

Abstract
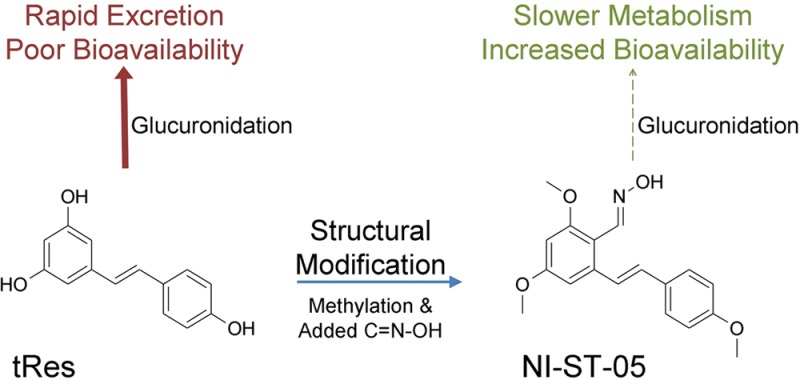
Trans-Resveratrol (tRes) has been shown to have powerful antioxidant, anti-inflammatory, anticarcinogenic, and antiaging properties; however, its use as a therapeutic agent is limited by its rapid metabolism into its conjugated forms by UDP-glucuronosyltransferases (UGTs). The aim of the current study was to test the hypothesis that the limited bioavailability of tRes can be improved by modifying its structure to create analogs which would be glucuronidated at a lower rate than tRes itself. In this work, three synthetic stilbenoids, (E)-3-(3-hydroxy-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (NI-12a), (E)-2,4-dimethoxy-6-(4-methoxystyryl)benzaldehyde oxime (NI-ST-05), and (E)-4-(3,5-dimethoxystyryl)-2,6-dinitrophenol (DNR-1), have been designed based on the structure of tRes and synthesized in our laboratory. UGTs recognize and glucuronidate tRes at each of the 3 hydroxyl groups attached to its aromatic rings. Therefore, each of the above compounds was designed with the majority of the hydroxyl groups blocked by methylation and the addition of other novel functional groups as part of a drug optimization program. The activities of recombinant human UGTs from the 1A and 2B families were examined for their capacity to metabolize these compounds. Glucuronide formation was identified using HPLC and verified by β-glucuronidase hydrolysis and LC–MS/MS analysis. NI-12a was glucuronidated at both the −COOH and −OH functions, NI-ST-05 formed a novel N–O-glucuronide, and no product was observed for DNR-1. NI-12a is primarily metabolized by the hepatic and renal enzyme UGT1A9, whereas NI-ST-05 is primarily metabolized by an extrahepatic enzyme, UGT1A10, with apparent Km values of 240 and 6.2 μM, respectively. The involvement of hepatic and intestinal UGTs in the metabolism of both compounds was further confirmed using a panel of human liver and intestinal microsomes, and high individual variation in activity was demonstrated between donors. In summary, these studies clearly establish that modified, tRes-based stilbenoids may be preferable alternatives to tRes itself due to increased bioavailability via altered conjugation.
Introduction
Resveratrol (trans-3,5,4′-trihydroxystilbene; tRes) is a well-known, natural polyphenol found in grapes, peanuts, red wine, and other foods.1,2 It has been shown to be a powerful antioxidant, neuroprotector, anticarcinogenic, and anti-inflammatory agent that lacks detectable toxicity.3 In clinical studies, resveratrol has demonstrated the ability to reduce tumor cell proliferation in patients with colorectal cancer.4 However, although initial preclinical data are encouraging, the oral bioavailability of native tRes is relatively low, due in large part to its rapid conjugation,5−7 which presents a significant limitation to future clinical development of this potentially valuable drug.
The in vitro glucuronidation of tRes has been relatively well characterized,1,2,5,8,9 and it has been shown that human hepatic and intestinal UGTs are actively involved in tRes glucuronidation and that both tissues contribute significantly to its biotransformation.2,8 The glucuronidation reactions are very region- and stereospecific, with a trans-3-O-glucuronide being the predominant product formed.2 In order to potentially circumvent the unfavorable pharmacokinetics of tRes, the search for compounds with enhanced bioavailability, such as synthetic or naturally occurring structural analogs of tRes that exhibit more favorable conjugation profiles, is of great significance.
Natural prenylated tRes analogs, trans-arachidin-1 and -3, produced in and purified from peanut hairy root culture, were previously tested toward several UGT1A enzymes and human hepatic and intestinal microsomes.9 These two derivatives, both prenylated at position 4, had glucuronidation rates that were slower in comparison to native tRes (decreased Vmax values). Therefore, the greater lipophilicity of prenylated tRes analogs changed the enzyme affinity, ultimately leading to a slower metabolism, which is expected to improve bioavailability. Although the isolation of compounds from medicinal plants is an exciting avenue for obtaining compounds that could be developed into drugs, this method is limited to naturally occurring compounds. Chemical synthesis opens the door for the production of compounds specifically designed to maximize effectiveness and improve bioavailability.
A number of nucleophilic functional groups are known to be targets for glucuronidation. Excluding extraordinary cases, glucuronides can be formed at carbon atoms with oxygen derived nucleophiles (O-glucuronides), and upon N-carbamyolation of primary and secondary amino groups (N-glucuronides).10 Sulfur- and selenium-linked glucuronides and di- and bis-glucuronides are less common but also occur.10 Considering the above, a number of tRes-based stilbenoid derivatives can be designed that have improved bioavailability due to restricted glucuronidation while maintaining biological activity.
