

Background: NAI-107 is a potent lantibiotic with an unknown mode of action.
Results: NAI-107 targets bactoprenol-bound cell envelope precursors, e.g. lipid II, and in addition affects the bacterial membrane.
Conclusion: Cell wall biosynthesis is blocked by sequestration of lipid II and functional disorganization of the cell wall machinery.
Significance: The dual mechanism of action may explain the potency of NAI-107 and related lantibiotics.
Keywords: Antibiotics, Cell Wall, Membrane, Nuclear Magnetic Resonance, Peptidoglycan, Staphylococcus aureus, Teichoic Acid, Lantibiotics, Lipid II, Nisin
Abstract
The lantibiotic NAI-107 is active against Gram-positive bacteria including vancomycin-resistant enterococci and methicillin-resistant Staphylococcus aureus. To identify the molecular basis of its potency, we studied the mode of action in a series of whole cell and in vitro assays and analyzed structural features by nuclear magnetic resonance (NMR). The lantibiotic efficiently interfered with late stages of cell wall biosynthesis and induced accumulation of the soluble peptidoglycan precursor UDP-N-acetylmuramic acid-pentapeptide (UDP-MurNAc-pentapeptide) in the cytoplasm. Using membrane preparations and a complete cascade of purified, recombinant late stage peptidoglycan biosynthetic enzymes (MraY, MurG, FemX, PBP2) and their respective purified substrates, we showed that NAI-107 forms complexes with bactoprenol-pyrophosphate-coupled precursors of the bacterial cell wall. Titration experiments indicate that first a 1:1 stoichiometric complex occurs, which then transforms into a 2:1 (peptide: lipid II) complex, when excess peptide is added. Furthermore, lipid II and related molecules obviously could not serve as anchor molecules for the formation of defined and stable nisin-like pores, however, slow membrane depolarization was observed after NAI-107 treatment, which could contribute to killing of the bacterial cell.
Introduction
Lanthipeptides are small (19–39 amino acids) ribosomally synthesized (1, 2) and post-translationally modified peptides, which are characterized by lanthionine and methyl-lanthionine containing intramolecular rings (3). The most prominent group of lanthipeptides are lantibiotics that are produced by and act on Gram-positive bacteria (4, 5). Lantibiotics are chemically and structurally very diverse; two subgroups of lantibiotics have been distinguished, based on differences in their structures and modes of action (6). Type A lantibiotics, such as nisin, are screw-shaped, elongated, flexible, and amphiphatic peptides with an overall positive net charge and pore-forming activities. Type B lantibiotics, e.g. mersacidin, are globular and carry either no net charge or are negatively charged. A more recent classification of lantibiotics in different subgroups has been proposed, based on the organization of their biosynthetic genes and enzymes, rather than on the activity profile or three-dimensional structure (3, 7). The activity of lantibiotics is based on different killing mechanisms, which may be combined in one molecule. The most extensively studied lantibiotic is nisin (8). It is produced by some Lactococcus lactis and widely used as a food preservative for more than 40 years. Like many lantibiotics nisin inhibits growth of Gram-positive bacterial strains by interfering with peptidoglycan through binding to the key intermediate lipid II (9–12).
Lipid II represents the central cell wall building block of peptidoglycan biosynthesis that is structurally conserved among eubacteria. The precursor consists of the bactoprenol carrier lipid (C55-P), and is linked to the peptidoglycan building block N-acetylmuramyl-pentapeptide-N-acetylglucosamine (MurNAc-pp-GlcNAc) via a pyrophosphate bridge. Lipid II synthesis occurs at the inner face of the cytoplasmic membrane, where the translocase MraY and the transferase MurG link the soluble, UDP-activated sugars UDP-N-acetylmuramic acid-pentapeptide and UDP-N-acetylglucosamine to produce lipid I and lipid II, respectively. Lipid II is characteristically modified in different species. In staphylococci, the peptidyltransferases FemXAB catalyze the attachment of a pentaglycine-interpeptide bridge (13, 14) and the glutamate in position 2 of the stempeptide is amidated to glutamine by the bienzyme complex MurT-GatD (15). The fully modified lipid II molecule is then translocated across the cytoplasmic membrane (16) where it is assembled into the growing peptidoglycan network through the activity of transglycosylases and transpeptidases (penicillin-binding protein (PBPs)), thereby releasing the C55P-carrier, which after dephosphorylation enters a new synthesis cycle. Binding of nisin to lipid II locks the cell wall precursor in a stable complex, thereby blocking the entire peptidoglycan synthesis cycle. Additionally, nisin combines this activity with a unique target-mediated pore formation, using lipid II as a docking molecule, and causing dissipation of the membrane potential, rapid efflux of small metabolites, and cessation of cellular biosynthetic processes (17–19).
The renewed interest in the chemotherapeutic potential of antibacterial lantibiotics that inhibit cell wall synthesis originates from the fact that they are effective in treating infections sustained by methicillin-resistant Staphylococcus aureus and that they do not show cross-resistance with glycopeptides (7, 20). In fact, binding of lantibiotics does not involve the glycopeptide binding site, i.e. the C-terminal d-alanyl-d-alanine (d-Ala-d-Ala) moiety of the pentapeptide side chain. Rather, lantibiotics containing the nisin-like double ring system at the N terminus bind to the pyrophosphate linkage unit of lipid II, which equally blocks access of the transglycosylase to its substrate (21–23). For the same group of lantibiotics it has been recently shown that besides binding to lipid II, they interact with the lipid intermediates lipid III (undecaprenol-pyrophosphate-N-acetylglucosamine) and lipid IV (undecaprenol-pyrophosphate-N-acetylglucosamine-N-acetylmannosamine) of the wall teichoic acid (WTA)4 biosynthesis pathway (24) all sharing the pyrophosphate moiety of the bactoprenol membrane anchor.
NAI-107 (Fig. 1), also known as microbisporicin or 107891 (20), is a new type AI lantibiotic produced by the actinomycete Microbispora s. ATCC PTA-5024, which is active against multidrug-resistant Gram-positive pathogens, including methicillin-resistant S. aureus, vancomycin-resistant Enterococcus sp., penicillin-resistant Streptococcus pneumoniae and unusually for a lantibiotic, also against some Gram-negative species (20, 21). The compound was discovered during a screening program designed to detect all classes of cell-wall inhibitors except for β-lactams and glycopeptides (4). In addition to one methyllanthionine and three lanthionine bridges and a C-terminal S-((Z)-2-aminovinyl)-d-cysteine, modifications found in other lantibiotics, NAI-107 contains two unusually modified amino acids: 5-chlorotryptophan and 3,4-dihydroxyproline (16, 25). These modifications are unique to NAI-107 within the lantibiotic class of compounds and are rare in other ribosomally synthesized peptides. Genetic analysis previously identified the biosynthetic gene cluster and revealed insights into the pathway-specific regulation by an extracytoplasmic function σ-factor·anti-σ-factor complex (26, 27).
FIGURE 1.

Structures of the lantibiotics NAI-107, gallidermin, nisin, and mersacidin. The nisin-like lipid II binding motif is highlighted in green and lipid II binding motifs similar to that found in mersacidin are marked red (22, 23).
NAI-107 is produced as a complex of two major structurally related 24-amino acid variants (A1, 2246 Da; and A2, 2230 Da), which differ in proline 14 being monohydroxylated in variant A2, or bishydroxylated in variant A1 (28). Overall NAI-107 seems to combine two known lipid II targeting motifs (Fig. 1) with its 1–11 N-terminal sequence being similar to nisin and its C-terminal ring system, which shares structural elements with epidermin and mersacidin (3, 29). NAI-107 is currently in late preclinical development and displays efficacy in animal models of multidrug-resistant infections superior to the drugs of last resort, linezolid and vancomycin (30). Interestingly, NAI-107-resistant mutants were not observed during these studies. Preliminary mode of action studies gave the first hints toward inhibition of cell wall biosynthesis (28). In the present study, we set out to identify the molecular target and the specific mechanism of action of the lantibiotic NAI-107.
MATERIALS AND METHODS
Susceptibility Testing
Determination of minimal inhibitory concentrations (MICs) was performed in 96-well polypropylene microtiter plates (Nunc) by standard broth microdilution in cation-adjusted Mueller-Hinton broth (Oxoid), according to the general guidelines provided by CLSI/NCCLS. NAI-107 was prepared essentially as described (31).
Killing Kinetics
S. aureus ATCC 29213 was grown overnight in half-concentrated Mueller Hinton Broth and diluted in fresh medium to an optical density (A600) of 0.1. NAI-107 was added in concentrations corresponding to 1× and 2× MIC (4 and 8 μg/ml, respectively). Controls were made either without peptide or with the corresponding amount of dimethyl sulfoxide in the culture. The viable count was monitored up to 24 h. Aliquots were taken at defined intervals, especially focusing on the first 60 min. Colony counts were performed on serially diluted samples in 0.9% NaCl and 100 μl of the dilutions were plated onto MH agar plates. The plates were incubated at 37 °C and the colony forming units (cfu) were read after 24 h.
