(1–42) aggregates prepared in the presence of both of these ions and at different metal ion ratios (Fig. 5). Fig. 5A shows how H2O2 levels are influenced by Zn(II):Cu(II):Aβ(1–42) molar ratios over incubation periods of up to 72 h. At a Cu(II):Aβ(1–42) ratio of 1:1 but varying Zn(II) ratios up to 1:1:1, the loss of H2O2 is substantial but declines as Zn(II) increases. The same observation is true at a Cu(II):Aβ(1–42) molar ratio of 1:4, but the deterioration in H2O2 is less noticeable than at 1:1. This may due to competition between redox-active Cu(II) and redox-inactive Zn(II) for binding sites, with those Cu(II) ions still bound to the peptide retaining their redox activity. It is possible, however, that both Cu(II) and Zn(II) could be bound via an alternate binding mode. Cu(II) is believed to have a second, albeit lower-affinity, binding site (49, 50). In addition, a number of alternative binding sites are reported for Zn(II) (discussed in Ref. 41), with evidence suggesting only a partial overlap between Cu(II) and Zn(II) binding sites (41, 50, 51). In this scenario, the binding of Zn(II) may prevent the redox events enabled by Cu(II) binding via disruption of the Aβ aggregate morphology that supports the redox capabilities of the Cu(II)-Aβ complex. Supporting this, in the complete absence of added Cu(II) there is little or no decline in H2O2 levels with incubation as the Zn(II):Aβ(1–42) ratio varies. We also found supporting evidence for some Cu(II) binding to Aβ(1–42) from our detection of carbonyl groups in the peptide as the Zn(II):Aβ(1–42) ratio varied (Fig. 5B). Here, the formation of carbonyl groups decreased as the ratio of Zn(II) ions to Cu(II) ions increased. Again, we believe that this oxidative damage is a consequence of the conversion of H2O2 to •OH (via redox activity) and the subsequent attack of this latter radical on the peptide. Notably, our results with both Cu(II) and Zn(II) together are compatible with a recent report showing that Zn(II) can “redox-silence” Aβ, in this case by suppressing the copper-dependent formation of H2O2 from Aβ(1–42) and rescuing cells from the damaging effects of this peptide (52).
Finally, we find that further supporting evidence for competition between Cu(II) and Zn(II) can be seen in the corresponding AFM images (Fig. 5C). When the Zn(II):Aβ(1–42) ratio is 1:1 (but in the absence of added Cu(II)), somewhat spherical amorphous aggregates are seen. This type of aggregate has been noted before in the case of Aβ(1–40) (27, 53, 54). However, the images obtained for samples prepared when both metal ions and the peptide are present at a 1:1:1 ratio show the coexistence of fibrillar and amorphous aggregates.
In the Introduction, we outlined a change in opinion regarding the amyloid cascade hypothesis, with a move toward the belief that the predominant toxic forms of Aβ are small, soluble oligomers. Support for this latter view has been mounting over recent years, and fibrillar forms of Aβ are now seen by many researchers as being relatively non-toxic compared with oligomeric forms, or even as a protective response of the brain. However, our new results, presented here, indicate that, in the presence of bound redox-active ions, particularly Cu(II), mature aggregates, including fibrils, retain their redox activity. Consequently, they remain capable of converting H2O2 to the very reactive •OH radical. This could subsequently result in oxidative damage to the peptide itself and also to surrounding areas of the brain that are immediately adjacent to senile plaque amyloid deposits. Mature fibrils are not, therefore, simply inert tombstones at the end of the aggregation process. This could help to explain the many “reactive” processes that have been found to occur in the brain in the vicinity of senile plaque amyloid deposits, including abnormal neuritic processes of nerve cells and the recruitment and activation of glial cells (55).
We have drawn together some of our main observations and illustrated them in Fig. 6. In the absence of any added Cu(II) (but in the presence of any Cu(II) that is unavoidably present in the materials used in the preaggregation of our samples), we find (Fig. 6, top left) some loss of H2O2 over a 144-h incubation period. Moving counterclockwise, we note that this loss is much more noticeable when the Cu(II):Aβ(1–42) ratio is 1:4 and even more obvious when it is 1:1. Changes in aggregate morphology and a decline in H2O2 levels and in carbonyl group formation are also noted as the ratio of copper ions increases. When the Cu(II):Zn(II) ratio is set at 1:1 in the preaggregation of the peptide, the loss of H2O2 is still considerable, but no loss is noted when only Zn(II) (and no Cu(II)) is present (Fig. 6, top right).
