

Abstract
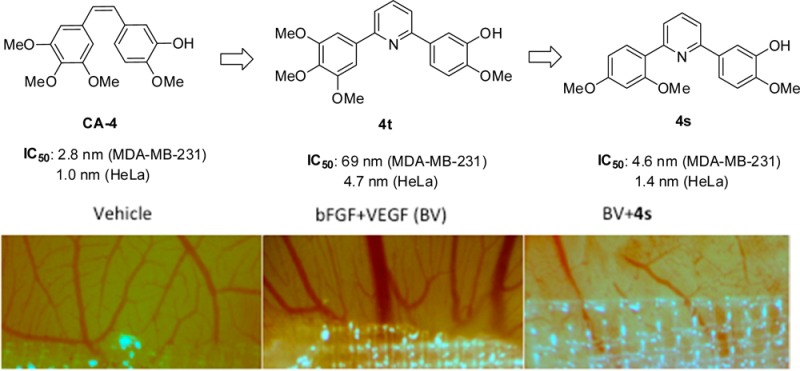
A series of novel pyridine-bridged analogues of combretastatin-A4 (CA-4) were designed and synthesized. As expected, the 4-atom linker configuration retained little cytotoxicities in the compounds 2e, 3e, 3g, and 4i. Activities of the analogues with 3-atom linker varied widely depending on the phenyl ring substitutions, and the 3-atom linker containing nitrogen represents the more favorable linker structure. Among them, three analogues (4h, 4s, and 4t) potently inhibited cell survival and growth, arrested cell cycle, and blocked angiogenesis and vasculature formation in vivo in ways comparable to CA-4. The superposition of 4h and 4s in the colchicine-binding pocket of tubulin shows the binding posture of CA-4, 4h, and 4s are similar, as confirmed by the competitive binding assay where the ability of the ligands to replace tubulin-bound colchicine was measured. The binding data are consistent with the observed biological activities in antiproliferation and suppression of angiogenesis but are not predictive of their antitubulin polymerization activities.
Introduction
Inhibition of tubulin polymerization disrupts the formation of tumor vasculature, making the microtubule cytoskeleton an effective target for cancer chemotherapy.1−3 Combretastatin-A4 (CA-4) is the prototype of a large group of vascular disrupting agents that have been designed, synthesized, and tested in various biological models as potential therapeutic candidates for cancer treatment.4,5 CA-4 binds to the colchicine binding site of tubulin to block microtubule assembly, causing rapid vascular shutdown and cell death in the tumor.6 The water-soluble phosphate prodrug form (CA-4P, also known as fosbretabulin) is in phase II/III clinical trials either alone or in combination with traditional chemotherapeutic agents or with radiotherapy.7−10 Meanwhile, over the past two decades, numerous novel derivatives of CA-4 have been discovered to confer cytotoxic potency and antitubulin activity that are comparable to CA-4, significantly expanding the arsenal of vascular disrupting agents that could be further explored for clinical applications. Despite the intense interest and the large number of potent derivatives of CA-4 that have been discovered that aimed at targeting the colchicine-binding site of tubulin, none of these inhibitors has reached the clinical stage. Thus, challenges remain in developing CA-4 analogues with improved pharmacological properties for eventual acceptance in the clinic.
Modifications made on the two phenyl rings, for example, have led to hundreds of active compounds that possess desirable cytotoxicity while retaining varying degrees of antitubulin activities.11 Most structural variations of the phenyl rings involve different combinations of hydroxyl and methoxy substitutions. These include various substituted phenyl rings12 and other aromatic rings.13 A few reports have attempted to modify the trimethoxy ring with mixed outcomes. For example, the m-methoxy group has been substituted by a fluoride to yield a similarly potent compound.14 In another example, when the trimethoxy ring was replaced by a trimethyl ring,15 the cytotoxicity of the compound was significantly reduced but the antitubulin activity was largely retained. This suggests that it might be possible to achieve disruption of tumor vasculature with fewer cytotoxic side effects.
Modifications of the double bond have also led to diverse structural variations that remain viable as cytotoxic and antitubulin compounds. The olefinic bond is believed to be critical in placing the two phenyl rings at an appropriate distance and giving the molecule the right dihedral angle to maximize the interaction with the target. As such, replacement of the double bond by rings that facilitate a cis-locked configuration has proven to be effective in retaining both cytotoxicity and antitubulin activity.16−20 Indeed, this strategy has led to the discovery of perhaps more active CA-4 analogues than any other types of structural modifications. On the other hand, the observation that two-carbon linkers are the optimal length of the bridge between the two phenyl rings has somewhat limited exploration in this strategy of modifications with some encouraging exceptions. For example, when the methylene bridge is replaced by a carbonyl group, the resulting analogue, phenstatin, actually retained much of the antitubulin activity.21 Interestingly, increasing the bridge length to three carbons such as a chalcone-like linker have been reported to strongly inhibit tubulin polymerization as well as cell survival.22 However, progress in this direction of structural modifications has been limited.
The current study was undertaken to investigate the effect of a novel variation of bridge length and structure on the anticancer activities of resulting CA-4 analogues. Specifically we have designed, synthesized, and evaluated a series of novel pyridine-linked CA-4 analogues (Figure 1) in which the distance between the two phenyl rings is configured to be three or four atoms, i.e., meta- or para- to each other. Pyridine has been introduced to replace the cis-double bond between the A ring and B ring.23 However, it was found that the antitubulin activities were largely lost in these pyridine-containing analogues. We show that cytotoxicity and antitubulin activities comparable to CA-4 can be obtained when the bridge length is fixed at three atoms (including the pyridine nitrogen) and substitutions on one or both of the phenyl rings are optimized. Here we describe the synthesis of 34 pyridine-bridged CA-4 analogues that were tested for their ability to inhibit cancer cell growth and proliferation by arresting cell cycles, their effect on tubulin polymerization as well as their activities in blocking angiogenesis. Molecular modeling and competitive binding assays were also performed to better understand the structural requirements for the pyridine-linked CA-4 analogues to retain antimitotic potency.
Figure 1.

Structures of CA-4 and three pyridine-linked analogues 4h, 4s, and 4t.
Results and Discussion
Chemistry
As shown in Scheme 1, the designed analogues were synthesized following the general procedures as detailed below and in the Experimental Section. Starting from commercially available dibromopyridines, palladium-catalyzed monocoupling reactions gave mainly the products of 2 with varying yields of the minor products of 3, some of which were isolated and purified successfully. The products of 2 were further transformed to symmetrically or unsymmetrically diaryl-substituted pyridines (3 and 4) by the application of aforementioned sequential Suzuki coupling. The reaction of 4s with hydrogen chloride in ether afforded its chloride salt 5.
Scheme 1. Synthesis of Novel Pyridine-Bridged Analogues of Combrestastatin-A4.

Reagents and conditions: (a) PdCl2(dppf) (0.40 g, 0.5 mmol), RB(OH)2, toluene–EtOH (4:1), Na2CO3, 80 °C; (b) PdCl2(dppf) (0.40 g, 0.5 mmol), RB(OH)2, toluene–EtOH (4:1), Na2CO3, 120 °C; (c) HCl in ether, rt.