Incorporation of Radioactive Metabolites
The effect of NAI-107 on the synthesis of macromolecules was studied monitoring the incorporation of 3H- or 14C-labeled precursors (5-[3H]thymidine, [3H]glucosamine hydrochloride, and l-[14C]isoleucine). An overnight culture of Staphylococcus simulans M22 grown in half-concentrated Mueller Hinton Broth containing 1 mm of the respective unlabeled metabolite was diluted 50-fold into fresh medium and cultured at 37 °C to an A600 of 0.5. Cultures were then split into two aliquots, diluted to an A600 of 0.04 and allowed to regrow to an A600 of 0.1. Subsequently, the respective labeled precursor was added to each culture (final concentration 1 μCi/ml); NAI-107 was added at 0.5× MIC or 1× MIC, and another aliquot was run as a control without adding peptide. Incorporation was monitored for up to 40 min. Macromolecules were precipitated with ice-cold TCA (10%) and incubated for at least 30 min on ice before being filtered through glass microfiber filters (Whatman). Filters were washed with 5 ml of TCA (2.5%) containing 10 mm unlabeled metabolite, dried, and counted.
Carboxyfluorescein Efflux from Lipid II Containing Liposomes
Large unilamellar vesicles were prepared by the extrusion technique, essentially as described by Wiedemann et al. (11). Vesicles were made of 1,2-dioleoyl-sn-glycerol-3-PC supplemented with 0.5 mol % of lipid II (referring to the total amount of phospholipid). Carboxyfluorescein-loaded vesicles were prepared with 50 mm carboxyfluorescein and then diluted in 1.5 ml of buffer (50 mm MES-KOH, 100 mm K2SO4, pH 6.0) at a final concentration of 25 μm phospholipid on a phosphorous base. After addition of the peptide, the increase of fluorescence intensity was measured at 520 nm (excitation at 492 nm) on an RF-5301 spectrophotometer (Shimadzu) at room temperature. Leakage was documented relative to the total amount of marker release after solubilization of the vesicles by addition of 10 μl of 20% Triton X-100.
Monitoring the Membrane Potential Using DiBAC4
Cells of Bacillus subtilis 168, S. aureus ATCC 29213, and Micrococcus luteus DSM 1790 were grown in half-concentrated Mueller Hinton Broth to an A600 of 0.5 and incubated for 5 min with 1 μm of the membrane potential-sensitive fluorescent probe bis-(1,3-dibutylbarbituric acid)trimethine oxonol (DiBAC4(3); Molecular Probes). NAI-107 was added at 10 times the MIC. 1 μm Nisin was used as positive control. Fluorescence was measured at the excitation and emission wavelengths of 492 and 515 nm, respectively.
Accumulation of N-Acetylmuramic Acid-Pentapeptide
S. aureus ATCC 29213 was grown in MH broth to an A600 of 0.7 and supplemented with 130 μg/ml of chloramphenicol. After 15 min antibiotics (NAI-107 or vancomycin) were added at 10× MIC and incubated for 30 min. Cells were harvested and extracted with boiling water. The suspension was then centrifuged (48,000 × g, 30 min) and the supernatant lyophilized. Nucleotide-linked cell wall precursors were analyzed by HPLC and corresponding fractions were confirmed by mass spectrometry.
Antagonization Assays
Antagonization of the antibiotic activity of NAI-107 by potential target molecules was performed by a MIC-based setup in microtiter plates. NAI-107 (20 μg/ml corresponding to 5× MIC) were mixed with potential HPLC-purified antagonists (UDP-MurNAc-pentapeptide, C55-P, lipid I, lipid II) in a 10-fold molar excess with respect to the antibiotic. S. aureus ATCC 29213 (5 × 105 cfu/ml) was added and samples were examined for visible bacterial growth after overnight incubation.
Potassium Efflux from Whole Cells
For potassium efflux experiments a microprocessor pH meter (pH 213; Hanna Instruments, Kehl, Germany) with a MI-442 potassium electrode and MI-409F reference electrode was used. To obtain stable results, the electrodes were pre-conditioned by immersing both the potassium selective and the reference electrodes in choline buffer (300 mm choline chloride, 30 mm MES, 20 mm Tris, pH 6.5) for at least 1 h before starting calibration or measurements. Calibration was carried out before each determination by immersing the electrodes in fresh standard solutions containing 0.01, 0.1, or 1 mm KCl in choline buffer. Cells of B. subtilis 168 were grown in Mueller-Hinton Broth and harvested at an optical density (A600) of 1.0 to 1.5 (3300 × g, 4 °C, 3 min), washed with 50 ml of cold choline buffer, and resuspended in the same buffer to an A600 of 30. The concentrated cell suspension was kept on ice and used within 30 min. For each measurement the cells were diluted in choline buffer (25 °C) to an A600 of about 3. Calculations of potassium efflux in percent were performed according to the equations established by Orlov et al. (32). Peptide-induced leakage was monitored for 3 min, with values taken every 10 s, and was expressed relative to the total amount of potassium release induced by addition of 1 μm nisin. NAI-107 was added at 10× MIC.
In Vitro Lipid I/Lipid II Synthesis and Purification
In vitro lipid II synthesis was performed using membranes of M. luteus as described (22, 33). In short, lipid I and lipid II were synthesized in vitro using membrane preparations of M. luteus DSM 1790. Membranes were isolated from lysozyme-treated cells by centrifugation (40,000 × g), washed twice in 50 mm Tris-HCl, 10 mm MgCl2, pH 7.5, and stored under liquid nitrogen until use. Analytical assays were performed in a total volume of 150 μl containing 300–400 μg of membrane protein, 10 nmol of undecaprenylphosphate, 100 nmol of UDP-N-acetylmuramic acid pentapeptide (UDPMurNAc-pp), 100 nmol of UDP-GlcNAc in 60 mm Tris-HCl, 5 mm MgCl2, pH 7.5, and 0.5% (w/v) Triton X-100. For purification of milligram quantities of lipid II, the analytical procedure was scaled up by a factor of 500. Reaction mixtures were incubated for 1 h at 28 °C, and lipids were extracted with the same volume of n-butanol, 6 m pyridine-acetate, pH 4.2. Lipid I was synthesized as described for lipid II with the omission of UDP-GlcNAc from the synthesis reaction. Purification of lipid I/lipid II was performed on a DEAE-FF column (5 ml; GE Healthcare) and eluted in a linear gradient from chloroform/methanol/water (2:3:1) to chloroform, methanol, 300 mm ammonium bicarbonate (2:3:1). Lipid I- and lipid II-containing fractions were identified by TLC (silica plates, 60F254; Merck) using chloroform/methanol/water/ammonia (88:48:10:1) as the solvent (13). Spots were visualized by phorbol 12-myristate 13-acetate staining reagent (34).
Analysis of Enzyme Activities
The overall lipid II synthesis reaction was performed as described above using membrane preparations of M. luteus DSM 1790 in a total volume of 150 μl. For quantitative analysis 0.5 nmol of [14C]UDP-GlcNAc (7.4 GBq mmol−1; Amersham Biosciences) was added to the reaction mixture. To determine the enzymatic activity of purified MraY-His6 the assay was carried out in a total volume of 50 μl containing 5 nmol of C55-P or [3H]C55-P (14.8 GBq/mmol; Biotrend), 50 nmol of UDP-MurNAc-pentapeptide in 100 mm Tris-HCl, 30 mm MgCl2, pH 7.5, and 10 mm N-lauroylsarcosine. The reaction was initiated by the addition of 7.5 μg of the enzyme and incubated for 1 h at 37 °C. The MurG activity assay was performed in a final volume of 30 μl containing 2.5 nmol of purified lipid I, 25 nmol of UDP-GlcNAc or [14C]UDP-GlcNAc in 200 mm Tris-HCl, 5.7 mm MgCl2, pH 7.5, and 0.8% Triton X-100 in the presence of 0.45 μg of purified MurG-His6 enzyme. The reaction mixture was incubated for 30 min at 30 °C. The assay for synthesis of lipid II-Gly1 catalyzed by FemX was performed as described previously without any modifications (13). Enzymatic activity of PBP2 was determined by incubating 2.5 nmol of lipid II in 100 mm MES, 10 mm MgCl2, pH 5.5, and 0.1% Triton X-100 in a total volume of 50 μl. The reaction was initiated by the addition of 7.5 μg of PBP2-His6 and incubated for 1.5 h at 30 °C. The inhibitory effect on NAI-107 on WTA-precursor formation lipid III (undecaprenol-pyrophosphate-N-acetyl-glucosamine) and lipid IV (undecaprenyl-pyrophosphate-N-acetyl-glucosamine-N-acetylmannosamine) was investigated as described by Müller et al. (24). The enzymatic activity of the TagO-catalyzed lipid III synthesis was determined using purified recombinant TagO protein incubated in the presence of 5 nmol of C55-P, 67.5 nmol of UDP-GlcNAc, and 0.75 nmol of [14C]UDP-GlcNAc in 83 mm Tris-HCl, pH 8.0, 6.7 mm MgCl2, 8.3% (v/v) dimethyl sulfoxide, and 10 mm N-lauroylsarcosine. The reaction was initiated by the addition of 6.15 μg of the enzyme TagO-His6 and incubated for 90 min at 30 °C. TagA- and MnaA-catalyzed synthesis of lipid IV was performed using HPLC-purified lipid III. About 1 nmol of lipid III was incubated with 15 nmol of UDP-GlcNAc and 0.2 nmol of [14C]UDP-GlcNAc in the presence of 0.2% Triton X-100, 100 mm Tris-HCl, pH 7.5, and 250 mm NaCl in a final volume of 50 μl. About 1.1 μg of TagA-His6 and MnaA-His6 was added to start the reaction. Synthesized lipid intermediates were extracted from the reaction mixtures with n-butanol/pyridine acetate, pH 4.2 (2:1; v/v), analyzed by thin layer chromatography (TLC) as described earlier (13). Radiolabeled spots were visualized by iodine vapor, excised from the silica plates and quantified by β-scintillation counting (1900 CA Tri-Carb scintillation counter; Packard). Analysis of the lipid II conversion catalyzed by PBP2 was carried out by applying reaction mixtures directly onto TLC plates developed in solvent B (butanol/acetic acid/water/pyridine (15:3:12:10, v/v/v/v)). Purification of WTA lipid intermediates was performed as described for lipid II with slight modifications (13). NAI-107 and nisin were added in various molar concentrations ranging from 0.5 to 2:1 with respect to the amount of purified C55-P, lipid I, lipid II, lipid III, and lipid IV, respectively, in all in vitro assays.