FIGURE 6.

Aβ42 incubated with copper and zinc forms distinct aggregates that have different capacities for hydrogen peroxide degradation. Aβ42 incubated with no exogenous copper, or at a 1:4 copper:Aβ molar ratio, forms typical amyloid fibrils, with H2O2 degradation being proportional to copper concentration. Increasing copper to 1:1 prevents fibril formation, with hydrogen peroxide degradation and hydroxyl radical formation increasing further. Addition of 1:1 zinc to Aβ also prevents fibril formation and eliminates H2O2 degradation. Equimolar copper and zinc generates atypical fibers that are capable of similar levels of hydrogen peroxide degradation to 1:4 copper. This is hypothesized to be due to competition for the metal ion binding site(s).
There is further support for our concepts in a recent publication by Sayre et al. (56), who have shown that β-amyloid plaques in situ in human brain sections can convert 3,3′-diaminobenzidine into its insoluble brown reaction product in the presence of H2O2 but in the complete absence of any peroxidase enzyme. This shows that the amyloid plaques themselves have peroxidase-like activity, which would be compatible with our observation that Aβ fibrils can destroy H2O2. Interestingly, neurofibrillary tangles also showed a similar property, suggesting that both plaques and tangles are sites of this type of redox activity (56).
Of course, for our hypothesis to be valid, H2O2 would have to be present in the vicinity of senile plaques in the human brain. However, this peroxide is very diffusible within and between cells and is formed in sizeable quantities by a number of biological systems, including mitochondria and, especially, dysfunctional mitochondria, which appear to be present in AD and other neurodegenerative conditions (for fuller details, see Ref. 57). Moreover, as noted above, some forms of prefibrillar Aβ are also capable of generating H2O2.
We believe that our new experiments help to emphasize the growing realization that metal ions (especially copper ions) are important in the pathogenesis of AD, supporting recent research like that by Singh et al. (58). It is now widely established that copper ions bind directly to the peptide (59), influence its aggregation, and modify the morphology of the aggregate (43). The redox activity of the copper-bound peptide converts any physiological H2O2 to •OH (60), thus acting as a source of ROS.
In conclusion, we have shown that the redox activity of the Cu(II)-bound peptide is retained in fibrillar Aβ(1–42) aggregates, which are capable of destroying H2O2. Amyloid fibrils are, therefore, not necessarily “inert bystanders” at the end of the disease process. It is possible that, in the future, new drugs could be developed that specifically target this Aβ redox activity, and these could be beneficial even in the more advanced stages of AD. Metal ion chelators are already in development as a possible AD therapy (61), but drugs that specifically bind to sites in Aβ fibrils that are involved in this redox activity could be a viable alternative option.
This work was supported by The Sir John Fisher Foundation.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- ROS
- reactive oxygen species
- PB
- phosphate buffer
- ThT
- Thioflavin T
- AFM
- atomic force microscopy
- DNPH
- 2,4-dinitrophenyl hydrazine.