In the current studies, three new stilbenoids, NI-ST-05, DNR-1, and NI-12A, have been designed based on the structure of tRes, with the majority of the hydroxyl groups blocked by methylation and novel functional groups added to the stilbene scaffold. It was expected that this would result in lower levels of UGT-mediated metabolism. The study aimed to elucidate: (i) whether the newly synthesized stilbene derivatives are substrates for human recombinant UGTs and (ii) what the level of glucuronidation activity of selected UGT enzymes is toward these novel stilbenes in comparison to the native tRes. Twelve major recombinant human UGT1A and 2B enzymes have been examined for their capacity to metabolize these compounds. The formation of glucuronide conjugates has been identified using HPLC–UV–vis analysis, and the structures of these metabolites have been elucidated by LC–MS/MS and β-glucuronidase hydrolysis. We have hypothesized that the selected UGT enzymes would have lower affinity for these modified analogs of tRes. Therefore, the synthesized stilbenoids would represent a useful scaffold for the design of selective and efficacious resveratrol analogs with improved bioavailability and which could be developed as chemotherapeutic agents.
Experimental Procedures
Materials
All chemicals used in this study were of at least reagent grade. Recombinant proteins UGT1A1, 1A3, 1A4, and 1A6–1A10 were provided by Dr. Moshe Finel (Drug Discovery and Development Technology Center, Faculty of Pharmacy, University of Helsinki, Helsinki, Finland). Recombinant proteins UGT2B4, 2B7, 2B15, and 2B17 were obtained from BD Biosciences (San Diego, CA). Spectrophotometric grade dimethyl sulfoxide (>99.9% pure) was purchased from Sigma-Aldrich (St. Louis, MO), and LC–MS grade methanol and reagent grade acetic acid were from Fisher Scientific (Pittsburgh, PA). Human hepatic and intestinal microsomes were obtained as described previously.11,12 UDP-glucuronic acid (-GlcUA) and all other chemicals and reagents, unless otherwise stated, were purchased from Sigma-Aldrich.
Chemical Synthesis of (E)-2,4-Dimethoxy-6-(4-methoxystyryl)benzaldehyde Oxime (NI-ST-05)
(E)-1,3-Dimethoxy-5-(4-methoxystyryl)benzene (1) (1 mmol) was dissolved in 5 mL of dimethylformamide (DMF) and cooled to 0 °C, and POCl3 (1.3 mmol) was added dropwise over 15 min. The reaction mixture was stirred for 30 min at ambient temperature. The purple-colored solution was then added to 20 mL of ice-cold water and the resulting mixture extracted three times with 10 mL portions of dichloromethane (DCM). The organic extracts were combined and washed with water, dried over anhydrous Na2SO4, and concentrated on a rotary evaporator to afford the desired crude product. (E)-2,4-Dimethoxy-6-(4-methoxystyryl)benzaldehyde (2) was recrystallized in DCM from the crude product in 65% yield. (E)-2,4-Dimethoxy-6-(4-methoxystyryl)benzaldehyde (2) (1 mmol), hydroxylamine HCl (1.1 mmol), and sodium acetate (AcONa; 1.5 mmol) were dissolved in 10 mL of ethanol (EtOH), and the mixture was heated under reflux for 4 h. A white precipitate was formed, which was filtered off to afford NI-ST-05 (3) in 80% yield (see Figure 1).
Figure 1.

Synthesis and chemical structures of novel stilbenoids. (a) POCl3, DMF, 0 °C, 69% yield; (b) hydroxylamine, AcONa, methanol, reflux, 4 h, 90% yield; (c) triphenylphosphine, toluene, reflux, 12 h, 95% yield; (d) concentrated H2SO4, fuming HNO3, 0 °C, 55% yield; (e) NaOMe, methanol, ambient temp., 12 h, 60% yield; (f) TEA, Ac2O, 140 °C, 40% yield. (3) NI-ST-05; (8) DNR-1; (11) NI-12a; (tRes) trans-resveratrol.
C18H19NO4. 1H NMR (DMSO-d6, ppm): δ 3.77 (s, 3H, −OCH3), 3.81 (s, 3H, −OCH3), 3.85 (s, 3H, −OCH3), 6.54 (s, 1H, −ArH), 6.89 (s, 1H, −ArH), 6.93–6.96 (d, 2H, −ArH, J = 8.4 Hz), 7.11–7.15 (d, 1H, −ArH, J = 16.4 Hz), 7.45–7.47 (d, 2H, −ArH, J = 8 Hz), 7.59–7.63 (d, 1H, −ArH, J = 16.4 Hz), 8.30 (s, 1H, −ArH), 11.15 (s, 1H, −OH). 13C NMR (DMSO-d6, ppm): δ 55.6, 55.8, 55.3, 98.0, 102.7, 110.0, 112.6, 114.6, 126.4, 128.3, 130.1, 130.5, 138.7, 145.2, 159.4, 159.7, 161.0.
Chemical Synthesis of (E)-4-(3,5-dimethoxystyryl)-2,6-dinitrophenol (DNR-1)
1-(Bromomethyl)-3,5-dimethoxybenzene (4) (1.0 mmol) and triphenylphosphine (1.5 mmol) were dissolved in toluene and the mixture refluxed overnight to yield (3,5-dimethoxybenzyl) triphenylphosphonium bromide (5) as a white precipitate, which was filtered off. The yield was 95%. In a separate reaction, 4-hydroxybenzaldehyde (6) (2 g) was dissolved in 12.5 mL of concentrated H2SO4 and cooled to 0 °C. A mixture of concentrated H2SO4 (5 mL) and fuming HNO3 (5 mL) was cooled to 0 °C and added slowly to the above reaction mixture. The resulting mixture was stirred for 4 h at ambient temperature. The reaction mixture was then slowly added to 50 mL of ice water and the resulting mixture stirred for 1 h at 0 °C. At this point, a yellow precipitate was formed, which was filtered off and purified through silica gel flash column chromatography with 2:3 ethyl acetate/hexane as the mobile phase, affording 4-hydroxy-3,5-dinitrobenzaldehyde (7) in 75% yield. (3,5-Dimethoxybenzyl)triphenylphosphonium bromide (5) (1.0 mmol) was dissolved in 5 mL of methanol, and 4-hydroxy-3,5-dinitrobenzaldehyde (7) (1.0 mmol) and sodium methoxide (NaOMe, 5.0 mmol) were added. The resulting solution was stirred at room temperature for 5 h. The reaction was monitored by TLC, and after consumption of starting materials, the reaction mixture was concentrated and extracted into ethyl acetate followed by concentration on a rotary evaporator and silica gel flash column chromatographic fractionation using methanol/DCM as the mobile phase to afford DNR-1 (8) in 45% yield (see Figure 1).