Complex Formation of NAI-107 with Lipid II as Analyzed by TLC
Purified lipid II or 14C-labeled lipid II (2 nmol) was incubated in 10 mm Tris-HCl, pH 7.5, in the presence of increasing NAI-107 concentrations (NAI-107:lipid II molar ratios ranging from 0.5 to 2:1) in a total volume of 30 μl. After incubation for 30 min at 25 °C, the mixture was analyzed by TLC using solvent B (butanol/acetic acid/water/pyridine; 15:3:12:10, v/v/v/v). Analysis was carried out by phorbol 12-myristate 13-acetate or ninhydrin staining or/and phosphoimaging (STORM Phosphorimager).
Microscopy
Sample preparation and bright field microscopy for examination of the cell shape after acetic acid/methanol fixation was performed as described by Schneider et al. (35) with minor modifications (36). Briefly, B. subtilis 168 was grown in Belitzky minimal medium and treated with antibiotics in early exponential growth phase. After 15 min of antibiotic stress cells were fixed in a 1:3 mixture of acetic acid and methanol, immobilized in low melting agarose, and microscopically examined.
Depolarization assays were performed with B. subtilis 1981 GFP-MinD (37) as reported recently (36). B. subtilis 1981 GFP-MinD was grown overnight in Belitzky minimal medium. Cells were then inoculated to an A500 of 0.1 in Belitzky minimal medium containing 0.1% xylose to induce expression of the GFP-MinD fusion protein. After reaching an A500 of 0.35, cells were exposed to 5 μg/ml of NAI-107. After 15 min of antibiotic stress, 0.5 μl of the bacterial culture was withdrawn and the nonfixed, nonimmobilized samples were imaged immediately in fluorescent mode. Pore formation was monitored in B. subtilis 168 using the live/dead BacLight bacterial viability kit (Invitrogen) as described previously (38). In short, exponentially growing cells were treated with antibiotics for 15 min. Subsequently, 2 ml of culture were incubated with 4 μl of a 1:1 mixture of the respective fluorescent dyes for another 15 min in the dark. Cells were washed and resuspended in TE buffer (100 mm Tris, 1 mm EDTA, pH 7.5). Five μl of the cell suspension were imaged without fixation or immobilization in fluorescent mode.
Proteome Analysis
MICs in Belitzky minimal medium were determined in test tubes under steady agitation. Growth inhibition experiments were performed to find an appropriate antibiotic concentration eliciting a sufficient proteome response. We selected a NAI-107 concentration that led to the up-regulation of marker proteins without completely inhibiting biosynthesis of household proteins. Protein synthesis rates were determined by measuring the incorporation of radioactively labeled methionine into proteins prior to two-dimensional PAGE. Incorporation rates between 20,000 and 100,000 cpm/μg of crude protein were considered appropriate for proteome analysis (data not shown). Proteomic profiling of the acute bacterial stress response was performed with 0.9 μg/ml of NAI-107 (0.1× MIC). Radioactive labeling of newly synthesized proteins and subsequent separation of the cytosolic proteome by two-dimensional PAGE was performed as described previously (37). In short, 5 ml of a B. subtilis culture were exposed to antibiotics in early exponential growth phase. After 10 min cells were pulse-labeled with l-[35S]methionine for 5 min. Radioactive incorporation was stopped by inhibition of protein biosynthesis by adding 1 mg/ml of chloramphenicol and an excess of non-radioactive methionine. Cells were harvested by centrifugation, washed three times with TE buffer, and disrupted by ultrasonication. Fifty-five μg of protein for analytical and 300 μg for preparative gels were loaded onto 24-cm immobilized pH 4–7 gradient strips (GE Healthcare) by passive rehydration for 18 h. Proteins were separated in a first dimension by isoelectric focusing and in a second dimension by SDS-PAGE using 12.5% acrylamide gels. Analytical gel images were analyzed as described previously (39, 40) using Decodon Delta 2D 4.1 image analysis software (Decodon, Greifswald, Germany). Proteins more than 2-fold up-regulated in three independent biological replicates were defined as marker proteins. Protein spots were identified either by MALDI-TOF measurements on a 4800 MALDI TOF/TOF Analyzer (Applied Biosystems, Foster City, CA) as described previously (PMID: 21383089) or by nanoUPLC-ESI-MS/MS as previously reported (38) using a Synapt G2S high definition mass spectrometer equipped with a lock spray source for electrospray ionization and a TOF detector (Waters, Milford, MA).
NMR Titration of Lipid II with NAI-107
A solution of lipid II in chloroform/methanol was dried under nitrogen and the residue was redissolved in a solution of 360 mm dodecylphosphocholine (DPC)-d38 (Cambridge Isotopes), 25 mm imidazole-d4, 1 mm NaN3, and 17.2 μm TSP-d4 (sodium 2,2′,3,3′-tetradeutero 3-trimethylsilylpropionate) in 90% H2O, 10% D2O. The pH of the solution was measured to 6.50, the total volume was 0.55 ml. The lipid II content was quantified by integrating lipid II and TSP-d4 NMR resonances to 2.65 mm.
1H and 31P NMR spectra were recorded, then NAI-107 was added as a dried aliquot and redissolved under vortexing. 1H and 31P NMR spectra were recorded for each titration point. Experiments were recorded on a BRUKER AVIII-600MHz spectrometer equipped with a BBFO+ RT-probe. All spectra were recorded at 310.1 K.
NAI-107 Structure in Micelle
3.5 mm NAI-107 was dissolved in 150 mm DPC-d38 in 5% D2O at pH 6.2. A NOESY spectrum with 80 ms mixing time and two natural abundance 13C-1H-heteronuclear single quantum coherence spectra, one for the aliphatic signals, one for the aromatic signals, were recorded on a BRUKER AV-900MHz spectrometer equipped with a TCI cryoprobe. A TOCSY spectrum with 80 ms mixing time was recorded on a BRUKER DRX-600 MHz spectrometer equipped with a TCI cryoprobe. Both spectrometers belong to the Centre for Biomolecular Magnetic Resonance, Frankfurt, Germany. Insertion depth and orientation of NAI-107 in DPC micelles was determined as described in Ref. 41. Briefly, Inversion-Recovery weighted NOESY spectra were recorded in the absence of paramagnetic agent and in the presence of 1 and 2 mm Gd(DTPA-BMA). Relaxation rates of 1H resonances were extracted and plotted versus the concentration of gadolinium. The slope of the line was taken as paramagnetic relaxation enhancement (PRE). The PREs were then converted to distance information as described in Ref. 41.
Structure Calculation
Integrated and assigned NOESY peak lists were used as input for the program CYANA (42) to calculate 20 structures. These 20 structures were further refined four times each in explicit solvent with YASARA (43) using the NOVA and YASARA force fields. For each of the four repetitions of a structures calculated, the one with the lowest residual violations was retained and thus, a final bundle of 20 structures was obtained.
RESULTS
Antimicrobial Activity of NAI-107
NAI-107 exhibits potent antibacterial activity against a number of Gram-positive pathogens, including multiresistant strains such as methicillin-resistant S. aureus, penicillin-resistant pneumococci, and vancomycin-resistant enterococci (Table 1). Growth kinetic data of S. aureus ATCC 29213 exposed to NAI-107 resembled those obtained with cell wall-interfering agents (such as vancomycin, penicillin, and bacitracin), rather than rapidly lytic membrane-active agents such as polymyxin and novispirin (35). Consistently, killing kinetics indicated that over a period of approximately one generation time (0.5 h), treated cells were just unable to multiply before the number of colony-forming units decreased (data not shown).
TABLE 1.