REFERENCES
- 1. Hardy J., Allsop D. (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 [DOI] [PubMed] [Google Scholar]
- 2. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 3. Walsh D. M., Selkoe D. J. (2007) Aβ oligomers: a decade of discovery. J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 4. Demuro A., Mina E., Kayed R., Milton S. C., Parker I., Glabe C. G. (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280, 17294–17300 [DOI] [PubMed] [Google Scholar]
- 5. Klein W. L., Stine W. B., Jr., Teplow D. B. (2004) Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging 25, 569–580 [DOI] [PubMed] [Google Scholar]
- 6. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 7. Chromy B. A., Nowak R. J., Lambert M. P., Viola K. L., Chang L., Velasco P. T., Jones B. W., Fernandez S. J., Lacor P. N., Horowitz P., Finch C. E., Krafft G. A., Klein W. L. (2003) Self-assembly of Aβ(1–42) into globular neurotoxins. Biochemistry 42, 12749–12760 [DOI] [PubMed] [Google Scholar]
- 8. Kim H. J., Chae S. C., Lee D. K., Chromy B., Lee S. C., Park Y. C., Klein W. L., Krafft G. A., Hong S. T. (2003) Selective neuronal degeneration induced by soluble oligomeric amyloid β protein. FASEB J. 17, 118–120 [DOI] [PubMed] [Google Scholar]
- 9. Mucke L., Masliah E., Yu G. Q., Mallory M., Rockenstein E. M., Tatsuno G., Hu K., Kholodenko D., Johnson-Wood K., McConlogue L. (2000) High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein W. L., Krafft G. A., Finch C. E. (2001) Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224 [DOI] [PubMed] [Google Scholar]
- 11. Tomic J. L., Pensalfini A., Head E., Glabe C. G. (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kayed R., Sokolov Y., Edmonds B., McIntire T. M., Milton S. C., Hall J. E., Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366 [DOI] [PubMed] [Google Scholar]
- 13. Bitan G., Kirkitadze M. D., Lomakin A., Vollers S. S., Benedek G. B., Teplow D. B. (2003) Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nunomura A., Hofer T., Moreira P. I., Castellani R. J., Smith M. A., Perry G. (2009) RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 118, 151–166 [DOI] [PubMed] [Google Scholar]
- 15. Riederer B. M., Leuba G., Elhajj Z. (2013) Oxidation and ubiquitination in neurodegeneration. Exp. Biol. Med. (Maywood) 238, 519–524 [DOI] [PubMed] [Google Scholar]
- 16. Allsop D., Mayes J., Moore S., Masad A., Tabner B. J. (2008) Metal-dependent generation of reactive oxygen species from amyloid proteins implicated in neurodegenerative disease. Biochem. Soc. Trans. 36, 1293–1298 [DOI] [PubMed] [Google Scholar]
- 17. Nunomura A., Perry G., Aliev G., Hirai K., Takeda A., Balraj E. K., Jones P. K., Ghanbari H., Wataya T., Shimohama S., Chiba S., Atwood C. S., Petersen R. B., Smith M. A. (2001) Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767 [DOI] [PubMed] [Google Scholar]
- 18. Markesbery W. R., Kryscio R. J., Lovell M. A., Morrow J. D. (2005) Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann. Neurol. 58, 730–735 [DOI] [PubMed] [Google Scholar]
- 19. Huang X., Atwood C. S., Hartshorn M. A., Multhaup G., Goldstein L. E., Scarpa R. C., Cuajungco M. P., Gray D. N., Lim J., Moir R. D., Tanzi R. E., Bush A. I. (1999) The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38, 7609–7616 [DOI] [PubMed] [Google Scholar]
- 20. Huang X., Cuajungco M. P., Atwood C. S., Hartshorn M. A., Tyndall J. D., Hanson G. R., Stokes K. C., Leopold M., Multhaup G., Goldstein L. E., Scarpa R. C., Saunders A. J., Lim J., Moir R. D., Glabe C., Bowden E. F., Masters C. L., Fairlie D. P., Tanzi R. E., Bush A. I. (1999) Cu(II) potentiation of Alzheimer Aβ neurotoxicity: correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274, 37111–37116 [DOI] [PubMed] [Google Scholar]
- 21. Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998) Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 158, 47–52 [DOI] [PubMed] [Google Scholar]
- 22. Smith M. A., Harris P. L., Sayre L. M., Perry G. (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. U.S.A. 94, 9866–9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tabner B. J., Turnbull S., El-Agnaf O. M., Allsop D. (2002) Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer's disease and Parkinson's disease. Free Radic. Biol. Med. 32, 1076–1083 [DOI] [PubMed] [Google Scholar]