Antiproliferative Activities
To evaluate the cytotoxicities of the pyridine-bridged combretastatin analogues, three human cancer cell lines were cultured and treated with the pyridine-bridged combretastatin analogues. Treatment at four different concentrations was used in order to determine the IC50 values for each compound along with the reference compound, CA-4. As shown in Table 1, when the two phenyl rings are para to each other on the pyridine linker, separated by a 4-carbon bridge, the cytotoxicity as measured by the antiproliferative activity is largely lost. Thus, analogues 3e, 3g, and 4i all showed high IC50 values, suggesting that such stretched-out configuration of the whole molecule does not bind well in the purported colchicine binding site to be effective. When the two phenyl rings are 2,4 substitutions on the pyridine linker, making the bridge a 3-carbon length where the pyridine nitrogen is not included (3f, 3h), results are mixed, with 3f showing modest cytotoxicity while 3h shows none. However, in the configuration where the 3-carbon linker contains the pyridine nitrogen, i.e., the two phenyl rings are now 2,6 meta to each other, significant improvement in the antiproliferative activity is seen in some analogues with further optimization of the phenyl ring substitutions. Indeed, when separated by three carbons, ring substitutions appear to be critical in conferring enhanced cytotoxicity. Trimethoxy substitution on one phenyl ring generally affords few options of substitutions on the other ring. For example, of the five analogues (4a, 4c, 4f, 4g, 4t) containing the trimethoxy phenyl group, 4a and 4f were inactive in all three cell lines, 4c was inactive in A549 cells, and 4g was inactive in MDA-MB-231 cells. The analogue 4t that differed from CA-4 in just the pyridine linker was found to be the only one in this group that potently inhibited growth of all three cancer cell lines that were tested.
Table 1. Growth Inhibition IC50 Values of Synthetic Pyridine-Bridged CA-4 Analogues in Three Cancer Cell Lines: MDA-MB-231, A549, and HeLa.
| growth inhibition IC50 (μM) |
|||
|---|---|---|---|
| combretastatin analogues | MDA-MB-231 | A549 | HeLa |
| 2a | 6.0 ± 3.5 | 9.0 ± 0.1 | 12.0 ± 0.2 |
| 2b | >25 | >25 | >50 |
| 2c | >100 | >50 | >50 |
| 2d | 10.0 ± 0.1 | 2.0 ± 0.2 | 2.0 ± 0 |
| 2e | 12.05 ± 0.02 | >25 | 14.0 ± 0.3 |
| 2f | >100 | >100 | >25 |
| 3a | 11.0 ± 1.8 | >50 | 1.0 ± 0.2 |
| 3b | >50 | >50 | >25 |
| 3c | 0.075 ± 0.001 | 0.079 ± 0.019 | 0.011 ± 0.001 |
| 3d | 9.0 ± 1.0 | >100 | 2.0 ± 0.4 |
| 3e | 11.0 ± 0.4 | >100 | 2.68 ± 0.08 |
| 3f | 2.63 ± 0.15 | 7.86 ± 0.233 | 0.79 ± 0.02 |
| 3g | >100 | >100 | >50 |
| 3h | >50 | >50 | >25 |
| 4a | 10.00 ± 1.56 | >25 | 8.0 ± 0.5 |
| 4b | >50 | >100 | 0.86 ± 0.01 |
| 4c | 2.49 ± 0.0022 | >50 | 0.026 ± 0.001 |
| 4d | >25 | >25 | >25 |
| 4e | 13.0 ± 0.1 | >25 | >50 |
| 4f | >25 | >50 | >50 |
| 4g | >100 | 0.69 ± 0.05 | 4.43 ± 0.08 |
| 4h | 0.0031 ± 0.0003 | 0.089 ± 0.008 | 0.0038 ± 0.0001 |
| 4i | >50 | >50 | >25 |
| 4j | >25 | >25 | >50 |
| 4k | 17.2 ± 2.6 | 4.22 ± 0.58 | >100 |
| 4l | >25 | >25 | >50 |
| 4m | 0.034 ± 0.004 | 0.33 ± 0.01 | 0.034 ± 0.002 |
| 4n | >50 | >50 | >50 |
| 4o | >100 | >100 | >50 |
| 4p | 9.02 ± 0.09 | 0.56 ± 0.01 | 0.067 ± 0.007 |
| 4q | >50 | >50 | >25 |
| 4r | >25 | >50 | >25 |
| 4s | 0.0046 ± 0.0001 | 0.044 ± 0.002 | 0.0014 ± 0.0003 |
| 4t | 0.069 ± 0.0028 | 2.64 ± 0.38 | 0.0047 ± 0.0009 |
| 5 | 0.0054 ± 0.0006 | 0.042 ± 0.008 | 0.0026 ± 0.0006 |
| CA-4 | 0.0028 ± 0.0004 | 0.0038 ± 0.0003 | 0.0009 ± 0.0003 |
The most significant enhancement of antiproliferative activity was observed in the pyridine-linked analogues where one phenyl ring accommodates the 2,4-dimethoxy substitutions. A symmetric analogue 3c with both phenyl ring adopting the 2,4-dimethoxy configuration gave low nanomolar IC50 values in all three cancer cell lines. However, when the dimethoxy substitution on one phenyl ring is in any other position than 2,4 (4e, 3,4-dimethoxy; 4l, 3,5-dimethoxy; 4n, 2,3-dimethoxy; 4o, 2,6-dimethoxy), there is significant loss of the antiproliferative activity with the only exception of 4m, having a 2,5-dimethoxyphenyl ring. Interestingly, 3,4,5-trimethoxy substitution (4g) on the second phenyl ring produced modest antiproliferative activity in A549 and HeLa cells, but 2,3,5-trimethoxy substitution (4q) abolished any such activity. When only monomethoxy substitution is introduced into the second ring while maintaining the 2,4-dimethoxy substitution on the first ring, 3-methoxy (4p), 4-methoxy (4h), but not 2-methoxy (4r) substitution affords nanomolar to micromolar cytotoxicity. In fact, 4h is one of the most potent analogue in the series. Finally, addition of a 3-hydroxy substitution to the second ring of 4h leads to yet the most potent analogue, 4s, that shows low nanomolar potency in all three cell lines. Thus, one phenyl ring of 4s is identical to that of CA-4 with 3-hydroxy-4-methoxy substitutions and the other phenyl ring contains 2,4-dimethoxy substitutions (Figure 1).
Antimicrotubule Effects in HeLa Cells
To determine the microtubule disrupting effects of the pyridine-linked analogues, we selected 4h as a representative compound in a cell-based phenotypic screening. HeLa and MDA-MB-231 cells were used to examine the effect of 4h on the reorganization of microtubules during mitosis. Cells were treated with either 4h or CA-4 for 24 h, and microtubules were visualized by indirect immunofluorescence techniques. An antibody for β-tubulin was used to visualize interphase and mitotic microtubule structures. Vehicle-treated cells exhibited a normal filamentous microtubule array, with microtubules extending from the central regions of the cell to the cell periphery (Figure 2A,D). Compound 4h caused dramatic reduction of the interphase microtubule network, as shown in Figure 2C in HeLa cells and in Figure 2F for MDA-MB-231 cells. As demonstrated in parts B and E of Figure 2 for the two cell lines, respectively, CA-4 induced very similar loss of the microtubule network.
Figure 2.

Effects of CA-4 and 4h on interphase microtubules. MDA-MB-231 and HeLa cells were treated with vehicle (A,D) or 1 μM compounds (B,C,E,F) as indicated for 24 h. Cells were then fixed and microtubules visualized by indirect immunofluorescence techniques. Normal interphase microtubules are visible in the control cells of HeLa and MDA-MB-231. The loss of interphase microtubules induced by CA-4 and 4h is shown in respective treatments as labeled.