Antibacterial activity of NAI-107 against bacterial strains used in this study
| Bacterial strain | MIC |
|---|---|
| μg/ml | |
| S. aureus ATCC 29213 | 4 |
| S. simulans 22 | 4 |
| B. subtilis 168 | 0.5 |
| M. luteus DSM 1790 | ≤0.062 |
Effect of NAI-107 on Whole Cells
NAI-107, at 0.5× MIC, inhibited the incorporation of radiolabeled glucosamine, an essential precursor of bacterial peptidoglycan, into cells of S. simulans M22 (Fig. 2A), whereas DNA, RNA, and protein biosynthesis were much less affected, suggesting cell wall biosynthesis as a potential target pathway. To distinguish whether NAI-107 interferes with the cytoplasmic or membrane-associated steps, we analyzed the cytoplasmic pool of UDP-linked peptidoglycan precursors in NAI-107-treated cells. Antibiotics that interfere with the late stages of peptidoglycan synthesis, such as vancomycin, are known to trigger accumulation of the ultimate soluble peptidoglycan precursor in the cytoplasm. Consistent with the results of the incorporation studies, NAI-107 treatment led to the intracellular accumulation of soluble precursor UDP-MurNAc-pentapeptide, however, to a lower extent, as compared with the vancomycin control (Fig. 2B), suggesting that the biosynthesis capacity of the cell was somewhat impaired, however, gross leakage of the precursor was not observed. This is in line with the fact that NAI-107 slightly affected the membrane potential of growing cells (Fig. 3B), suggesting an interaction with the membrane without formation of stable pores. Consistently, in contrast to the positive control nisin, no potassium leakage (Fig. 3C), nor carboxyfluorescein efflux from lipid II-containing liposomes was detected (Fig. 3A). Whole cell assays were repeated with different strains (M. luteus DSM 1790, S. simulans, B. subtilis 168, and S. aureus ATCC 29213) to exclude membrane effects dependent on membrane thickness and lipid composition of the target strain as described for gallidermin or epidermin (21, 29).
FIGURE 2.

A, impact of NAI-107 on macromolecular biosyntheses of S. simulans M22. Untreated (circles) and NAI-107-treated (squares) cells were incubated with tritium-labeled precursors to monitor their incorporation into proteins (I), cell wall (II), DNA (III), and RNA (IV). B, intracellular accumulation of the soluble cell wall precursor UDP-MurNAc-pentapeptide in S. aureus ATCC 29213. Cells were treated with NAI-107 (dotted line) and vancomycin (dashed line), both at 10-fold MIC, or left untreated (solid line) for 60 min. Cells were harvested and extracted with boiling water. The suspension was then centrifuged and the supernatant lyophilized. C, UDP-MurNAc-pentapeptide was identified by means of mass spectrometry using the negative mode and 6-aza-2-thiothymine (in 50% (v/v) ethanol, 20 mm ammonium citrate) as matrix; the calculated monoisotopic mass is 1149.35.
FIGURE 3.

Impact of NAI-107 on the integrity of bacterial and liposome membranes. A, carboxyfluorescein efflux from lipid II containing liposomes. B, influence of NAI-107 on the membrane potential of S. aureus ATCC 29213 as analyzed using DiBAC4. Antibiotics were added at concentrations corresponding to 10-fold MIC. Under these experimental conditions the δ-ψ of the untreated cells amounts to −140 mV, of nisin-depolarized cells to −60 mV (57) and of NAI-107 or gallidermin-treated cells to −120 to −110 mV, δ-ψ was determined using the 3H-labeled tetraphenylphosphonium bromide ion as described (57). C, potassium efflux from B. subtilis 168 cells was monitored with a potassium-sensitive electrode. Ion leakage is expressed relative to the total amount of potassium released after addition of 1 μm pore-forming lantibiotic nisin (100%, circles). NAI-107 was added at 10× MIC (triangles); controls were without peptide antibiotics (squares).
To better estimate the contribution of the slow membrane depolarization (Fig. 3B) and potassium leakage (Fig. 3C) to the overall killing activity, we applied fluorescent microscopy techniques and studied the impact of NAI-107 on localization of a GFP fusion to the cell division protein MinD. MinD is part of the cell division regulation machinery of B. subtilis. In energized cells, MinD is attached to the membrane and localizes at the cell poles and in the cell division plain, whereas in the presence of membrane-depolarizing agents like valinomycin or proton ionophore carbonyl cyanide m-chlorophenylhydrazone, MinD delocalizes appearing in irregular clusters (37). After treatment with NAI-107 for 15 min, such irregular dispersion of GFP-labeled MinD was observed (Fig. 4), demonstrating sufficient membrane depolarizing for MinD delocalization. A threshold potential, necessary for keeping MinD in place, has not been reported, however, it is likely that MinD delocalization after 15 min is in agreement with the level of depolarization observed after 5 min measured with DIBAC4 (Fig. 3B). Using the green fluorescent dye SYTO 9 and the red fluorescing propidium iodide, we could also confirm that NAI-107 does not form stable pores or membrane holes. SYTO 9 diffuses through intact membranes, whereas propidium iodide can only enter bacterial cells through large pores or membrane holes. In a fluorescence overlay, perforated cells appear yellow, whereas intact cells appear green. Obviously NAI-107 did not efficiently facilitate penetration of propidium iodide into B. subtilis (Fig. 4), which is in line with our previous findings. Similar results were also reported for gallidermin and mersacidin (Table 2) (36).
FIGURE 4.

Microscopy of B. subtilis cells treated with NAI-107 or nisin. A, examination of the cell shape after acetic acid/methanol fixation. B, depolarization assays using B. subtilis 1981 GFP-MinD. C, light microscopy of cells in B. D and E, membrane depolarization as monitored with the live/dead BacLight bacterial viability kit: (D) individual cells and (E) overview. Images of control and nisin-treated cells in D were taken from Ref. 58.
TABLE 2.
Table of protein spots identified in two-dimensional gel and their corresponding functions
| Protein ID | Protein function | Functional category |
|---|---|---|
| CysC | Adenylyl-sulfate kinase | Sulfur metabolism |
| GamA | Glucosamine-6-phosphate deaminase | Cell wall |
| LiaH | Similar to phage shock protein | Cell wall |
| LiaR | Two-component response regulator, regulation of the liaI-liaH-liaG-liaF-liaS-liaR operon | Cell wall |
| NadE | NAD synthase | Energy |
| NrfA | Spx-dependent FMN-containing NADPH-linked nitro/flavin reductase | Energy |
| PbpC | Penicillin-binding protein | Cell wall |
| PspA | Phage shock protein | Membrane |
| RpsB | Ribosomal protein | Translation |
| YceC | Similar to tellurium resistance protein | Cell envelope |
| YceH | Similar to toxic anion resistance protein | Cell envelope |
| YpuA | Unknown | Cell envelope |
| YqiG | Similar to NADH-dependent flavin oxidoreductase | Energy |
| YtrE | ABC transporter (ATP-binding protein) | Transport |
| YvcR | ABC transporter (ATP-binding protein) for the export of lipid II-binding lantibiotics | Transport |
| YvIB | Unknown | Cell envelope |
Proteomic Response to NAI-107 Treatment
Depending on structural features, the level of interaction with the cytoplasmic membrane varies significantly among lipid II-targeting lantibiotics and range from mere binding to lipid II (mersacidin) to defined and stable pore formation (nisin) (10, 17, 23). In previous studies (36, 44–46) the impact of nisin, gallidermin, and mersacidin on cell wall integrity, pore formation, and membrane depolarization in B. subtilis was compared, and correlated to stress responses. Setting up a proteomic response reference library, including these lantibiotics, as well as other envelope-targeting antibiotics such as bacitracin, vancomycin, gramicidin S, or valinomycin, YtrE could be identified as the most reliable marker protein for interfering with membrane-bound steps of cell wall biosynthesis. Furthermore, NadE and PspA were identified as markers for antibiotics interacting primarily with the cytoplasmic membrane. In such an experiment NAI-107 treatment led to up-regulation of 20 protein spots compared with the untreated control, 15 of which could be identified by mass spectrometry (Fig. 5, supplemental Table S1). Among the identified proteins were LiaH, YtrB, YtrE, and YpuA (formerly referred to as NGM1 (35)), which are specific markers for inhibition of membrane-bound steps of cell wall biosynthesis and typically up-regulated by antibiotics interfering with lipid II or undecaprenylphosphate (36). LiaH is also up-regulated in response to compounds interfering with membrane architecture and localization of peripheral membrane proteins like MurG. Furthermore, YceC and YceH, which had been defined as cell envelope stress markers, and PspA and NadE, were found to be up-regulated in NAI-107-treated cells. Hence, NAI-107 elicits a typical dual cell wall biosynthesis and membrane stress response. The response strongly overlaps with that of gallidermin-treated cells, suggesting high mechanistic similarity between these two lantibiotics. Gallidermin integrates into membranes, with pore formation occurring only in some bacteria and correlating with membrane thickness and fatty acid branching (29, 36, 47). In contrast to nisin, which needs to dock to lipid II for membrane integration and pore formation, gallidermin shows very high association and low dissociation constants for binding to phospholipid bilayers, indicating that it readily integrates into the membrane independently of lipid II binding. Similarity of the proteome response profile of NAI-107 was also observed with MP196 and gramicidin S, both of which also integrate into the cytoplasmic membrane and disturb membrane architecture (36). Less overlap was observed with the response to nisin, which is dominated by rapid pore formation and membrane depolarization.