C16H14N2O7. 1H NMR (DMSO-d6, ppm): δ 3.76 (s, 6H, −OCH3), 6.33 (s, 1H, −ArH), 6.71 (s, 2H, −ArH), 6.83–6.88 (d, 1H, −ArH, J = 16.4 Hz), 7.12–7.16 (d, 1H, −ArH J = 16 Hz), 8.10 (s, 2H, −ArH) 13C NMR (DMSO-d6, ppm): δ 55.5, 99.6, 104.2, 110.0, 113.5, 124.2, 128.1, 128.5, 140.7, 143.6, 159.7, 161.0.
Chemical Synthesis of (E)-3-(3-hydroxy-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid (NI-12a)
3-Hydroxy-4-methoxybenzaldehyde (9) (1.0 mmol), 2-(3,4,5-trimethoxyphenyl)acetic acid (10) (2.0 mmol) and triethylamine (3.2 mmol) were added to 5 mL of acetic anhydride. The resulting reaction mixture was refluxed at 140 °C for 4 h and monitored by TLC. When the reaction was complete, 10 mL of ice water was added and extracted with 10 mL of ethyl acetate. The organic phase was concentrated on a rotary evaporator and the desired product purified by silica gel flash column chromatography using methanol/DCM as mobile phase to afford NI-12a (11) in 35% yield (see Figure 1).
C18H20O5. 1H NMR (CDCl3-d6, ppm): δ 3.69–3.72 (d, 12H, −OCH3, J = 15.2 Hz), 6.43–6.44 (d, 2H, −ArH, J = 2.8 Hz), 6.54 (s, 1H, −ArH), 6.61 (s, 1H, −ArH), 6.80 (s, 1H, −ArH), 8.93 (s, 1H, −ArH), 12.41 (s, 1H, −COOH). 13C NMR (DMSO-d6, ppm): δ 55.9, 56.4, 60.6, 107.2, 110.0, 112.0, 118.0, 123.3, 127.4, 131.0, 132.5, 137.4, 139.5, 146.2, 149.3, 153.5, 169.0.
Source of Human Microsomes and Recombinant UGTs
Human hepatic microsomes were obtained from 10 donors, and human intestinal microsomes were obtained from 13 donors. Recombinant proteins UGT1A1, 1A3, 1A4, and 1A6–1A10 were cloned and expressed in baculovirus-infected insect cells, as described previously.13,14 Human UGT2B4, 2B7, 2B15, and 2B17 were purchased from BD Biosciences (Woburn, MA) and assayed according to manufacturer protocols. Each enzyme tested in this study is known to be active toward substrates specific for that enzyme.
Screening of Human Microsomes and Recombinant UGTs
Screening experiments for glucuronidation activity were also performed with human hepatic and intestinal microsomes from 10 and 13 donors, respectively, one pooled liver sample, and commercially available hepatosomes (Human Biologics International). A 250 μM aliquot of substrate was added to the incubation tube dissolved in ethanol. This was allowed to air dry, and DMSO was added to solubilize the substrate and to act as an activator for membrane protein. The samples were sonicated to ensure substrate solubilization. Reaction buffer and human microsomes (50 mg of total protein) or recombinant UGT membranes (5 μg of total protein) were then added. UDP-GlcUA in molar excess was added, and the samples were incubated at 37 °C for 60 min. Final reaction concentrations were as follows: 100 μM Tris–HCl (pH 7.4)/5 mM MgCl2/5 mM saccharolactone/2% DMSO/250 μM substrate/2 mM UDPGA/50 or 5 μg of total protein, respectively. The total reaction volume was 30 μL. Reactions omitting substrates were run under the same conditions as controls. No additional detergents or activators were used in the incubations. The rate of glucuronidation with these enzymes has been shown to be linear for a maximum of 3 h (data not shown). The reactions were stopped by addition of 30 μL of ethanol. This was followed by centrifugation of the samples at 14 000 rpm for 8 min to collect the protein. The supernatants were separated by HPLC using an HP1050 HPLC system equipped with a UV–vis diode array detector. Instrument function and data acquisition were evaluated using Agilent ChemStation software.
Enzyme Kinetics Assays
Kinetic parameters were established by incubating recombinant UGT protein with varying concentrations of NI-12a (10–1000 μM) or NI-ST-05 (1–1000 μM) with a molar excess of UDP-GlcUA for 60 min. All kinetic assays were performed in duplicate under conditions identical to those utilized for screening experiments.