Effect on Cell Cycle Arrest
To investigate the effect of the pyridine-linked CA-4 analogues on cell cycle arrest, we used flow cytometry to analyze the cell cycle distribution of HeLa cells following treatment with 4h and 4s at 1 μM. Untreated cells were used as a negative control, and cells treated with CA-4 were used as a positive control. As shown in Figure 3, the two most potent CA-4 analogues, 4h and 4s, were found to be as effective in arresting the cell cycle at G2/M phase as CA-4. With the untreated cells, the percentage of cells in the G0/G1 phase was at 61.6% with only 15.70% in the G2/M phase. After treatment with 4h or 4s, the percentage of cells in the G2/M phase increased to 73.4% and 70.6%, respectively. These results compare favorably to 44.6% in the G2/M phase for cells treated with CA-4.
Figure 3.

Flow cytometric analysis of cell cycle distributions of HeLa cells treated with pyridine-bridged CA-4 analogues 4h and 4s.
Similar results were obtained when another cancer cell line, MDA-MB-231, was used to test the effect of these compounds on cell cycle arrest. At 1 μM concentration, 4h and 4s again showed strong cell cycle inhibition. As indicated in Table 2, after treatment with 4h and 4s, 73.4% and 70.6% of HeLa cells were arrested in the G2/M phase, respectively, whereas treatment with CA-4 resulted in 44.6% G2/M phase cells. Similarly, the percentage of MDA-MB-231 cells in the G2/M phase increased from 15.5% (control) to 57.8% (CA-4 treated), 70.5 (4h treated), and 54.1 (4s treated).
Table 2. HeLa and MDA-MB-231 Cell Cycle Distribution for after Treatment with CA-4, 4h, and 4s.
| G0/G1 (%) | S (%) | G2/M (%) | |
|---|---|---|---|
| HeLa Cells | |||
| control (−) | 61.60 | 22.10 | 15.70 |
| treated with CA-4 | 37.00 | 16.70 | 44.60 |
| treated with 4h | 10.30 | 15.40 | 73.40 |
| treated with 4s | 12.10 | 17.10 | 70.60 |
| MDA-MB-231 Cells | |||
| control (−) | 58.60 | 25.70 | 15.50 |
| treated with CA-4 | 27.50 | 11.80 | 57.80 |
| treated with 4h | 13.10 | 16.40 | 70.50 |
| treated with 4s | 18.60 | 17.10 | 54.10 |
Inhibition of Tubulin Polymerization in Vitro
The inhibition of tubulin polymerization by the pyridine-linked CA-4 analogues 4h, 4s, and 4t were tested using bovine brain tubulin. As shown in Figure 4, incubation with either vehicle (DMSO), CA-4, 4h, 4s, or 4t resulted in various degrees of inhibition of tubulin polymerization, depending on the compound and the dose. At 1 μM, all pyridine-linked analogues (4h, 4s, and 4t) failed to inhibit tubulin polymerization. Compared to vehicle, 4h at this concentration appeared to be slightly stimulating tubulin polymerization. In contrast, CA-4 at 1 μM inhibited tubulin polymerization by 35%. When analogues concentrations were increased to 10 μM, 4h remained ineffective against tubulin polymerization, whereas 4s and 4t were seen to inhibit tubulin polymerization by 57% and 32%, respectively. In comparison, CA-4 at 10 μM nearly completely blocked tubulin polymerization (Figure 4). These data suggest that it is possible for the pyridine-linked analogues to have potent cytotoxicity but very little antitubulin polymerization activity, such as 4h.
Figure 4.
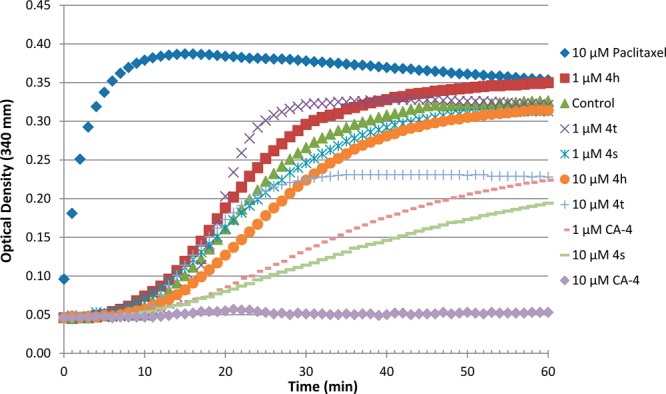
Effects of pyridine-linked CA-4 analogues on microtubule dynamics. Polymerization of tubulin at 37 °C in the presence of paclitaxel (10 μM), CA-4 (1 and 10 μM), 4h (1 and 10 μM), 4s (1 and 10 μM), and 4t (1 and 10 μM) and were monitored continuously by measuring the absorbance at 340 nm over 60 min. The reaction was initiated by the addition of tubulin to have a final concentration of 3.0 mg of tubulin per mL of incubation solution.
Antiangiogenesis Assay Using Chick Embryo Chorioallantoic Membrane (CAM)
The three most potent pyridine-linked combretastatin analogues, 4h, 4s, and 4t, were then tested for antiangiogenic and vasculature disrupting properties using the CAM assay. In this test, the vascular system of a fertilized chicken embryo is used as a model. Figure 5 demonstrates the effects of 4h, 4s, and 4t along with CA-4 on the development of embryonal blood vessels compared to a negative control (PBS) and a positive control (10 ng/plug basic fibroblast growth factor (bFGF) and 25 ng/plug vascular endothelial growth factor (VEGF)). The antiangiogenesis activities of CA-4 and the three pyridine-bridged analogues were determined by the suppression of angiogenic action of BV (bFGF + VEGF) when the compound was added to a collagen plug containing BV and placed on the chorioallantoic membrane of 10-day old embryos for 3 days. All compounds, 4h, 4s, 4t, and CA-4, led to inhibition of new vessel growth 4 days after treatment scored on a scale of 0–3 in a blinded manner. The negative control (vehicle) and the test compounds 4s and 4t scored significantly lower than the positive control (BV). The CAM assay results suggest that the pyridine-bridged analogues 4h, 4s, and 4t are potent inhibitors of angiogenesis, with 4s being more effective than CA-4 in suppressing new vessel growth.
Figure 5.