FIGURE 5.
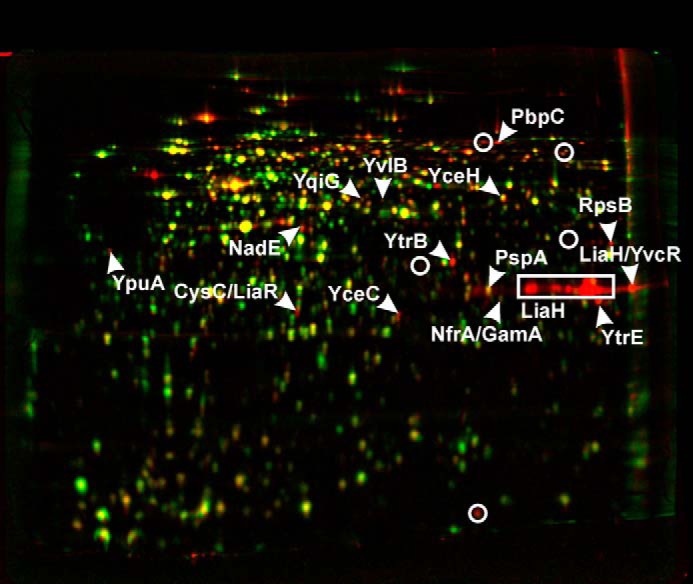
Differential proteome analysis of B. subtilis 168 in response to NAI-107. Two-dimensional gel-based protein synthesis profiles of the controls false colored in green were overlaid with those of the antibiotic-treated samples false colored in red. In the overlays, down-regulated proteins appear green, up-regulated proteins appear red, and proteins expressed at equal rates appear yellow.
Impact of NAI-107 on in Vitro Peptidoglycan Biosynthesis
Membrane preparations of M. luteus have sufficient enzyme activity (MraY, MurG) for the formation of cell wall precursors lipid I and lipid II in vitro. Testing the overall lipid II synthesis reactions with such membranes, to which defined amounts of C55-P, as well as the soluble precursors UDP-MurNAc-pentapeptide and UDP-GlcNAc, were added, we found that NAI-107 blocked the formation of lipid II as observed by TLC (Fig. 6A). In the positive control, where no inhibitor was present, the complete conversion of C55-P to lipid II was achieved. Quantitative analysis using [14C]UDP-GlcNAc to specifically label lipid II revealed a concentration-dependent reduction of the lipid II amount. Addition of equimolar concentrations of NAI-107 with respect to C55-P resulted in a 69% decrease in extractable lipid II, and a 2-fold molar excess of the lantibiotic completely blocked lipid II synthesis (Fig. 6B).
FIGURE 6.
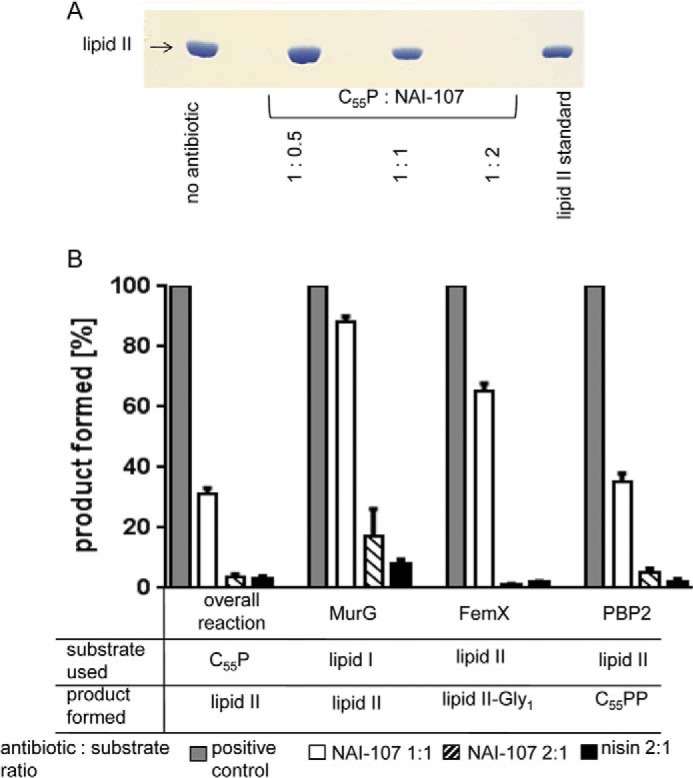
Effect of NAI-107 on membrane-associated stages of peptidoglycan synthesis in vitro. A, in vitro lipid II synthesis catalyzed by M. luteus membrane preparations. Thin layer chromatography of n-butanol/pyridine acetate extracts and detection by phosphomolybdic acid staining. NAI-107 was added to the reaction mixtures at molar ratios in respect to C55P as indicated. 2 nmol of purified lipid II was run as a standard. B, synthesis and analysis of the individual biosynthesis steps were performed as described under “Materials and Methods.” Bactoprenol containing products were analyzed by TLC. Product containing bands were visualized by iodine vapor, excised, and counted. Radiolabeling was based on 3H-labeled C55P (for lipid I), [14C]GlcNAc for lipid II, and [14C]glycine for lipid II-Gly1. For all reactions, the amount of products synthesized by control reactions in the absence of antibiotic was set to 100%. NAI-107 was added at molar ratios of 1:1 and 2:1 with respect to the lipid substrates as indicated. The lantibiotic nisin in a 2-fold molar excess served as a control. Mean values from three independent experiments are shown. Error bars indicate the standard deviation.
For detailed analysis, we focused on the individual cell-wall biosynthesis steps in vitro using purified recombinant enzymes from S. aureus (MurG, FemX, PBP2) in the presence of NAI-107. For these enzymes, lipid I (MurG) or lipid II (FemX, PBP2) are substrates, and significant inhibition of the reactions was observed when NAI-107 was added in 2-fold molar excess with respect to lipid I or lipid II (Fig. 6B), strongly suggesting that NAI-107 may form a stoichiometric complex with the substrates rather than inhibiting the enzymes.
To confirm that the activity of NAI-107 is primarily based on the formation of a complex with lipid I/lipid II, we performed conventional MIC determinations in the presence of C55-P, lipid I, lipid II, UDP-GlcNAc, and UDP-MurNAc-pentapeptide at 5-fold molar concentrations with respect to the concentration of NAI-107. The activity of NAI-107 was only antagonized in the presence of lipid I and lipid II (data not shown), which agrees well with its inhibition of the MurG-, FemX-, and PBP2-catalyzed reactions, in which lipid I and lipid II are substrates. Binding of NAI-107 and lipid II was further validated by incubating purified lipid II together with NAI-107 in various molar concentrations ranging from 0.5 to 2 in respect to the lipid precursor. Subsequent TLC was used to analyze the migration behavior (Fig. 7). Free lipid II migrated to a defined position (Fig. 7, A and B, first lane), whereas free NAI-107 was only detectable by ninhydrin staining (Fig. 7A, second lane). However, in complex with NAI-107, lipid II remained at the start point, appearing as a ring-like structure (Fig. 7, A, third lane, and B, second to fourth lanes). As has been observed in the cell-free assays, only at a 2:1 ratio, no free lipid II was detectable, substantiating the formation of a 2:1 stoichiometric NAI-107·lipid II complex in our in vitro test system.
FIGURE 7.

Complex formation of NAI-107 with lipid II and estimation of binding stoichiometry. Lipid II was incubated with NAI-107 at the indicated molar ratios. The stable complex of NAI-107 with lipid II remains close to the application spot, whereas free lipids and NAI-107 migrate to the indicated positions. A, ninhydrin staining. First lane, free lipid II (2 nmol); second lane, NAI-107 (4 nmol); third lane, NAI-107:lipd II molar ratio 2:1. B, phorbol 12-myristate 13-acetate staining of lipid II incubated with increasing molar ratios of NAI-107. Free NAI-107 could not be detected with phorbol 12-myristate 13-acetate staining. Spots were separated using Solvent B (see “Materials and Methods”).
Interaction of NAI-107 with WTA Precursors
Binding of nisin and related lantibiotics to lipid II mainly involves the pyrophosphate and the MurNAc moiety (23), whereas the second sugar (GlcNAc) does not appear to significantly contribute. Because lipid III and lipid IV, the first lipid intermediates of the WTA biosynthesis pathway (48, 49), have similar pyrophosphate sugar linkage moieties, we investigated the impact of NAI-107 on these reactions using recombinantly purified enzymes TagO, TagA, and MnaA (24). Only about 35% of the product 14C-lipid III could be detected when NAI-107 was added at a 2-fold molar excess with respect to C55-P, suggesting that NAI-107 forms a stable complex with lipid III, which escapes extraction of the lipid intermediate from the synthesis mixture, as described also for the nisin·lipid III complex (24). Quantitative analysis of the TagA-catalyzed lipid IV synthesis reaction using radiolabeled UDP-GlcNAc also revealed dose-dependent inhibition by NAI-107 and nisin (Fig. 8). Added in equimolar concentrations, NAI-107 inhibited the formation of lipid IV, compared with a positive control, where no antibiotic was added, to about 45%. At a molar ratio of 2:1 (NAI-107:lipid), NAI-107 inhibited the synthesis of radiolabeled lipid IV to about 80%.