Data Analysis
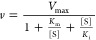
Kinetic data for the glucuronidation of NI-12a and NI-ST-05 by UGTs were estimated by plotting the measured initial reaction velocity values as a function of substrate concentration. Curve-fitting and statistical analyses were conducted utilizing GraphPad Prism v4.0b (GraphPad Software, Inc., San Diego, CA). Kinetic constants were obtained by fitting the experimental data to the following kinetic models using the nonlinear regression (Curve Fit) function
-
1.Michaelis–Menten (M–M) equation for the one-enzyme model
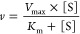
-
2.Hill equation, which describes sigmoidal autoactivation kinetics, where S50 is the substrate concentration at 50% Vmax (analogous to Km in M–M kinetics) and n is the Hill coefficient, which can be considered to be a measure of autoactivation, and reflects the extent of cooperativity among multiple binding sites15
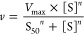
-
3.Uncompetitive substrate inhibition (USI) model, where Ki is the inhibition constant describing the reduction in rate
The fit of the data for each model was assessed from the standard error, 95% confidence intervals, and R2 values. Kinetic curves were also analyzed as Eadie–Hofstee plots to support kinetic models (graphs not shown). Kinetic constants are reported as the mean ± standard error.
Glucuronidation by human hepatic and intestinal microsomes is presented as the line of median value and the error bars represent the range of values. Statistical comparisons were performed using the Kruskal–Wallis nonparametric analysis of variance followed by Dunn’s multiple comparison test.
Glucuronide Product Analysis
HPLC–UV analysis of the supernatants was carried out on an HP1050 LC system equipped with a variable wavelength UV–DAD detector. Instrument control and data collection was accomplished using Agilent ChemStation software. The samples were separated on a reversed-phase 5 μm Suplex pKb-100 analytical column (0.46 cm × 25 cm, C18) (Supelco, USA) maintained at 25 °C. HPLC analyses were carried out at a flow rate of 1 mL/min with the following elution system: a linear gradient from 15 to 80% methanol in ammonium formate buffer (0.05 M, pH 3.4) for 25 min followed by a linear gradient from 80 to 100% methanol in ammonium formate for 3 min. The column was then re-equilibrated to initial conditions for 10 min between runs. The elution of each metabolite of NI-12a was monitored at 335 nm, whereas the elution of the metabolite of NI-ST-05 was monitored at 325 nm.
Analysis of Glucuronide Product Structures by ESI–HPLC–MS
HPLC–MS analysis of the glucuronide products were conducted using an Acquity uHPLC system interfaced to a Quattro Premier triple quadrupole mass analyzer (Waters Corporation, Beverly, MA) with an electrospray probe operating in the positive ion mode. LC conditions were identical to those used for LC–UV analysis with 10 μL of sample injected onto the column. The mass spectrometer was calibrated using sodium cesium iodide (NaCsI) over a range of 20–1974 Da. The electrospray capillary voltage was 3.5 kV, and the cone voltage was 25 V. The desolvation gas (nitrogen) flow rate was 600 L/h and was maintained at 500 °C. The source temperature was 150 °C.
Results
HPLC–UV–vis and HPLC–MS/MS Analysis of Glucuronide Metabolites
Preliminary studies on the glucuronidation of NI-ST-05, DNR-1, and NI-12a were aimed at evaluating their glucuronidation by UGT1A and UGT2B enzymes. HPLC analysis of the incubation mixtures demonstrated the formation of glucuronidation products for NI-ST-05 and NI-12a but not for DNR-1. Representative HPLC analyses indicates the formation of two metabolites at tR = 17.6 and tR = 17.8 min for NI-12a, and one product at tR 23.2 min for NI-ST-05 (Figure 2). Retention time values of these metabolites are lower than those of their respective parent compounds.
Figure 2.

Metabolism of NI-12a (A) and NI-ST-05 (B) with human liver microsomes. Representative HPLC analyses are shown from 60 min incubations of 50 μg of human liver proteins with 0.25 mM substrate and 3 mM UDP-GlcUA. DNR-1 was not metabolized under the studied conditions.
LC–(+)-ESI–MS analysis, presented in Figure 3, indicated that the two NI-12a glucuronides (tR = 16.0 and 16.3 min) had an [M + NH4]+ peak m/z 554, and a major peak at tR =18.9 min, (m/z 361, 100%) corresponded to NI-12a. The two slightly different in-source fragmentation patterns in the spectra of the NI-12a metabolites indicated the presence of two types of glucuronides. We suggest that these are COO- and O-glucuronides. In the studies presented below, β-glucuronidase, which selectively hydrolyzes only O-glucuronides, was used to identify which of the two possible glucuronide metabolites was present. The major fragment observed for both glucuronides resulted from neutral loss of the glucuronide moiety. The higher intensity of the m/z 361 peak relative to the m/z 554 is evidence that the chromatographic peak at tR = 16.0 min is an acyl glucuronide. The acyl glucuronide should be a better leaving group than the corresponding O-glucuronide.
Figure 3.

Extracted ion chromatogram (m/z 361) and (+)-ESI mass spectra of NI-12a and its glucuronide conjugate NH4+ adducts. Spectra of NI-12a glucuronides (two isomers of tR = 16.0 and 16.3 min) showed an [M + NH4]+ peak (m/z 554) plus a fragment ion resulting from neutral loss of the glucuronide (m/z 361).
Ammonium adducts of NI-ST-05 glucuronides were not observed in MS/MS spectra, but the proton adduct ([M + H]+) was observed with a retention time of 23.2 min and an m/z 490 (Figure 4). The parent compound (NI-ST-05) eluted at 25.5 min and had a base peak at m/z 314 [M + H]+. NI-ST-05 has only one available group for glucuronidation, allowing unequivocal determination that the product was the C=N–O-glucuronide. In-source fragmentation was not observed for these compounds. Retention times for LC–MS runs (Figures 3 and 4) were slightly different from LC–UV runs, despite analyses being run using the same chromatographic column and mobile phase. The small differences can be attributed to different LC systems with unique system dead volumes.
Figure 4.