- 24. Turnbull S., Tabner B. J., El-Agnaf O. M., Moore S., Davies Y., Allsop D. (2001) α-Synuclein implicated in Parkinson's disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 30, 1163–1170 [DOI] [PubMed] [Google Scholar]
- 25. Tabner B. J., El-Agnaf O. M., Turnbull S., German M. J., Paleologou K. E., Hayashi Y., Cooper L. J., Fullwood N. J., Allsop D. (2005) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 280, 35789–35792 [DOI] [PubMed] [Google Scholar]
- 26. Harper J. D., Wong S. S., Lieber C. M., Lansbury P. T. (1997) Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 4, 119–125 [DOI] [PubMed] [Google Scholar]
- 27. Yoshiike Y., Tanemura K., Murayama O., Akagi T., Murayama M., Sato S., Sun X., Tanaka N., Takashima A. (2001) New insights on how metals disrupt amyloid β-aggregation and their effects on amyloid-β cytotoxicity. J. Biol. Chem. 276, 32293–32299 [DOI] [PubMed] [Google Scholar]
- 28. Tabner B. J., Turnbull S., El-Agnaf O., Allsop D. (2001) Production of reactive oxygen species from aggregating proteins implicated in Alzheimer's disease, Parkinson's disease and other neurodegenerative diseases. Curr. Top. Med. Chem. 1, 507–517 [DOI] [PubMed] [Google Scholar]
- 29. Tabner B. J., Mayes J., Allsop D. (2011) Hypothesis: soluble Aβ oligomers in association with redox-active metal ions are the optimal generators of reactive oxygen species in Alzheimer's disease. Int. J. Alzheimers Dis. 10.4061/2011/546380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tabner B. J., Turnbull S., El-Agnaf O. M. A., Allsop D. (2003) Direct production of reactive oxygen species from aggregating proteins and peptides implicated in the pathogenesis of neurodegenerative diseases. Curr. Med. Chem. 3, 299–308 [DOI] [PubMed] [Google Scholar]
- 31. Manzoni C., Colombo L., Messa M., Cagnotto A., Cantù L., Del Favero E., Salmona M. (2009) Overcoming synthetic Aβ peptide aging: a new approach to an age-old problem. Amyloid 16, 71–80 [DOI] [PubMed] [Google Scholar]
- 32. LeVine H., 3rd. (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou M., Diwu Z., Panchuk-Voloshina N., Haugland R. P. (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 253, 162–168 [DOI] [PubMed] [Google Scholar]
- 34. Horcas I., Fernández R., Gómez-Rodríguez J. M., Colchero J., Gómez-Herrero J., Baro A. M. (2007) WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 78, 013705 [DOI] [PubMed] [Google Scholar]
- 35. Kiselyova O. I., Yaminsky I. V. (2004) Atomic force microscopy of protein complexes. Methods Mol. Biol. 242, 217–230 [DOI] [PubMed] [Google Scholar]
- 36. Dalle-Donne I., Rossi R., Giustarini D., Milzani A., Colombo R. (2003) Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 329, 23–38 [DOI] [PubMed] [Google Scholar]
- 37. Manevich Y., Held K. D., Biaglow J. E. (1997) Coumarin-3-carboxylic acid as a detector for hydroxyl radicals generated chemically and by γ radiation. Radiat. Res. 148, 580–591 [PubMed] [Google Scholar]
- 38. Tabner B. J., El-Agnaf O. M., German M. J., Fullwood N. J., Allsop D. (2005) Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 33, 1082–1086 [DOI] [PubMed] [Google Scholar]
- 39. Hewitt N., Rauk A. (2009) Mechanism of hydrogen peroxide production by copper-bound amyloid β peptide. A theoretical study. J. Phys. Chem. B 113, 1202–1209 [DOI] [PubMed] [Google Scholar]
- 40. Hureau C., Faller P. (2009) Aβ-mediated ROS production by Cu ions: structural insights, mechanisms and relevance to Alzheimer's disease. Biochimie 91, 1212–1217 [DOI] [PubMed] [Google Scholar]
- 41. Faller P., Hureau C. (2009) Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-β peptide. Dalton Trans. 7, 1080–1094 [DOI] [PubMed] [Google Scholar]
- 42. Davies M. J. (2005) The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 [DOI] [PubMed] [Google Scholar]
- 43. Smith D. P., Ciccotosto G. D., Tew D. J., Fodero-Tavoletti M. T., Johanssen T., Masters C. L., Barnham K. J., Cappai R. (2007) Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer's disease amyloid-β peptide. Biochemistry 46, 2881–2891 [DOI] [PubMed] [Google Scholar]