FIGURE 8.

Top, schematic representation of peptidoglycan and WTA biosynthesis. Bottom, impact of NAI-107 on the lipid III and lipid IV synthesis. For lipid III synthesis purified recombinant TagO protein was incubated in the presence of 5 nmol of C55-P and [14C]UDP-GlcNAc. The reaction was initiated by the addition of 6.15 μg of the enzyme TagO-His6 and incubated for 90 min at 30 °C. For lipid IV synthesis, TagA was incubated with purified lipid III and [14C]UDP-GlcNAc. The reaction was initiated by the addition of 1.1 μg of MnaA and TagA and incubated for 4 h at 30 °C. NAI-107 was added at molar ratios of 1 and 2 with respect to the substrate C55P or lipid III. Reaction products were extracted with BuOH/PyrAc (2:1) and separated by TLC. Detection and quantification were carried out using a Storm Phosphorimager.
NMR Studies
To gain further insight into the nature of the NAI-107/lipid II interaction at the membrane interface, we attempted to follow the process by NMR spectroscopy (Fig. 9). At first, lipid II was titrated with NAI-107. To keep aggregate sizes as low as possible, we utilized micelles of DPC as membrane models (supplemental Fig. S1). The 31P NMR resonances of lipid II were followed through the titration (supplemental Fig. S2A). In the course of the titration, signals of free lipid II disappear up to a molar ratio of 1:1, however, no signals of any bound form seem to appear. At a molar ratio of 1:1, only NMR signals of buffer components are seen. Further addition of NAI-107 leads to the reappearance of 31P NMR signals. The same is seen when looking at 1H NMR spectra throughout the titration (supplemental Fig. S2B). When going from 0:1 (NAI-107:lipid II) to 1:1, lipid II signals gradually disappear. No new signals appear, nor are there visible signals of NAI-107. Visible signals appeared only at molar ratios for NAI-107:lipid II >1:1. Integration of 31P spectra can be used to follow the amounts of different species throughout the titration. The resonance at −16.3 ppm was integrated, assumed to stem from one phosphorous atom of free lipid II, the region from −17.0 to −19.0 was integrated, assumed to stem from two phosphorous atoms of bound lipid II in a NAI-107·lipid II 2:1 complex. The 31P resonance of DPC was used as an internal standard for integration between spectra. The integral of the first sample of pure lipid II was assumed to be 100% free lipid II and used to calculate free lipid II concentrations at all titration points. The integral of the last titration sample was assumed to be 100% NAI-107·lipid II 2:1 complex and was used to calculate NAI-107·lipid II 2:1 complex concentrations at all titration points. The NAI-107·lipid II 1:1 complex does not yield any quantifiable NMR signals, thus its concentration was back-calculated by subtracting the concentrations of free and 2:1 complex from the total concentration of lipid II in the sample. The concentrations of the three different species of lipid II during the course of titration are shown in Fig. 10.
FIGURE 9.
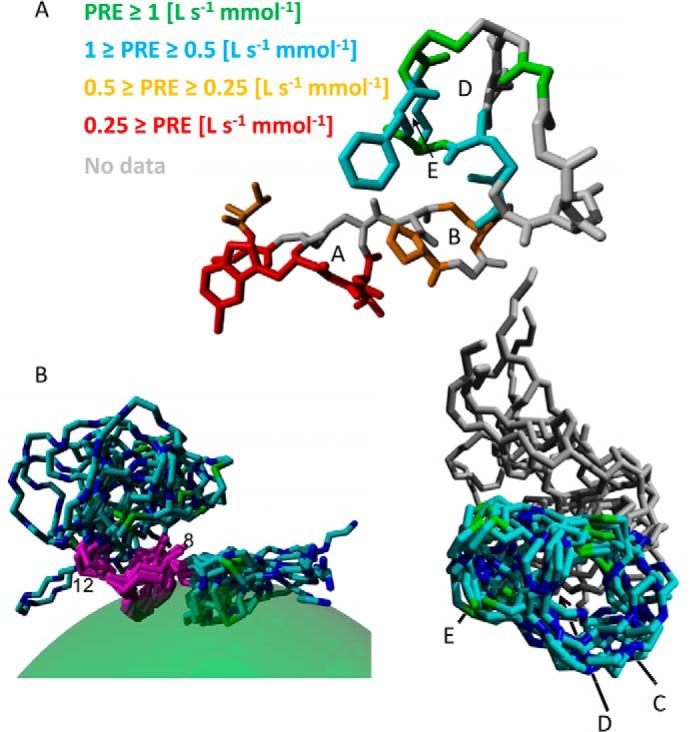
A, structure of NAI-107 in DPC micelles (conformer with lowest energy of constraint violation) with residues colored according to PRE. Letters A–E indicate the lanthionine rings. For better visibility, the whole residue was colored even though an experimental PRE was obtained only for one or two hydrogen atoms fixed to the backbone. Lower PRE values indicate less contact with the solvent/larger distance to the solvent, i.e. deeper penetration into the micelle. As a rule of thumb, residues with PRE ≈ 0.5 (liters s−1 mmol−1) are located at the micelle-water interface, residues in orange are located in the polar part of the micelle, residues in red are buried in the hydrophobic part of the micelle, whereas residues with higher PREs (cyan and green) are located outside the micelle. B, structure of NAI-107 in DPC micelles. The bundle of 10 structure with lowest root mean square deviation, superimposed on residues 8–12 is shown with the DPC micelle of the first structure indicated as semi-transparent green sphere. Right panel, bundle of 10 structures with the lowest root mean square deviation superimposed on residues 12–24, letters C, D, and E indicate lanthionine rings.
FIGURE 10.
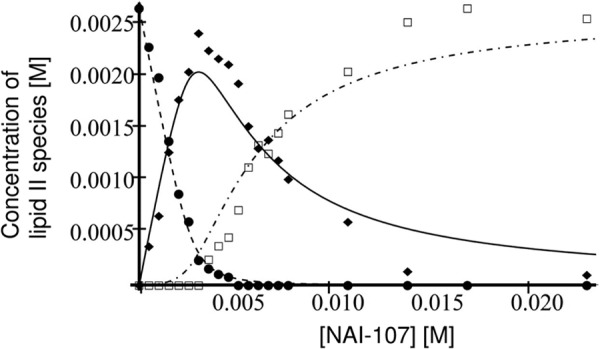
Titration of 2.65 mm lipid II with NAI-107: concentration of free lipid II (●), concentration of NAI-107·lipid II 1:1 complex (♦), concentration of NAI-107·lipid II 2:1 complex (□). The theoretical concentrations, assuming sequential binding with Ka1 = 2 × 104 (m−1) and Ka2 = 450 (m−1) are shown as a dashed line for free lipid II, full line for the 1:1 complex, and dot-dashed line for the 2:1 complex.
Subsequently, we studied the interaction of NAI-107 with membrane mimetic, DPC micelles. NAI-107, which is insoluble in pure water, readily dissolves in water in the presence of 150 mm DPC. The appearance of 1H NMR spectra also suggests that the correlation time of NAI-107 in the presence of micelles is higher than that of NAI-107 dissolved in 30% acetonitrile, indicating substantial interaction with the micelle.
Chemical shifts of NAI-107 were assigned in the presence of DPC micelles and NOEs were collected. In addition, paramagnetic relaxation enhancement of NAI-107 backbone atoms by Gd(DTPA-BMA) in the bulk solution were determined and converted to distances from the micellar center. These data together were then used to simultaneously calculate the structure of NAI-107 in DPC micelles and its insertion into the micelle. Fig. 9A illustrates the distribution of paramagnetic relaxation enhancements throughout the structure. Fig. 9B shows the structure of NAI-107 bound to the micelle. Statistical data on the structure can be found in supplemental Table S2. As can be expected for short peptides, the structure is quite dynamic, resulting in elevated root mean square deviation values. NMR assignments and molecular coordinates have been deposited at the BMRB (accession number 19619) and PDB databases (Protein Data Bank code 2mh5), respectively.
DISCUSSION
The increasing level of antibiotic resistance is a threat to human health, and existing classes of antibiotics need to be further developed or entirely new classes need to be identified. For the most challenging groups of multiresistant Gram-positive bacteria, the lipid II-binding antibiotics offer excellent possibilities for development. Generally, targets such as the sugar-pyrophosphate moiety in lipid II, which is recognized by many lantibiotics (23), cannot be altered as easily as protein targets, or more variable sugar moieties, and the d-Ala-d-Ala terminus of lipid II. Detailed mode-of-action studies of lantibiotics, such as NAI-107, demonstrate the multiplicity of activities that can be achieved when high affinity binding of lipid II is combined with direct effects on the cytoplasmic membrane, be it defined pore formation like with nisin or insertion and subsequent impact on membrane functionality, like with NAI-107. The integration of NAI-107 into the membrane certainly affects protein localization and promotes disorganization of the highly dynamic biosynthesis and energy generating protein machineries. Such membrane activities are intrinsic features of cationic amphiphilic compounds and are also characteristic for some of the glyco(lipo)peptides and glycodepsipeptides like telavancin, dalbavancin, and oritavancin (50, 51) that are on the market or in late-stage clinical investigations. Notably, the increase in amphiphilicity, at least in some cases, not only enhances antibiotic potency and bactericidal activity, but also impacts positively on pharmacokinetic parameters and enables favorable once daily or even once weekly dosage schemes.