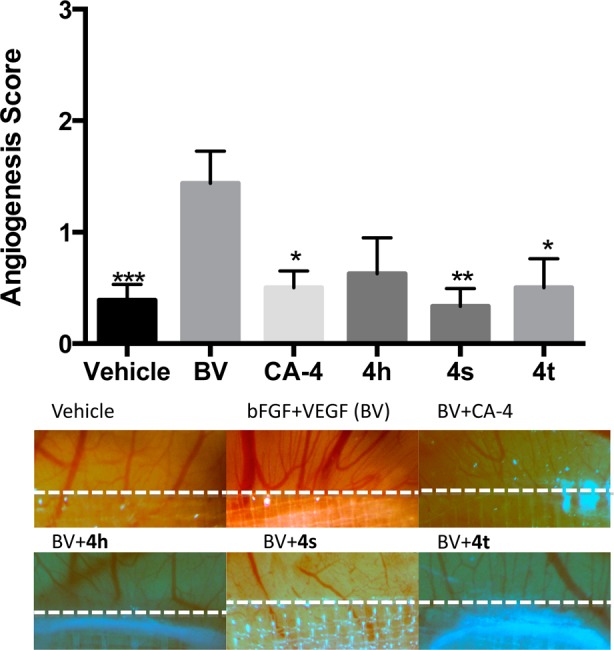
Effect of 4h, 4s, 4t, and CA-4 on angiogenesis in chick embryo. CA-4 and pyridine-bridged analogues 4h, 4s, and 4t potently inhibited the induction of angiogenesis by bFGF and VEGF (BV). Angiogenesis was scored by two scorers in a blinded fashion with similar results. *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Mouse Plasma Concentrations of 4h, 4s, and 4t vs CA-4
To evaluate the possible effect of the pyridine bridge on the analogues’s bioavailability, we measured the plasma concentration levels of 4h, 4s, and 4t in comparison with CA-4 in mice that were given a single dose of 5 mg/kg of the compound (Figure 6). After oral administration, blood samples were collected from the orbital sinus of the mice at 1, 3, 6, and 24 h. At 1 and 3 h, CA-4 reached plasma concentration of 2.3 and 2.1 ng/mL, respectively. At 6 and 24 h, plasma concentration of CA-4 was no longer detectable. Analogue 4h and 4t showed lower peak concentrations than CA-4. Analogue 4s was detected at the highest plasma concentration of 9.2 ng/mL at the first hour, with its level dropping rapidly at 6 h and going below detection limit at 24 h. These preliminary results indicate that the pyridine-bridged analogues varied widely in their bioavailability, as reflected in their respective plasma concentrations. It appears that 4s demonstrated the best bioavailability of the three analogues tested.
Figure 6.

Plasma concentrations of CA-4, 4h, 4s, and 4t in mice after the administration of a single oral dose of 5 mg/kg. Four blood samples were collected, each at 1, 3, 6, and 24 h, respectively, after oral intake.
Molecular Modeling
To elucidate the binding mode of pyridine-linked combretastatin analogues, we postulated that the analogues have the same binding site as colchicine and CA-4. We have visually analyzed 20 top scoring postures for each compound, and most of the top scoring poses of each compound changed very little. The Surflex docking scores were 7.35 for CA-4, 7.79 for 4h, 7.53 for 4s and 5.75 for 4t, where higher scores indicate greater binding affinity. The order of the docking scores appears to be consistent with the IC50 values of the compounds for growth inhibition of MDA-MB-231 human breast cancer cells. Here we examine the docking details of three most potent pyridine-linked analogues, 4h, 4s, and 4t (IC50 values are 2.75 nM for CA-4, 3.13 nM for 4h, 4.56 nM for 4s, and 68.7 nM for 4t in MDA-MB-231 cells) as compared to CA-4. The structures of these compounds are shown in Figure 1.
The binding modes of these compounds in the colchicine-binding site of tubulin are depicted in Figure 7. The binding postures of CA-4, 4h, and 4s (Figure 7A–C) in the binding pockets are similar. CA-4 formed a hydrogen bond with the backbone carbonyl oxygen of Val238, which was absent when 4h and 4s were bound. The close proximity of the functional groups of CA-4 (Figure 7A) to the polar amino acids Asn258, Lys352, Met259, and Cys241 suggests a likely stronger electrostatic interaction with the protein. In addition, the hydrophobic moiety of the CA-4 is well embedded in a pocket interacting with several hydrophobic residues making CA-4 bind tightly to tubulin. Despite the absence of one hydroxyl group in 4h, its binding pose is essentially superimposable to 4s, and they both bind in a fashion similar to CA-4. The close proximity of the functional groups of 4h (Figure 7B) to the side chain polar residues Asn258, Met259, Tyr202, Cys241, and Asn167 allows stronger interactions with the protein. The additional hydroxyl group in 4s (Figure 7C) seems to be stabilized by its interaction with the side chain of Lys352. Here again the hydrophobic moieties of these compounds are well shielded by several hydrophobic residues in the binding pocket. The binding posture of 4t, however, is slightly different from CA-4, 4h, and 4s. The three bulky methoxy groups on the para and meta positions of one of the phenyl rings of 4t present steric hindrance to occupying the exact colchicine binding site, causing it to bind to a better position in the nearby colchicine binding site. Even though CA-4 also has three methoxy groups on one phenyl ring, the cis conformation of CA-4 makes it more compact than 4t, thus it is less impeded by steric hindrance to binding into the colchicine binding site. The functional groups of 4t (Figure 7D) are in close proximity to the side chains of Asn349 and Asn258, and the hydrophobic moieties interact with the neighboring hydrophobic side chains. The overall interactions between 4t and tubulin are weaker than CA-4, 4h, and 4s.
Figure 7.

Molecular modeling of CA-4 and three pyridine-linked analogues in complex with tubulin. Shown is the proposed binding mode and interaction between tubulin and selected compounds, (A) CA-4, (B) 4h, (C) 4s, and (D) 4t. The compounds and important amino acids in the binding pockets are shown in stick model, whereas tubulin is depicted in the ribbon model.
The superposition of all four compounds in the binding pocket is depicted in Figure 8. It clearly shows the binding posture of CA-4, 4h, and 4s are similar, whereas the binding location of 4t is slightly different. The free energy of binding of these compounds with the receptor protein tubulin was calculated by employing the MMGB/SA approach. The binding energies are −77.46, −71.24, −66.72, and −49.54 kcal/mol for CA-4, 4h, 4s, and 4t, respectively. The binding energies correlate with the experimental IC50 values of these compounds in MDA-MB-231 human breast cancer cells. Both the docking scores and the free energy of binding show weaker binding of 4t, which is in agreement with their observed cytotoxic potencies (IC50: 4h < 4s < 4t) but not the experimentally measured antitubulin polymerization activities where 4h was not active in inhibiting tubulin polymerization and 4s and 4t were minimally active. This inconsistency suggests that cytotoxicity of the pyridine-linked CA-4 analogues could be separated from their antitubulin polymerization activities.
Figure 8.
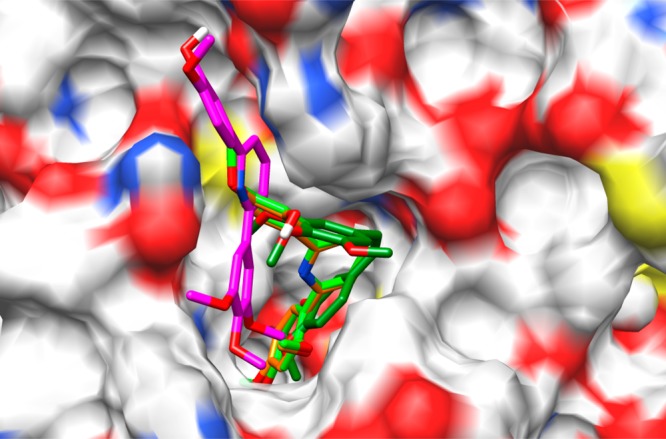
Superposition of CA-4, 4h, 4s, and 4t in the colchicine-binding pocket of tubulin in surface representation. The compounds are shown in stick model with carbon atoms in dark-green (CA-4), light-green (4h), orange (4s), or purple (4t).