Mass spectra and structures of NI-ST-05 and its glucuronide conjugates. Spectrum of NI-ST-05 glucuronide (tR = 21.4 min) showed an [M + H]+ peak (m/z 490) plus a major peak corresponding to the NI-ST-05 substrate (m/z 314).
Screening of Recombinant UGTs for Activity toward NI-12a and NI-ST-05
Eight human recombinant UGT1A enzymes expressed as His-tag proteins in baculovirus-infected Sf9 insect cells, and UGT2B4, 2B7, 2B15, and 2B17 from BD Biosciences were evaluated for their ability to glucuronidate NI-12a and NI-ST-05. Activities at a substrate concentration of 250 μM were determined using HPLC–UV/vis analysis (Figure 5). Beta-glucuronidase hydrolysis was used to differentiate between COO-glucuronides and O-glucuronides.
Figure 5.

Glucuronidation of NI-12a (A) and NI-ST-05 (B) by human recombinant UGTs. UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B15, 2B17 (5 μg of protein) were evaluated for their ability to glucuronidate DNR-1, NI-12a, and NI-ST-05 (250 μm). No activity was observed toward DNR-1, and UGT2B7, 2B4, 2B15, and 2B17 were not active toward any compound. Activities are expressed in nanomoles per milligram of protein per minute.
Data for NI-12a indicated that UGT1A3 catalysis led to both COO- and O-glucuronides. UGT1A1 produced only the carboxyl metabolite, whereas UGTs 1A7–1A10 produced only the hydroxyl metabolite. UGT1A4 and 1A6 were not active toward this compound. None of the UGT2B enzymes screened produced any measurable metabolite under our conditions (data not shown). UGT1A9 (a hepatic enzyme) had the highest activity toward NI-12a, that is, close to 10 nmol/mg/min.
With NI-ST-05, UGT1A1, 1A9, and 1A10 catalyzed the formation of the C=N–O-glucuronide with the extrahepatic enzyme, with UGT1A10 having the highest activity. All the remaining UGTs screened were inactive toward this compound.
Glucuronidation of NI-12a and NI-ST-05 by Human Hepatic and Intestinal Microsomes
Screening experiments for glucuronidation activity were also carried out with human hepatic and intestinal microsomes from 10 and 13 donors, respectively, one pooled liver sample, and commercially available hepatosomes (Figure 6). All hepatic samples were shown to glucuronidate both NI-12a and NI-ST-05 producing two and one metabolic products, respectively. In assays with intestinal samples, donors HI27, HI28, HI29, and HI54 failed to display glucuronidation activity toward NI-12a. Additionally, donors HI34, HI36, HI40, and HI41 produced only one (the hydroxyl) metabolite, whereas the remaining donors with activity produced two (hydroxyl and carboxyl) metabolites. For NI-ST-05, donors HI27, HI29, and HI30 failed to display glucuronidation activity, whereas all other donors produced one metabolite. In general, liver microsomes had significantly higher O-glucuronidation activity for NI-12a than for COO-glucuronidation of this compound or for C=N–O-glucuronidation of NI-ST-05. In contrast, the glucuronidation activity of intestinal microsomes did not significantly differ with the type of metabolites.
Figure 6.

Glucuronidation activities of human hepatic (A) and human intestinal (B) microsomes toward NI-12a and NI-ST-05. Human liver microsomes from 10 different donors, a pooled liver sample, and purchased “hepatosomes” as well as and human intestinal microsomes from 13 different donors were analyzed. Each substrate concentration was 0.25 mM, with a molar excess of UDP-GlcUA (2 mM); the reactions were incubated for 60 min. Activities are expressed in nanomoles per milligram of protein per minute.
Kinetic Analysis of Selected Recombinant UGTs with NI-12a and NI-ST-05
On the basis of the activity screening data, selected human recombinant UGT enzymes (UGT1A7–1A10 for NI-12a and UGT1A1, 1A9, and 1A10 for NI-ST-05) were subjected to steady state kinetic analysis (Table 1 and Figures 7 and 8). For NI-12a, UGT1A9 was the most active recombinant UGT (Vmax = 8.3 nm/mg protein/min) and had a relatively low affinity, with a Km value of 240 μM. UGT1A7 demonstrated the highest affinity for NI-12a (Km = 62 μM), and UGT1A10 had the lowest affinity for NI-12a (Km = 500 μM). The catalytic efficiency of UGT1A9 toward NI-12a, measured by the ratio Vmax/Km (35 μL/min/mg) was also the highest, likely because of the high Vmax. Both, UGT1A9 and UGT1A10 fit Michaelis–Menten kinetics. Data generated for UGT1A7 and 1A8 both fit the uncompetitive substrate inhibition kinetic model. This model suggests the existence of multiple binding sites in these enzymes.
Table 1. Glucuronidation Kinetics for NI-12a and NI-ST-05 Metabolitesa.
| UGT1A1 | UGT1A7 | UGT1A8 | UGT1A9 | UGT1A10 | |
|---|---|---|---|---|---|
| NI-12a | |||||
| NI-12a-O-gluc | not produced | ||||
| Km (μM) | 62 ± 43 | 240 ± 76 | 240 ± 40 | 500 ± 75 | |
| Vmax (nmol/mg/min) | 2.2 ± 0.77 | 6.0 ± 1.3 | 8.3 ± 0.52 | 3.5 ± 0.24 | |
| Ks (μM) | 242 ± 133 | 950 ± 449 | |||
| CLint or CLmax (μL/mg/min) | 35 | 25 | 35 | 7 | |
| kinetic model | USI | USI | M–M | M–M | |
| R2 | 0.82 | 0.98 | 0.97 | 0.98 | |
| NI-12a-COO-gluc | too low to be assessed | not produced | not produced | not produced | not produced |
| NI-ST-05 | |||||
| NI-ST-05–N–O-gluc | not produced | not produced | |||
| Km or S50 (μM) | <5b | <5b | 6.2 ± 0.35 | ||
| Vmax (nmol/mg/min) | 0.21 ± 0.02b | undetermined | 0.65 ± 0.017 | ||
| n | 3.4 | ||||
| CLint or CLmax (μL/mg/min) | 105 | ||||
| kinetic model | M–Mb | undetermined | Hill | ||
| R2 | 0.61b | 0.87 | |||
Glucuronidation activities of selected recombinant UGTs were measured by incubating membrane fractions with increasing concentrations of substrate (see Figures 7 and 8) at a constant concentration of UDP-GlcUA (3 mM). Reactions were centrifuged, supernatants separated by HPLC, and curve fits and kinetic constants determined using GraphPad Prism 4 software.Values estimated based on an incomplete data set.