NAI-107 interacts with cell envelope precursors attached to the undecaprenylphosphate (C55-P) carrier molecule, such as lipid I and lipid II and those for WTA biosynthesis (Fig. 8). In this context, the accessibility of WTA precursors must be taken into account to evaluate their importance as targets in vivo. As lipid III and lipid IV are further processed to the linear WTA polymer on the cytoplasmic side, they could only function as targets after internalization of the lantibiotic. Nisin translocation across lipid bilayers, which may occur in the course of pore formation, has been observed in several studies (9, 52–54). Whether this is true for NAI-107 remains to be determined. The fully assembled C55P-linked WTA polymer, exported to the outside, seems to be the most likely candidate for a relevant target, as it is the only WTA building block exposed at the cellular surface (Fig. 8). Although NAI-107 has the highest affinity for lipid II, which we consider its primary target, it is possible that, if supplied at high concentrations, the antibiotic will recognize the other targets as well, simultaneously trapping the bactoprenol carrier at several sites and causing general depletion of the undecaprenyl pool. Interaction with multiple targets is beneficial in light of reduced resistance development, and therefore an advantage of NAI-107. In general, bacterial cells, which are exposed to antibiotic binding to the lipid II pyrophosphate linkage, do not merely suffer from inhibition of the transglycosylation step in peptidoglycan synthesis. Such lantibiotics appear to have a profound impact on the correct localization of lipid II (55) and presumably disrupt the functional organization of cell envelope biosynthesis machineries that rely on the C55P carrier.
The 2:1 binding stoichiometry observed in most assay systems raised the question whether NAI-107 forms dimers in solution, or whether one peptide binds to lipid II followed by recruitment of another molecule of NAI-107. The latter mechanism is known from some two-peptide lantibiotics, such as lacticin 3147, where significant antibiotic activity is only achieved through the cooperative action of two peptides. Peptide A contains a mersacidin-like lipid II binding motif and binds to the CW precursor; the second peptide is then able to dock to the peptide·lipid II complex, allowing the complex to insert into the membrane to form a defined pore (56). We did not find evidence for a dimerization in solution, but the data showed clear evidence for two separate events in NAI-107·lipid II binding, a relatively strong initial interaction leading to the disappearance of free lipid II resonances and a weaker subsequent interaction leading to the appearance of bound lipid II and NAI-107 resonances (supplemental Fig. S2). It is puzzling that we do not observe any resonances of the complex at 1:1 stoichiometry. This could be explained by either conformational exchange at a time scale comparable with NMR chemical shift differences (≈10−3–10−2 s−1) or by the formation of larger aggregates.
31P NMR shows a change in chemical shifts of lipid II phosphorous resonances upon interaction with NAI-107. The two atoms change their shift by approximately −2 and −4 ppm. This is similar to the changes observed upon binding of nisin to lipid II (23). Therefore, it is reasonable to assume that also NAI-107 binds to the pyrophosphate moiety. Because the pyrophosphate moiety can be expected to locate at the interface between the solution and the cellular membrane, it makes sense to look for the binding site of NAI-107 to lipid II around this interface. NMR data clearly show that NAI-107 interacts with the membrane mimetic at rings A and, to a lesser extent, B. The pyrophosphate group carries a negative charge, and the only positive charge in the whole NAI-107 molecule is located at the N terminus preceding ring A, and also this charge is located at the interface between bulk water and membrane mimetic. Taken together, the data point at an N-terminal interaction site of NAI-107 with lipid II, as is also the case for nisin. The sequence similarity between nisin and NAI-107 around rings A and B (Fig. 1) supports this interpretation, however, nisin has one more positive charge in ring A (Lys-4) compared with NAI-107, which has the unusual 5-chloro-Trp at this position.
Supplementary Material
Acknowledgments
We thank Dirk Albrecht (University of Greifswald, Germany) for MALDI-TOF analysis. We thank the Centre for Biomolecular Magnetic Resonance, Frankfurt, Germany, for access to NMR equipment and Dr. Frank Löhr for expert assistance. Michaele Josten provided excellent technical help. The NMR laboratory at Aalborg University is supported by the Obel, SparNord, and Carlsberg Foundations as well as part of the Danish Center for Antibiotic Research and Development (DanCARD) financed by Danish Council for Strategic Research Grant 09-067075. The Synapt G2S mass spectrometer was supported by the state of North Rhine Westphalia (Forschungsgroßgeräte der Länder).
This work was supported in part by grants from the European Commission within the LAPTOP-project (contract number 245066 for FP7-KBBE-2009-3) to the University of Bonn and NAICONS SrI and by the BONFOR program of the Medical Faculty of the University of Bonn.

This article contains supplemental Tables S1 and S2 and Figs. S1 and S2.
- WTA
- wall teichoic acid
- MIC
- minimal inhibitory concentration
- DPC
- dodecylphosphocholine
- PRE
- paramagnetic relaxation enhancement
- PBP
- penicillin-binding protein
- BMA
- 1,7-bis(methylcarbamoylmethyl)-1,4,7-triazaheptane-1,4,7-triacetic acid.
REFERENCES
- 1. Arnison P. G., et al. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schnell N., Entian K.-D., Schneider U., Götz F., Zähner H., Kellner R., Jung G. (1988) Prepetide sequence of epidermin a ribosomally synthesized antibiotic with four sulphide rings. Nature 333, 276–278 [DOI] [PubMed] [Google Scholar]
- 3. Chatterjee C., Paul M., Xie L., van der Donk W. A. (2005) Biosynthesis and mode of action of lantibiotics. Chem. Rev. 105, 633–684 [DOI] [PubMed] [Google Scholar]
- 4. Jabes D., Donadio S. (2010) Strategies for the isolation and characterization of antibacterial lantibiotics. Methods Mol. Biol. 618, 31–45 [DOI] [PubMed] [Google Scholar]
- 5. Willey J. M., van der Donk W. A. (2007) Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 [DOI] [PubMed] [Google Scholar]
- 6. Sahl H. G., Bierbaum G. (1998) Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 52, 41–79 [DOI] [PubMed] [Google Scholar]
- 7. Cotter P. D., Hill C., Ross R. P. (2005) Bacterial lantibiotics: strategies to improve therapeutic potential. Curr. Protein Pept. Sci. 6, 61–75 [DOI] [PubMed] [Google Scholar]
- 8. Rogers L. (1928) The inhibiting effect of Streptococcus lactis on Lactobacillus bulgaricus. J. Bacteriol. 16, 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Breukink E., Wiedemann I., van Kraaij C., Kuipers O. P., Sahl H. G., de Kruijff B. (1999) Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286, 2361–2364 [DOI] [PubMed] [Google Scholar]
- 10. Brötz H., Josten M., Wiedemann I., Schneider U., Götz F., Bierbaum G., Sahl H. G. (1998) Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol. 30, 317–327 [DOI] [PubMed] [Google Scholar]
- 11. Wiedemann I., Breukink E., van Kraaij C., Kuipers O. P., Bierbaum G., de Kruijff B., Sahl H. G. (2001) Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 276, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 12. Breukink E., de Kruijff B. (2006) Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 5, 321–332 [DOI] [PubMed] [Google Scholar]
- 13. Schneider T., Senn M. M., Berger-Bächi B., Tossi A., Sahl H. G., Wiedemann I. (2004) In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 53, 675–685 [DOI] [PubMed] [Google Scholar]
- 14. Rohrer S., Berger-Bächi B. (2003) FemABX peptidyl transferases: a link between branched-chain cell wall peptide formation and β-lactam resistance in Gram-positive cocci. Antimicrob. Agents Chemother. 47, 837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Münch D., Roemer T., Lee S. H., Engeser M., Sahl H. G., Schneider T. (2012) Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. Plos Pathog. 8, e1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Heijenoort Y., Derrien M., Van Heijenoort J. (1978) Polymerization by transglycosylation in the biosynthesis of the peptidoglycan of Escherichia coli K12 and its inhibition by antibiotics. FEBS Lett. 89, 141–144 [DOI] [PubMed] [Google Scholar]
- 17. Ruhr E., Sahl H. G. (1985) Mode of action of the peptide antibiotic nisin and influence on the membrane potential of whole cells and on cytoplasmic and artificial membrane vesicles. Antimicrob. Agents Chemother. 27, 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sahl H. G., Brandis H. (1982) Mode of action of the staphylococcin-like peptide Pep 5 and culture conditions affecting its activity. Zentralbl Bakteriol Mikrobiol Hyg A 252, 166–175 [PubMed] [Google Scholar]