Competitive Binding to Colchicine Binding Site of Tubulin
To determine if the CA-4 analogues indeed bind to the colchicine-binding site of tubulin, we next performed a binding experiment based on an HPLC-MS based method.26 In this method, colchicine was incubated with tubulin in the absence or presence of various concentrations of the test compounds, CA-4, 4h, 4s, and 4t in the incubation buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, pH 6.9) at 37 °C for 1 h. After incubation, the concentration of the displaced colchicine was measured by HPLC-MS/MS as previously described.26 The ability of the analogue to inhibit the binding of colchicine was expressed as a percentage of control binding in the absence of any competitor. As shown in Figure 9, CA-4 demonstrated the highest relative binding capacity, reaching 78% of colchicine binding. All three analogues showed similar, but slightly weaker binding affinity than CA-4. The binding results are largely consistent with the molecular modeling predictions, with minor differences in the order of binding scores where 4h and 4s scored higher than 4t.
Figure 9.
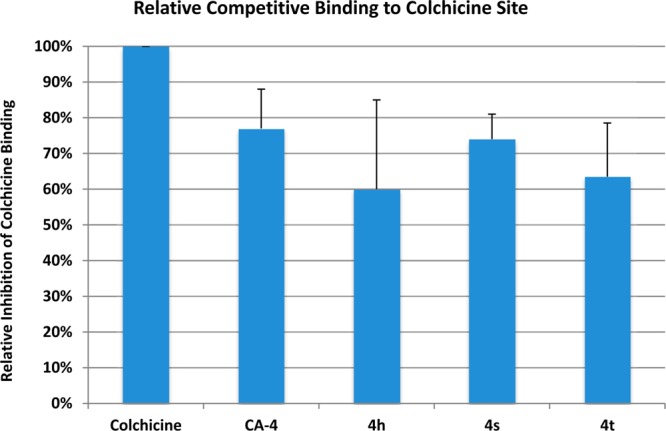
Competitive mass spectrometry-based binding assay for characterization of CA-4, 4h, 4s, and 4t binding to the colchicine binding site of tubulin.
Conclusion
We have designed and synthesized a series of combretastatin analogues with a pyridine linker that exceeds the standard two-atom linker length. Structurally, these compounds represented nonisomerizable analogues of CA-4 with three linker configurations: a 3-atom linker containing the pyridine nitrogen, a 3-atom linker without the nitrogen, and a 4-atom linker with the two phenyl rings para to each other on pyridine. As expected, the 4-atom linker configuration retained little cytotoxicities in the compounds (2e, 3e, 3g, and 4i). Activities of the analogues with 3-atom linkers varied widely depending on the phenyl ring substitutions, but the most potent compounds are exclusively with the nitrogen-containing linker configuration. The progressive improvement of activity from 3e to 3f to 4c demonstrates, at least qualitatively, that the 3-atom linker containing nitrogen represents the more favorable linker structure. More importantly, this novel 3-atom pyridine linker led us to discover at three analogues (4h, 4s, and 4t) that potently inhibited cell survival and growth, arrested cell cycle, and blocked angiogenesis and vasculature formation in vivo in ways comparable to CA-4. However, unlike CA-4, 4h was inactive in inhibiting tubulin polymerization and 4s and 4t were significantly weaker than CA-4 in blocking antitubulin polymerization. Molecular modeling analysis indicates all three analogues bind to the colchicine site of tubulin with 4h and 4s, showing greater binding affinity. Competitive binding assay confirmed that the pyridine-linked analogues bind to the colchicine site with affinities similar to CA-4.
Experimental Section
General Chemistry
All reagents and solvents were purchased from CombiPhos Catalyst, Combi-Block, AK Scientific, Sigma-Aldrich Chemical Co., Acros, and Pharmco-AAPER and were used as received. 1H and 13C NMR spectra were obtained on a Bruker-300 NMR spectrometer. Chemical shifts are reported as parts per million (ppm) relative to TMS. Mass spectral data were collected on an Agilent 2010 instrument, and HRMS spectra data were collected on a Thermo LTQ Orbitrap-XL mass spectrometer in positive ion modes. Unless specified otherwise, all tested compounds were confirmed to be >95% pure by HPLC and GC-MS. Melting points were obtained on a Haake Buchler melting point apparatus and are uncorrected.
General Procedure for Synthesis of Compounds 2 and 3
The mixture of the dibromopyridines 1a–c (11.8 g, 0.05 mol), the phenyl boronic acids (0.075 mol), sodium carbonate (15.9 g, 0.15 mol), and PdCl2(dppf) (0.40 g, 0.5 mmol) in toluene–ethanol (4:1, 100 mL) was stirred at 80 °C until completion of reaction (about 3–4 days). After cooling down to room temperature, the reaction mixture was filtered through Celite and the filtrate was concentrated. The residue crude was purified to afford products 2 and 3 (minor) by flash column chromatography on silica gel with hexane–ethyl acetate (9:1) as eluant.
2-Bromo-6-(3,4,5-trimethoxyphenyl)pyridine (2a)
Yield 43%, 7.0 g. Solid, mp 100–102 °C. 1H NMR (CDCl3, 300 Hz): 7.64–7.55 (2H, m), 7.39 (1H, d, J = 7.2 Hz), 7.21 (2H, s), 3.96 (6H, s), 3.90 (3H, s). 13C NMR (CDCl3, 75 Hz): 158.3, 153.5, 142.0, 139.6, 139.0, 133.3, 126.2, 118.9, 104.3, 61.0, 56.3. MS-EI: 323, 325 (M+). HRMS (ESI(+)) calcd for C14H15BrNO3 (M + H): 324.0235, 326.0215; found 324.0225, 326.0204.
2-Bromo-6-(3,4-dimethoxyphenyl)pyridine (2b)
Yield 43%, 6.3 g. Solid, mp 78–80 °C. 1H NMR (CDCl3, 300 Hz): 7.64–7.61 (2H, m), 7.56 (1H, d, J = 7.8 Hz), 7.51 (1H, dd, J = 2.1 and 8.4 Hz), 7.35 (1H, dd, J = 0.9 and 7.8 Hz), 6.93 (1H, d, J = 8.4 Hz), 3.99 (3H, s), 3.93 (3H, s). 13C NMR (CDCl3, 75 Hz): 158.2, 150.4, 149.2, 142.0, 138.9, 130.5, 125.6, 119.6, 118.3, 110.9, 109.9, 56.0, 55.9. MS-EI: 293, 295 (M+). HRMS (ESI(+)) calcd for C13H13BrNO2 (M + H): 294.0129, 296.0109; found 294.0123, 296.0104.
2-Bromo-6-(2,4-dimethoxyphenyl)pyridine (2c)
Yield 75%, 11.0 g. Solid, mp 48–50 °C. 1H NMR (CDCl3, 300 Hz): 7.86 (2H, m), 7.50 (1H, m), 7.30 (1H, d, J = 7.8 Hz), 6.60 (1H, dd, J = 2.1 and 8.4 Hz), 6.52 (1H, d, J = 2.1 Hz), 3.85 (6H, s). 13C NMR (CDCl3, 75 Hz): 161.8, 158.3, 156.7, 141.2, 138.0, 132.3, 127.0, 125.1, 123.2, 105.2, 98.7, 55.5, 55.4. MS-EI: 293, 295 (M+). HRMS (ESI(+)) calcd for C13H13BrNO2 (M + H): 294.0129, 296.0109; found 294.0126, 296.0100.