- 44. Dasgupta A., Zheng J., Bizzozero O. A. (2012) Protein carbonylation and aggregation precede neuronal apoptosis induced by partial glutathione depletion. ASN Neuro 4, e00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao L., Buxbaum J. N., Reixach N. (2013) Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 52, 1913–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen W. T., Liao Y. H., Yu H. M., Cheng I. H., Chen Y. R. (2011) Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-β stability, oligomerization, and aggregation: amyloid-β destabilization promotes annular protofibril formation. J. Biol. Chem. 286, 9646–9656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greenough M. A., Camakaris J., Bush A. I. (2013) Metal dyshomeostasis and oxidative stress in Alzheimer's disease. Neurochem. Int. 62, 540–555 [DOI] [PubMed] [Google Scholar]
- 48. Ryu J., Girigoswami K., Ha C., Ku S. H., Park C. B. (2008) Influence of multiple metal ions on β-amyloid aggregation and dissociation on a solid surface. Biochemistry 47, 5328–5335 [DOI] [PubMed] [Google Scholar]
- 49. Syme C. D., Nadal R. C., Rigby S. E., Viles J. H. (2004) Copper binding to the amyloid-β (Aβ) peptide associated with Alzheimer's disease: folding, coordination geometry, pH dependence, stoichiometry, and affinity of Aβ-(1–28): insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 279, 18169–18177 [DOI] [PubMed] [Google Scholar]
- 50. Atwood C. S., Scarpa R. C., Huang X., Moir R. D., Jones W. D., Fairlie D. P., Tanzi R. E., Bush A. I. (2000) Characterization of copper interactions with Alzheimer amyloid-β peptides: identification of an attomolar-affinity copper binding site on amyloid-β 1–42. J. Neurochem. 75, 1219–1233 [DOI] [PubMed] [Google Scholar]
- 51. Syme C. D., Viles J. H. (2006) Solution 1H NMR investigation of Zn2+ and Cd2+ binding to amyloid-β peptide (Aβ) of Alzheimer's disease. Biochim. Biophys. Acta 1764, 246–256 [DOI] [PubMed] [Google Scholar]
- 52. Cuajungco M. P., Goldstein L. E., Nunomura A., Smith M. A., Lim J. T., Atwood C. S., Huang X., Farrag Y. W., Perry G., Bush A. I. (2000) Evidence that the β-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of Aβ by zinc. J. Biol. Chem. 275, 19439–19442 [DOI] [PubMed] [Google Scholar]
- 53. Raman B., Ban T., Yamaguchi K., Sakai M., Kawai T., Naiki H., Goto Y. (2005) Metal ion-dependent effects of clioquinol on the fibril growth of an amyloid-β peptide. J. Biol. Chem. 280, 16157–16162 [DOI] [PubMed] [Google Scholar]
- 54. Tõugu V., Karafin A., Zovo K., Chung R. S., Howells C., West A. K., Palumaa P. (2009) Zn(II)-and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1–42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 110, 1784–1795 [DOI] [PubMed] [Google Scholar]
- 55. Smith M. A., Perry G., Richey P. L., Sayre L. M., Anderson V. E., Beal M. F., Kowall N. (1996) Oxidative damage in Alzheimer's. Nature 382, 120–121 [DOI] [PubMed] [Google Scholar]
- 56. Sayre L. M., Perry G., Harris P. L., Liu Y., Schubert K. A., Smith M. A. (2000) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals. J. Neurochem. 74, 270–279 [DOI] [PubMed] [Google Scholar]
- 57. Mancuso C., Scapagini G., Currò D., Giuffrida Stella A. M., De Marco C., Butterfield D. A., Calabrese V. (2007) Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front. Biosci. 12, 1107–1123 [DOI] [PubMed] [Google Scholar]
- 58. Singh I., Sagare A. P., Coma M., Perlmutter D., Gelein R., Bell R. D., Deane R. J., Zhong E., Parisi M., Ciszewski J., Kasper R. T., Deane R. (2013) Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. U.S.A. 110, 14771–14776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tõugu V., Karafin A., Palumaa P. (2008) Binding of zinc(II) and copper(II) to the full-length Alzheimer's amyloid-β peptide. J. Neurochem. 104, 1249–1259 [DOI] [PubMed] [Google Scholar]
- 60. Barnham K. J., Masters C. L., Bush A. I. (2004) Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214 [DOI] [PubMed] [Google Scholar]
- 61. Duce J. A., Bush A. I. (2010) Biological metals and Alzheimer's disease: implications for therapeutics and diagnostics. Prog. Neurobiol. 92, 1–18 [DOI] [PubMed] [Google Scholar]