Values estimated based on an incomplete data set.
Figure 7.

Steady state kinetic curves for the glucuronidation of NI-12a by selected human recombinant UGTs. Glucuronidation activities for wild-type UGT1A7, 1A8, 1A9, and 1A10 were measured by incubating membrane fractions containing recombinant UGTs with increasing concentrations (shown in figure) of the substrates at a constant concentration of UDP-GlcUA (3 mM) for 60 min at 37 °C. Curve fits and kinetic constants were determined using GraphPad Prism 4 software. The graphical fits of the data (mean ± SD) are shown.
Figure 8.

Steady state kinetic curves for the glucuronidation of NI-ST-05 by selected human recombinant UGTs. Glucuronidation activities for wild-type UGT1A1, 1A9, and 1A10 were measured by incubating membrane fractions containing recombinant UGTs with increasing concentrations (shown in figure) of the substrates at a constant concentration of UDP-GlcUA (3 mM) for 60 min at 37 °C. Curve fits and kinetic constants were determined using GraphPad Prism 4 software. The graphical fits of the data (mean ± SD) are shown.
The C=N–O-glucuronidation of NI-ST-05 resulted in very little product formation and therefore our ability to perform kinetic analyses were limited by the sensitivity of our HPLC–UV/vis method. Each of the active enzymes exhibited high affinity and relatively low activity for this compound. Because of this, the data generated with UGT1A1 cannot be used to estimate Km values because of a lack of measurable product formation at substrate concentrations below 5 μM. Thus, we have indicated that the Km value for this reaction is <5 μM. UGT1A1 activities at concentrations above 5 μM were used to estimate a Vmax value of 0.2 nmol/mg/min assuming M–M kinetics. UGT1A10 fit the Hill equation, indicating cooperative binding. This reaction produced very low S50 and Vmax values of 6.2 μM and 0.65 nm/mg protein/min, respectively. The catalytic efficiency of this reaction (Vmax/Km = 105 μL/min/mg) was 3 to 10 times higher than that of NI-12a. Data generated with UGT1A9 did not fit any of the kinetic models tested. This assay showed the highest level of activity at the 5 μM concentrations, indicating a high affinity for this enzyme-compound set, and this activity decreased in a concentration-dependent manner up to 15 μM when the velocity plateaued and remained constant up to 75 μM (the highest concentration assayed).
Discussion
The aim of the current study was to test the hypothesis that the limited bioavailability of tRes, resulting from its rapid UGT-mediated metabolism, can be improved by modifying its structure to create analogs which would be glucuronidated at a lower rate than tRes itself. tRes has been the focus of great attention because it is a very efficacious antioxidant16 and anti-inflammatory17 agent with potential therapeutic applications.18 It is recognized that resveratrols induce a large number of different downstream effects in vitro and in vivo. However, the exact mechanism of their action has yet to be revealed. Moreover, it is documented that the presence of the 3 hydroxyl groups in its structure may actually decreases the cytotoxicity of these compounds because they are targets for rapid metabolism leading to poor bioavailability. In fact, it has been shown that when these groups are substituted with methoxy groups the cytotoxic activity of these compounds is much improved.19−21 This phenomenon was the inspiration behind the design of our compounds.
In designing the structures of the new compounds studied here, it was essential to decrease the number of free hydroxyl groups and to modulate the electron density both in the aromatic rings and in the molecule as a whole. Thus, three new stilbenoid derivatives were synthesized by replacing hydroxyl groups with methoxyl groups and by adding N-hydroxyl, aromatic nitro, and carboxyl substituents to create NI-ST-05, DNR-1, and NI-12a, respectively. Such chemical structures are assumed to be less susceptible to glucuronidation, thus improving their bioavailability, while hopefully preserving their biological properties. In previous studies carried out in our laboratory, we have used the same recombinant and microsomal enzyme preparations to determine tRes glucuronidation rates,9 allowing us to directly compare the effects of these structural changes on the glucuronidation of these compounds.
We first demonstrated that only two of the synthesized stilbenoids, NI-ST-05 and NI-12a, were substrates for recombinant UGT enzymes and human hepatic and intestinal microsomes. The absence of DNR-1 glucuronidation is not surprising. Although a phenolic hydroxyl substituent remains in the structure of DNR-1, this lack of DNR-1 glucuronidation may result from the presence of two nitro groups at ortho positions to this phenolic hydroxyl group, which changes its acid–base and nucleophilic properties. Therefore, the nucleophilic attack of the charged oxygen atom on C1 of glucuronic acid in the presence of the two negatively charged phosphatic groups of UDP would be limited. This, combined with the large steric hindrance of the two polar nitro groups limits the glucuronidation potential of DNR-1.