2-Bromo-6-(4-methoxyphenyl)pyridine (2d)
Yield: 62%, 8.1 g. Solid, mp 99–100 °C. 1H NMR (CDCl3, 300 Hz): 7.95 (2H, d, J = 9.0 Hz), 7.61 (1H, dd, J = 0.9 and 7.8 Hz), 7.55 (1H, m), 7.34 (1H, dd, J = 1.9 and 7.5 Hz), 6.98 (2H, d, J = 9.0 Hz), 3.87 (3H, s). 13C NMR (CDCl3, 75 Hz): 160.9, 158.2, 142.0, 138.9, 130.2, 128.3, 125.4, 118.1, 114.1, 55.3. MS-EI: 263, 265 (M+). HRMS (ESI(+)) calcd for C12H11BrNO (M + H): 264.0024, 266.0004; found 264.0011, 266.0000.
5-Bromo-2-(3,4,5-trimethoxyphenyl)pyridine (2e)
Yield 39%, 6.3 g. Solid, mp 49–50 °C. 1H NMR (CDCl3, 300 Hz): 8.70 (1H, d, J = 2.4 Hz), 7.85 (1H, dd, J = 2.4 and 8.7 Hz), 7.58 (1H, d, J = 8.4 Hz), 7.21 (2H, s), 3.96 (6H, s), 3.91 (3H, s). 13C NMR (CDCl3, 75 Hz): 155.4, 153.5, 150.5, 139.3, 139.2, 133.8, 121.4, 119.1, 103.9, 60.9, 56.2. MS-EI: 323, 325 (M+). HRMS (ESI(+)) calcd for C14H15BrNO3 (M + H): 324.0235, 326.0215; found 324.0224, 326.0203.
5-Bromo-2-(2,4-dimethoxyphenyl)pyridine (2f)
Yield 49%, 7.2 g. Solid, mp 90–92 °C. 1H NMR (CDCl3, 300 Hz): 8.70 (1H, m), 7.79–7.75 (3H, m), 6.61 (1H, dd, J = 2.4 and 8.7 Hz), 6.55 (1H, d, J = 2.4 Hz), 3.86 (3H, s), 3.85 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.6, 158.1, 154.2, 150.1, 138.2, 131.8, 125.8, 120.7, 118.0, 105.1, 98.8, 55.6, 55.5. MS-EI: 293, 295 (M+). HRMS (ESI(+)) calcd for C13H13BrNO2 (M + H): 294.0129, 296.0109; found 294.0121, 296.0099.
2,6-Bis(3,4,5-trimethoxyphenyl)pyridine (3a)
Yield 4%, 0.4 g. Solid, mp 175–176 °C. 1H NMR (CDCl3, 300 Hz): 7.81 (1H, m), 7.64 (2H, d, J = 7.8 Hz), 7.41 (4H, s), 3.98 (12H, s), 3.92 (6H, s). 13C NMR (CDCl3, 75 Hz): 156.3, 153.5, 139.1, 137.5, 135.0, 118.3, 104.2, 61.1, 56.2. MS-EI: 411 (M+). HRMS (ESI(+)) calcd for C23H26NO6 (M + H): 412.1760; found 412.1745.
2,6-Bis(3,4-dimethoxyphenyl)pyridine (3b)
Yield 14%, 2.4 g. Solid, mp 140–141 °C. 1H NMR (CDCl3, 300 Hz): 7.84 (2H, d, J = 1.8 Hz), 7.76 (1H, m), 7.65 (2H, dd, J = 1.8 and 8.1 Hz), 7.60 (2H, d, J = 7.8 Hz), 6.98 (2H, d, J = 8.1 Hz), 4.02 (6H, s), 3.96 (6H, s). 13C NMR (CDCl3, 75 Hz): 156.2, 150.0, 149.1, 137.4, 132.5, 119.4, 117.5, 111.0, 110.0, 56.0, 55.9. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1538.
2,5-Bis(3,4,5-trimethoxyphenyl)pyridine (3e)
Yield: 7%, 1.5 g. Solid, mp 176–177 °C. 1H NMR (CDCl3, 300 Hz): 8.88 (1H, dd, J = 0.9 and 2.4 Hz), 7.90 (1H, dd, J = 2.4 and 8.1 Hz), 7.76 (1H, dd, J = 0.9 and 8.4 Hz), 7.30 (2H, s), 6.81 (2H, s), 3.99 (6H, s), 3.95 (6H, s), 3.922 (3H, s), 3.915 (3H, s). 13C NMR (CDCl3, 75 Hz): 155.7, 153.7, 153.5, 147.8, 139.1, 138.2, 135.0, 134.5, 133.4, 120.0, 104.2, 103.9, 61.03, 61.00, 56.2. MS-EI: 411 (M+). HRMS (ESI(+)) calcd for C23H26NO6 (M + H): 412.1760; found 412.1744.
2,4-Bis(3,4,5-trimethoxyphenyl)pyridine (3f)
Yield 9%, 1.9 g. Solid, mp 116–118 °C. 1H NMR (CDCl3, 300 Hz): 8.70 (1H, d, J = 5.1 Hz), 7.79 (1H, s), 7.41 (1H, d, J = 5.1 Hz), 7.26 (2H, s), 6.86 (2H, s), 3.98 (6H, s), 3.96 (6H, s), 3.93 (3H, s), 3.92 (3H, s). 13C NMR (CDCl3, 75 Hz): 157.7, 153.8, 153.6, 149.9, 149.7, 139.2, 139.1, 135.0, 134.4, 120.4, 118.8, 104.5, 104.4, 61.0, 60.9, 56.39, 56.35. MS-EI: 411 (M+). HRMS (ESI(+)) calcd for C23H26NO6 (M + H): 412.1760; found 412.1746.
2,5-Bis(2,4-dimethoxyphenyl)pyridine (3g)
Yield 36%, 6.2 g. Solid, mp 124–125 °C. 1H NMR (CDCl3, 300 Hz): 8.79 (1H, m), 7.84–7.81 (3H, m), 7.30 (1H, d, J = 8.1 Hz), 6.65–6.57 (4H, m), 3.68 (9H, s), 3.83 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.1, 160.8, 158.1, 157.7, 153.5, 149.5, 136.3, 131.9, 131.3, 131.0, 123.8, 122.0, 120.0, 105.0, 104.8, 99.0, 98.8, 55.59, 55.53, 55.48, 55.45. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1538.
2,4-Bis(2,4-dimethoxyphenyl)pyridine (3h)
Yield 12%, 2.2 g. Solid, mp 95–97 °C. 1H NMR (CDCl3, 300 Hz): 8.65 (d, J = 5.1 Hz, 1H), 7.92 (dd, J = 1.5 and 0.6 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.36 (dd, J = 5.1 and 1.5 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 6.66–6.56 (m, 4H), 3.880 (s, 3H), 3.878 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H). 13C NMR (CDCl3, 75 Hz): 161.2, 161.1, 158.0, 157.8, 155.7, 148.8, 145.7, 132.0, 131.2, 125.1, 122.6, 121.8, 121.1, 104.9, 98.99, 98.87, 55.6, 55.5, 55.43, 55.41. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1537.
Procedure for Synthesis of Compounds 3d
The mixture of 2-bromo-6-(4-methoxyphenyl)pyridine (2d, 1.3 g, 5 mmol), 4-methoxyphenyl boronic acids (1.1 g, 7.5 mol), sodium carbonate (1.6 g, 0.015 mol), and PdCl2(dppf) (0.1 g, 0.1 mmol) in toluene–ethanol (4:1, 10 mL) was stirred overnight at 120 °C. After cooling down to room temperature, the reaction mixture was filtered through Celite followed by concentration of the filtrate. The residue crude was purified to afford product 3d by flash column chromatography on silica gel with hexane–ethyl acetate (9:1) as eluant.