- 19. Sahl H. G., Kordel M., Benz R. (1987) Voltage-dependent depolarization of bacterial membranes and artificial lipid bilayers by the peptide antibiotic nisin. Arch. Microbiol. 149, 120–124 [DOI] [PubMed] [Google Scholar]
- 20. Castiglione F., Lazzarini A., Carrano L., Corti E., Ciciliato I., Gastaldo L., Candiani P., Losi D., Marinelli F., Selva E., Parenti F. (2008) Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol. 15, 22–31 [DOI] [PubMed] [Google Scholar]
- 21. Brötz H., Bierbaum G., Reynolds P. E., Sahl H. G. (1997) The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 246, 193–199 [DOI] [PubMed] [Google Scholar]
- 22. Brötz H., Bierbaum G., Leopold K., Reynolds P. E., Sahl H. G. (1998) The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42, 154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsu S. T., Breukink E., Tischenko E., Lutters M. A., de Kruijff B., Kaptein R., Bonvin A. M., van Nuland N. A. (2004) The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 11, 963–967 [DOI] [PubMed] [Google Scholar]
- 24. Müller A., Ulm H., Reder-Christ K., Sahl H. G., Schneider T. (2012) Interaction of type A lantibiotics with undecaprenol bound cell envelope precursors. Microb. Drug Resist. 18, 261–270 [DOI] [PubMed] [Google Scholar]
- 25. Lazzarini A., Gastaldo L., Cadiani G., Ciciliato I., Losi D., Marinelli F., Selva E., Parenti F. (April 1, 2008) US Patent US7351687 B2 Antibiotic 107891, its factors A1 and A2, pharmaceutically acceptable salts and compositions, and use thereof
- 26. Foulston L. C., Bibb M. J. (2010) Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U.S.A. 107, 13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Foulston L., Bibb M. (2011) Feed-forward regulation of microbisporicin biosynthesis in Microbispora corallina. J. Bacteriol. 193, 3064–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jabés D., Brunati C., Candiani G., Riva S., Romanó G., Donadio S. (2011) Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrob. Agents Chemother. 55, 1671–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonelli R. R., Schneider T., Sahl H. G., Wiedemann I. (2006) Insights into in vivo activities of lantibiotics from gallidermin and epidermin mode-of-action studies. Antimicrob. Agents Chemother. 50, 1449–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castiglione F., Cavaletti L., Losi D., Lazzarini A., Carrano L., Feroggio M., Ciciliato I., Corti E., Candiani G., Marinelli F., Selva E. (2007) A novel lantibiotic acting on bacterial cell wall synthesis produced by the uncommon actinomycete Planomonospora sp. Biochemistry 46, 5884–5895 [DOI] [PubMed] [Google Scholar]
- 31. Vasile F., Potenza D., Marsiglia B., Maffioli S., Donadio S. (2012) Solution structure by nuclear magnetic resonance of the two lantibiotics 97518 and NAI-107. J. Pept. Sci. 18, 129–134 [DOI] [PubMed] [Google Scholar]
- 32. Orlov D. S., Nguyen T., Lehrer R. I. (2002) Potassium release, a useful tool for studying antimicrobial peptides. J. Microbiol. Methods 49, 325–328 [DOI] [PubMed] [Google Scholar]
- 33. Umbreit J. N., Strominger J. L. (1972) Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J. Bacteriol. 112, 1306–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rick P. D., Hubbard G. L., Kitaoka M., Nagaki H., Kinoshita T., Dowd S., Simplaceanu V., Ho C. (1998) Characterization of the lipid-carrier involved in the synthesis of enterobacterial common antigen (ECA) and identification of a novel phosphoglyceride in a mutant of Salmonella typhimurium defective in ECA synthesis. Glycobiology 8, 557–567 [DOI] [PubMed] [Google Scholar]
- 35. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 36. Wenzel M., Kohl B., Münch D., Raatschen N., Albada H. B., Hamoen L., Metzler-Nolte N., Sahl H. G., Bandow J. E. (2012) Proteomic response of Bacillus subtilis to lantibiotics reflects differences in interaction with the cytoplasmic membrane. Antimicrob. Agents Chemother. 56, 5749–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strahl H., Hamoen L. W. (2010) Membrane potential is important for bacterial cell division. Proc. Natl. Acad. Sci. U.S.A. 107, 12281–12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wenzel M., Patra M., Senges C. H. R., Ott I., Stepanek J. J., Pinto A., Prochnow P., Vuong C., Langklotz S., Metzler-Nolte N., Bandow J. E. (2014) Analysis of the mechanism of action of potent antibacterial hetero-tri-organometallic compounds: a structurally new class of antibiotics. ACS Chem. Biol. 8, 1442–1450 [DOI] [PubMed] [Google Scholar]
- 39. Wenzel M., Patra M., Albrecht D., Chen D. Y., Nicolaou K. C., Metzler-Nolte N., Bandow J. E. (2011) Proteomic signature of fatty acid biosynthesis inhibition available for in vivo mechanism-of-action studies. Antimicrob. Agents Chemother. 55, 2590–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bandow J. E., Baker J. D., Berth M., Painter C., Sepulveda O. J., Clark K. A., Kilty I., VanBogelen R. A. (2008) Improved image analysis workflow for 2D gels enables large-scale 2D gel-based proteomics studies: COPD biomarker discovery study. Proteomics 8, 3030–3041 [DOI] [PubMed] [Google Scholar]
- 41. Franzmann M., Otzen D., Wimmer R. (2009) Quantitative use of paramagnetic relaxation enhancements for determining orientations and insertion iepths of peptides in micelles. ChemBioChem 10, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 42. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 43. Krieger E., Koraimann G., Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA: a self-parameterizing force field. Proteins 47, 393–402 [DOI] [PubMed] [Google Scholar]
- 44. Bandow J. E., Brötz H., Leichert L. I., Labischinski H., Hecker M. (2003) Proteomic approaches to antibiotic drug discovery. Antimicrob. Agents Chemother. 47, 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wenzel M., Bandow J. E. (2011) Proteomic signatures in antibiotic research. Proteomics 11, 3256–3268 [DOI] [PubMed] [Google Scholar]
- 46. Brötz-Oesterhelt H., Beyer D., Kroll H. P., Endermann R., Ladel C., Schroeder W., Hinzen B., Raddatz S., Paulsen H., Henninger K., Bandow J. E., Sahl H. G., Labischinski H. (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 11, 1082–1087 [DOI] [PubMed] [Google Scholar]
- 47. Christ K., Al-Kaddah S., Wiedemann I., Rattay B., Sahl H. G., Bendas G. (2008) Membrane lipids determine the antibiotic activity of the lantibiotic gallidermin. J. Membr. Biol. 226, 9–16 [DOI] [PubMed] [Google Scholar]
- 48. Brown S., Zhang Y. H., Walker S. (2008) A revised pathway proposed for Staphylococcus aureus wall teichoic acid biosynthesis based on in vitro reconstitution of the intracellular steps. Chem. Biol. 15, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xia G., Kohler T., Peschel A. (2010) The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 300, 148–154 [DOI] [PubMed] [Google Scholar]
- 50. Barrett J. F. (2005) Recent developments in glycopeptide antibacterials. Curr. Opin. Investig. Drugs. 6, 781–790 [PubMed] [Google Scholar]
- 51. Kim S. J., Cegelski L., Stueber D., Singh M., Dietrich E., Tanaka K. S., Parr T. R., Jr., Far A. R., Schaefer J. (2008) Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 377, 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wiedemann I., Benz R., Sahl H. G. (2004) Lipid II-mediated pore formation by the peptide antibiotic nisin: a black lipid membrane study. J. Bacteriol. 186, 3259–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Breukink E., Ganz P., de Kruijff B., Seelig J. (2000) Binding of nisin Z to bilayer vesicles as determined with isothermal titration calorimetry. Biochemistry 39, 10247–10254 [DOI] [PubMed] [Google Scholar]
- 54. Scherer K., Wiedemann I., Ciobanasu C., Sahl H. G., Kubitscheck U. (2013) Aggregates of nisin with various bactoprenol-containing cell wall precursors differ in size and membrane permeation capacity. Biochim. Biophys. Acta 1828, 2628–2636 [DOI] [PubMed] [Google Scholar]
- 55. Hasper H. E., Kramer N. E., Smith J. L., Hillman J. D., Zachariah C., Kuipers O. P., de Kruijff B., Breukink E. (2006) Alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 313, 1636–1637 [DOI] [PubMed] [Google Scholar]
- 56. Wiedemann I., Böttiger T., Bonelli R. R., Wiese A., Hagge S. O., Gutsmann T., Seydel U., Deegan L., Hill C., Ross P., Sahl H. G. (2006) The mode of action of the lantibiotic lacticin 3147: a complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol. Microbiol. 61, 285–296 [DOI] [PubMed] [Google Scholar]
- 57. Schneider T., Gries K., Josten M., Wiedemann I., Pelzer S., Labischinski H., Sahl H. G. (2009) The lipopeptide antibiotic friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 53, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wenzel M., Chiriac A. I., Otto A., Zweytick D., May C., Schumacher C., Gust R., Albada H. B., Penkova M., Kramer U., Erdmann R., Metzler-Nolte N., Straus S. K., Bremer E., Becher D., Brotz-Oesterhelt H., Sahl H. G., Brandow J. E. (2014) Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc Natl. Acad. Sci. U.S.A., in press, 10.1073/pnas.131199001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.