The structures of the NI-12a and NI-ST-05 glucuronides formed were determined by HPLC–MS/MS analysis. MS spectra of the molecular proton adduct were not observed in the case of NI-12a, but the ammonium adduct was detected. These data suggest that the ammonium ion, which is present in the mobile phase, chelates with the ortho methoxy/hydroxy pair to form the ammonium adduct observed in the MS spectrum of the NI-12a glucuronide. This proposed interaction is supported by the absence of an ammonium adduct in the MS spectrum of NI-ST-05, which does not contain the ortho substituted methoxy/hydroxy moiety.
Glucuronidation assays with NI-12a resulted in multiple products and atypical reaction kinetics specific for each enzyme (Table 1), which point to a complex mechanism of metabolism for this compound. The hepatic enzyme UGT1A9 had the highest O-glucuronidation activity toward NI-12a, whereas UGT1A7, 1A8, and 1A10 transformation rates were lower. It is important to note that in our previous work,9 the enzyme that was determined to have the highest rate of tRes-3-O-glucuronidation (the primary metabolite in humans) was UGT1A1 (Vmax = 31 nmol/mg/min) and that this enzyme has very little activity toward this compound (too low to reliably determine kinetic parameters using our system). Steady state kinetic analyses demonstrated that UGT1A9 catalyzed the formation of the NI-12a–O-glucuronide at a slightly higher rate than the tRes-3-O-glucuronide (Vmax = 8.3 and 2.5 nmol/mg/min, respectively); however, the affinity of this enzymes for NI-12a (Km = 240 μM) is significantly less than for tRes (Km = 2.5 μM) or for the naturally occurring tRes analogs tA1 (Km = 190 μM) and tA3 (Km = 21 μM).9 Taken together, these parameters indicate that the modified stilbene structure of NI-12a most likely will have improved bioavailability compared to that of tRes and to those of the naturally occurring prenylated stilbenoids.
Glucuronidation assays with NI-ST-05 resulted in the formation of a rare C=N–O-glucuronide, and the rate of this reaction was significantly lower than the formation of either of the tRes or tA glucuronides.9 NI-ST-05 is characterized by C=N–OH group, which may be responsible for the high affinity of this compound for each of the UGTs assayed, and the as yet unidentified kinetic mechanism observed with UGT1A9. NI-ST-05 shows promise for further development as a stilbenoid with low glucuronidation potential; however, the resulting atypical reaction kinetics indicate a complex mechanism for the metabolic pathway for this compound, which is specific for selected UGT enzymes.
In conclusion, the glucuronidation of the novel stilbenoids studied in this report revealed that these compounds demonstrate improved glucuronidation profiles as compared to tRes. The selected structural modifications have resulted in them acting as poorer substrates for recombinant and microsomal UGT enzymes. Thus, these tRes analogs can be predicted to have better bioavailability in vivo. In addition to glucuronidation, tRes is also extensively sulfated in vitro and in vivo.5−7 It is anticipated that we can extrapolate the conclusions from this work from UGTs to sulfotransferases. It is also possible that these structural changes could affect the metabolism of these compounds by other enzymes such as cytochromes P450s. These additional metabolic pathway should be the topic of future studies.
These resveratrol analogs have also been evaluated as anticancer agents against human cancer cell lines MCF-7 (breast), A549 (lung), KB-3-1 (epidermoid carcinoma), and IGROV (ovarian).22−24 Compound NI-12A was the most potent analog and was found to be an effective antitubilin agent.22 On the basis of these data, the synthesized stilbenoids likely represent a useful scaffold for the design of highly selective and efficacious analogs which potentially could be developed as therapeutics. These preliminary enzymatic studies have provided useful information for a better understanding of the involvement of conjugative metabolism in the biotransformation of synthetic stilbenoids in humans and for assessing the potential utility of these molecules as novel chemotherapeutic agents.
Acknowledgments
The authors would like to acknowledge Dr. Moshe Finel for his generously provided recombinant UGT proteins.
Glossary
Abbreviations
- UGTs
uridine diphosphate glucuronosyltransferases
- NI-12a
(E)-3-(3-hydroxy-4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)acrylic acid
- NI-ST-05
(E)-2,4-dimethoxy-6-(4-methoxystyryl)benzaldehyde oxime
- DNR-1
(E)-4-(3,5-dimethoxystyryl)-2,6-dinitrophenol
This work was funded by a National Institutes of Health Grant [GM075893] and a Department of Defense grant [X81XWH-11-1-0795] funded by USAMRMC to A.R.-P., as well as an Arkansas Research Alliance grant to P.A.C.
The authors declare no competing financial interest.
Notes
Both laboratories at the University of Arkansas for Medicinal Sciences contributed equally to this work.