2,6-Bis(4-methoxyphenyl)pyridine (3d)
Yield 89%,1.3 g. Solid, mp 156–158 °C. 1H NMR (CDCl3, 300 Hz): 8.10 (4H, d, J = 9.0 Hz), 7.74 (1H, m), 7.57(2H, dd, J = 0.6 and 7.5 Hz), 7.01 (4H, d, J = 9.0 Hz), 3.87 (6H, s). 13C NMR (CDCl3, 75 Hz): 160.4, 156.3, 137.3, 132.3, 128.2, 117.2, 114.0, 55.4. MS-EI: 291 (M+). HRMS (ESI(+)) calcd for C19H18NO2 (M + H): 292.1337; found 292.1327.
Procedure for Synthesis of Compounds 3c
The mixture of 2,6-dibromopyridines 1a (5.9 g, 0.025 mol), 2,4-dimethoxyphenyl boronic acids (13.7 g, 0.075 mol), sodium carbonate (15.9 g, 0.15 mol), and PdCl2(dppf) (0.20 g, 0.25 mmol) in toluene–ethanol (4:1, 50 mL) was stirred overnight at 120 °C. After cooling down to room temperature, the reaction mixture was filtered through Celite and the filtrate was concentrated. The residue crude was purified to afford product 3c by flash column chromatography on silica gel with hexane–ethyl acetate (9:1) as eluant.
2,6-Bis(2,4-dimethoxyphenyl)pyridine (3c)
Yield 92%, 8.0 g. Solid, mp 113–114 °C. 1H NMR (CDCl3, 300 Hz): 7.93 (2H, d, J = 8.4 Hz), 7.73–7.62 (3H, m), 6.62 (2H, dd, J = 2.1 and 8.4 Hz), 6.55 (2H, d, J = 2.1 Hz), 3.85 (12H, s). 13C NMR (CDCl3, 75 Hz): 161.1, 158.3, 155.0, 135.2, 132.3, 122.7, 122.2, 105.1, 98.8, 55.6, 55.5. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H) 352.1549; found 352.1537.
General Procedure for Synthesis of Compounds 4
The mixture of the monobromopyridines 2a–f (3 mmol), the phenyl boronic acids (4 mmol), sodium carbonate (0.5 g, 5 mmol), and PdCl2(dppf) (0.1 g, 0.1 mmol) in toluene–ethanol (4:1, 100 mL) was stirred overnight at 120 °C. After cooling down to room temperature, the reaction was quenched with saturated ammonium chloride solution; the organic layer was separated and extracted with ethyl acetate. The combined organic solution was dried over MgSO4, filtered, and concentrated. The residue crude was purified to afford products 4 by flash column chromatography on silica gel with hexane–ethyl acetate as eluant.
4-(6-(3,4,5-Trimethoxyphenyl)pyridin-2-yl)phenol (4a)
Eluant: hexane–ethyl acetate (7:3). Yield 41%, 0.42 g. Solid, mp 92–94 °C (d). 1H NMR (DMSO-d6, 300 Hz): 9.79 (1H, s), 8.05 (2H, d, J = 8.1 Hz), 7.85–7.62 (3H, m), 7.47 (2H, s), 6.90 (2H, d, J = 8.1 Hz), 3.90 (6H, s), 3.72 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 158.6, 155.5, 155.1, 153.1, 138.5, 137.9, 134.5, 129.6, 128.0, 117.7, 117.5, 115.5, 104.0, 60.1, 56.0. MS-EI: 337 (M+). HRMS (ESI(+)) calcd for C20H20NO4 (M + H): 338.1392; found 338.1390.
4-(6-(3,4-Dimethoxyphenyl)pyridin-2-yl)phenol (4b)
Eluant: hexane–ethyl acetate (7:3). Yield 40%, 0.37 g. Solid, mp 70–72 °C (d). 1H NMR (DMSO-d6, 300 Hz): 9.78 (1H, s), 8.05 (2H, d, J = 8.1 Hz), 7.85–7.71 (5H, m), 7.06 (1H, m), 6.90 (2H, d, J = 8.1 Hz), 3.88 (3H, s), 3.82 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 158.5, 155.5, 155.1, 149.8, 148.8, 137.8, 131.6, 129.7, 128.0, 119.2, 117.1, 116.9, 115.5, 111.7, 110.0, 55.5. MS-EI: 307 (M+). HRMS (ESI(+)) calcd for C19H18NO3 (M + H): 308.1285; found 308.1278.
2-(3,4-Dimethoxyphenyl)-6-(3,4,5-trimethoxyphenyl)pyridine (4c)
Eluant: hexane–ethyl acetate (8:2). Yield 80%, 0.91 g. Solid, mp 107–108 °C. 1H NMR (CDCl3, 300 Hz): 7.83 (1H, d, J = 1.8 Hz), 7.78 (1H, m), 7.67–7.59 (3H, m), 7.40 (2H, s), 6.98 (1H, d, J = 8.4 Hz), 4.01 (3H, s), 3.98 (6H, s), 3.96 (3H, s), 3.92 (3H, s). 13C NMR (CDCl3, 75 Hz): 156.2, 153.4, 150.0, 149.1, 139.0, 137.4, 135.2, 132.3, 119.4, 117.8, 111.0, 109.9, 104.1, 61.0, 56.1, 56.0, 55.8. MS-EI: 381 (M+). HRMS (ESI(+)) calcd for C22H24NO5 (M + H): 382.1654; found 382.1639.
2-(3,4-Dimethoxyphenyl)-6-(4-methoxyphenyl)pyridine (4d)
Eluant: hexane–ethyl acetate (8:2). Yield 97%, 0.94 g. Solid, mp 114–115 °C. 1H NMR (CDCl3, 300 Hz): 8.10 (2H, d, J = 9.0 Hz), 7.81 (1H, d, J = 1.8 Hz), 7.75 (1H, m), 7.66 (1H, dd, J = 1.8 and 8.4 Hz), 7.58 (2H, d, J = 7.8 Hz), 7.02 (2H, d, J = 9.0 Hz), 6.97 (1H, d, J = 8.4 Hz), 4.02 (3H, s), 3.95 (3H, s), 3.88 (3H, s). 13C NMR (CDCl3, 75 Hz): 160.4, 156.3, 149.9, 149.1, 137.3, 132.6, 132.2, 128.2, 119.5, 117.4, 114.0, 111.0, 110.0, 55.9, 55.3. MS-EI: 321 (M+). HRMS (ESI(+)) calcd for C20H20NO3 (M + H): 322.1443; found 322.1432.
2-(2,4-Dimethoxyphenyl)-6-(3,4-dimethoxyphenyl)pyridine (4e)
Eluant: hexane–ethyl acetate (8:2). Yield 95%, 1.0 g. Solid, mp 100–101 °C. 1H NMR (CDCl3, 300 Hz): 8.02 (1H, d, J = 8.7 Hz), 7.80–7.78 (2H, m), 7.72 (1H, m), 7.63 (1H, dd, J = 2.1 and 8.4 Hz), 7.58 (1H, dd, J = 1.2 and 7.5 Hz), 6.97 (1H, d, J = 8.4 Hz), 6.68 (1H, dd, J = 2.4 and 8.7 Hz), 6.59 (1H, d, J = 2.4 Hz), 4.01 (3H, s), 3.96 (3H, s), 3.89 (6H, s). 13C NMR (CDCl3, 75 Hz): 161.3, 158.4, 156.2, 155.0, 149.7, 149.1, 136.2, 132.9, 132.2, 122.5, 122.3, 119.4, 117.2, 110.9, 110.1, 105.1, 98.8, 55.9, 55.6, 55.4. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1537.