Funding Statement
National Institutes of Health, United States
References
- Aumont V.; Krisa S.; Battaglia E.; Netter P.; Richard T.; Merillon J. M.; Magdalou J.; Sabolovic N. (2001) Regioselective and stereospecific glucuronidation of trans- and cis-resveratrol in human. Arch. Biochem. Biophys. 393, 281–289. [DOI] [PubMed] [Google Scholar]
- Sabolovic N.; Humbert A. C.; Radominska-Pandya A.; Magdalou J. (2006) Resveratrol is efficiently glucuronidated by UDP-glucuronosyltransferases in the human gastrointestinal tract and in Caco-2 cells. Biopharm. Drug Dispos. 27, 181–189. [DOI] [PubMed] [Google Scholar]
- Baur J. A.; Sinclair D. A. (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discovery 5, 493–506. [DOI] [PubMed] [Google Scholar]
- Patel K. R.; Brown V. A.; Jones D. J.; Britton R. G.; Hemingway D.; Miller A. S.; West K. P.; Booth T. D.; Perloff M.; Crowell J. A.; Brenner D. E.; Steward W. P.; Gescher A. J.; Brown K. (2010) Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 70, 7392–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel E.; Somoza V. (2005) Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 49, 472–481. [DOI] [PubMed] [Google Scholar]
- Vitaglione P.; Sforza S.; Galaverna G.; Ghidini C.; Caporaso N.; Vescovi P. P.; Fogliano V.; Marchelli R. (2005) Bioavailability of trans-resveratrol from red wine in humans. Mol. Nutr. Food. Res. 49, 495–504. [DOI] [PubMed] [Google Scholar]
- Cottart C. H.; Nivet-Antoine V.; Laguillier-Morizot C.; Beaudeux J. L. (2010) Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food. Res. 54, 7–16. [DOI] [PubMed] [Google Scholar]
- Brill S. S.; Furimsky A. M.; Ho M. N.; Furniss M. J.; Li Y.; Green A. G.; Bradford W. W.; Green C. E.; Kapetanovic I. M.; Iyer L. V. (2006) Glucuronidation of trans-resveratrol by human liver and intestinal microsomes and UGT isoforms. J. Pharm. Pharmacol. 58, 469–479. [DOI] [PubMed] [Google Scholar]
- Brents L. K.; Medina-Bolivar F.; Seely K. A.; Nair V.; Bratton S. M.; Nopo-Olazabal L.; Patel R. Y.; Liu H.; Doerksen R. J.; Prather P. L.; Radominska-Pandya A. (2012) Natural prenylated resveratrol analogs arachidin-1 and -3 demonstrate improved glucuronidation profiles and have affinity for cannabinoid receptors. Xenobiotica 42, 139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argikar U. A. (2012) Unusual glucuronides. Drug. Metab. Dispos. 40, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Antonio L.; Xu J.; Little J. M.; Burchell B.; Magdalou J.; Radominska-Pandya A. (2003) Glucuronidation of catechols by human hepatic, gastric, and intestinal microsomal UDP-glucuronosyltransferases (UGT) and recombinant UGT1A6, UGT1A9, and UGT2B7. Arch. Biochem. Biophys. 411, 251–261. [DOI] [PubMed] [Google Scholar]
- Sabolovic N.; Heydel J. M.; Li X.; Little J. M.; Humbert A. C.; Radominska-Pandya A.; Magdalou J. (2004) Carboxyl nonsteroidal anti-inflammatory drugs are efficiently glucuronidated by microsomes of the human gastrointestinal tract. Biochim. Biophys. Acta 1675, 120–129. [DOI] [PubMed] [Google Scholar]
- Kurkela M.; Garcia-Horsman J. A.; Luukkanen L.; Morsky S.; Taskinen J.; Baumann M.; Kostiainen R.; Hirvonen J.; Finel M. (2003) Expression and characterization of recombinant human UDP-glucuronosyltransferases (UGTs). UGT1A9 is more resistant to detergent inhibition than other UGTs and was purified as an active dimeric enzyme. J. Biol. Chem. 278, 3536–3544. [DOI] [PubMed] [Google Scholar]
- Kuuranne T.; Kurkela M.; Thevis M.; Schanzer W.; Finel M.; Kostiainen R. (2003) Glucuronidation of anabolic androgenic steroids by recombinant human UDP-glucuronosyltransferases. Drug Metab. Dispos. 31, 1117–1124. [DOI] [PubMed] [Google Scholar]
- Weiss J. N. (1997) The Hill equation revisited: uses and misuses. FASEB J. 11, 835–841. [PubMed] [Google Scholar]
- Fraga C. G. (2007) Plant polyphenols: how to translate their in vitro antioxidant actions to in vivo conditions. IUBMB Life 59, 308–315. [DOI] [PubMed] [Google Scholar]
- Surh Y. J.; Kundu J. K.; Na H. K.; Lee J. S. (2005) Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J. Nutr. 135, 2993S–3001S. [DOI] [PubMed] [Google Scholar]
- Baur J. A.; Sinclair D. A. (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discovery 5, 493–506. [DOI] [PubMed] [Google Scholar]
- Roberti M.; Pizzirani D.; Simoni D.; Rondanin R.; Baruchello R.; Bonora C.; Buscemi F.; Grimaudo S.; Tolomeo M. (2003) Synthesis and biological evaluation of resveratrol and analogues as apoptosis-inducing agents. J. Med. Chem. 46, 3546–3554. [DOI] [PubMed] [Google Scholar]
- Lee S. K.; Nam K. A.; Hoe Y. H.; Min H. Y.; Kim E. Y.; Ko H.; Song S.; Lee T.; Kim S. (2003) Synthesis and evaluation of cytotoxicity of stilbene analogues. Arch. Pharmacal Res. 26, 253–257. [DOI] [PubMed] [Google Scholar]
- Piotrowska H.; Myszkowski K.; Ziolkowska A.; Kulcenty K.; Wierzchowski M.; Kaczmarek M.; Murias M.; Kwiatkowska-Borowczyk E.; Jodynis-Liebert J. (2012) Resveratrol analogue 3,4,4′,5-tetramethoxystilbene inhibits growth, arrests cell cycle and induces apoptosis in ovarian SKOV-3 and A-2780 cancer cells. Toxicol. Appl. Pharmacol. 263, 53–60. [DOI] [PubMed] [Google Scholar]
- Borrel C.; Thoret S.; Cachet X.; Guenard D.; Tillequin F.; Koch M.; Michel S. (2005) New antitubulin derivatives in the combretastatin A4 series: synthesis and biological evaluation. Bioorg. Med. Chem. 13, 3853–3864. [DOI] [PubMed] [Google Scholar]
- Madadi N. R. (2013) University of Arkansas for Medical Sciences, Little Rock, AR. Unpublished data.
- Yong Z., and Xu X. (2009) Styrene-acid derivative and use in manufacturing blood-vessel targeted-agent drugs. PCT Int. Appl. WO2007CN03127 20071105.