2-(4-Methoxyphenyl)-6-(3,4,5-trimethoxyphenyl)pyridine (4f)
Eluant: hexane–ethyl acetate (8:2). Yield 70%, 0.74 g. Solid, mp 112–114 °C. 1H NMR (CDCl3, 300 Hz): 8.09 (2H, d, J = 9.0 Hz), 7.77 (1H, m), 7.63–7.56 (2H, m), 7.37 (2H, s), 7.02 (2H, d, J = 9.0 Hz), 3.99 (6H, s), 3.92 (3H, s), 3.88 (3H, s). 13C NMR (CDCl3, 75 Hz): 160.5, 156.4, 153.4, 139.0, 137.4, 135.4, 132.0, 128.2, 117.8, 114.0, 104.2, 60.9, 56.2, 55.3. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1538.
2-(2,4-Dimethoxyphenyl)-6-(3,4,5-trimethoxyphenyl)pyridine (4g)
Eluant: hexane–ethyl acetate (8:2). Yield 79%, 0.9 g. Solid, mp 115–117 °C. 1H NMR (CDCl3, 300 Hz): 7.99 (1H, d, J = 8.4 Hz), 7.80 (1H, dd, J = 0.9 and 7.8 Hz), 7.72 (1H, m), 7.55 (1H, dd, J = 0.9 and 7.5 Hz), 7.33 (2H, s), 6.66 (1H, dd, J = 2.1 and 8.4 Hz), 6.58 (1H, d, J = 2.1 Hz), 3.97 (6H, s), 3.90 (3H, s), 3.88 (6H, s). 13C NMR (CDCl3, 75 Hz): 161.3, 158.4, 156.3, 155.1, 153.4, 138.8, 136.3, 135.7, 132.2, 123.0, 122.1, 117.6, 105.1, 104.2, 98.8, 60.9, 56.2, 55.6, 55.4. MS-EI: 381 (M+). HRMS (ESI(+)) calcd for C22H24NO5 (M + H): 382.1654; found 382.1644.
2-(2,4-Dimethoxyphenyl)-6-(4-methoxyphenyl)pyridine (4h)
Eluant: hexane–ethyl acetate (8:2). Yield 90%, 0.87 g. Solid, mp 97–98 °C. 1H NMR (CDCl3, 300 Hz): 8.04 (2H, d, J = 8.7 Hz), 8.00 (1H, d, J = 8.4 Hz), 7.75 (1H, dd, J = 1.2 and 8.1 Hz), 7.69 (1H, m), 7.54 (1H, dd, J = 1.5 and 7.5 Hz), 6.99 (1H, d, J = 9.0 Hz), 6.65 (1H, dd, J = 2.4 and 8.7 Hz), 6.56 (1H, d, J = 2.4 Hz), 3.864 (3H, s), 3.861 (3H, s), 3.856 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.3, 160.2, 158.4, 156.3, 155.1, 136.3, 132.5, 132.3, 128.2, 12.4, 122.3, 117.1, 116.1, 114.8, 114.0, 105.1, 98.9, 55.6, 55.4, 55.3. MS-EI: 321 (M+). HRMS (ESI(+)) calcd for C20H20NO3 (M + H): 322.1443; found 322.1434.
5-(3,4-Dimethoxyphenyl)-2-(3,4,5-trimethoxyphenyl)pyridine (4i)
Eluant: hexane–ethyl acetate (7:3). Yield 88%, 1.0 g. Solid, mp 131–132 °C. 1H NMR (CDCl3, 300 Hz): 8.88 (1H, d, J = 2.4 Hz), 7.91 (1H, dd, J = 2.4 and 8.1 Hz), 7.75 (1H, d, J = 8.4 Hz), 7.29 (2H, s), 7.20 (1H, dd, J = 2.4 and 8.1 Hz), 7.13 (1H, d, J = 2.1 Hz), 7.00 (1H, d, J = 8.4 Hz), 3.99 (6H, s), 3.98 (3H, s), 3.95 (3H, s), 3.92 (3H, s). 13C NMR (CDCl3, 75 Hz): 155.2, 153.5, 149.4, 149.2, 147.6, 139.0, 134.7, 134.6, 130.4, 120.0, 119.4, 111.7, 110.0, 103.9, 60.9, 56.2, 56.0. MS-EI: 381 (M+). HRMS (ESI(+)) calcd for C22H24NO5 (M + H): 382.1654; found 382.1638.
4-(6-(4-Methoxyphenyl)pyridine-2-yl)phenol (4j)
Eluant: hexane–ethyl acetate (7:3). Yield 73%, 0.60 g. Solid, mp 133–134 °C. 1H NMR (DMSO-d6, 300 Hz): 9.78 (1H, s), 8.15 (2H, d, J = 8.7 Hz), 8.05 (2H, d, J = 8.4 Hz), 7.81 (1H, m), 7.74–7.70 (2H, m), 7.05 (2H, d, J = 8.7 Hz), 6.90 (2H, d, J = 8.4 Hz), 3.82 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 160.2, 158.6, 155.6, 155.1, 138.0, 131.4, 129.8, 128.04, 127.95, 116.9, 115.6, 114.1, 55.3. MS-EI: 277 (M+). HRMS (ESI(+)) calcd for C18H16NO2 (M + H): 278.1181; found 278.1171.
4-(6-(2,4-Dimethoxyphenyl)pyridine-2-yl)phenol (4k)
Eluant: hexane–ethyl acetate (7:3). Yield 48%, 0.44 g. Solid, mp 112–114 °C (d). 1H NMR (DMSO-d6, 300 Hz): 9.74 (1H, s), 7.98 (2H, d, J = 8.7 Hz), 7.90 (1H, d, J = 9.0 Hz), 7.77–7.66 (3H, m), 6.88 (2H, d, J = 8.7 Hz), 6.70–6.68 (2H, m), 3.85 (3H, s), 3.83 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 161.1, 158.5, 158.2, 155.6, 154.3, 136.8, 131.7, 130.0, 128.0, 121.9, 121.3, 116.6, 115.5, 105.7, 98.7. MS-EI: 307 (M+). HRMS (ESI(+)) calcd for C19H18NO3 (M + H): 308.1285; found 308.1276.
2-(2,4-Dimethoxyphenyl)-6-(3,5-dimethoxyphenyl)pyridine (4l)
Eluant: hexane–ethyl acetate (9:1). Yield 63%, 0.66 g. Solid, mp 96–98 °C. 1H NMR (CDCl3, 300 Hz): 8.02 (1H, d, J = 8.7 Hz), 7.83 (1H, d, J = 7.5 Hz), 7.72 (1H, m), 7.57 (1H, d, J = 7.8 Hz), 7.26 (2H, s), 6.66 (1H, dd, J = 2.1 and 8.4 Hz), 6.57 (1H, d, J = 1.8 Hz), 6.53 (1H, m), 3.88 (12H, s). 13C NMR (CDCl3, 75 Hz): 161.3, 161.0, 158.4, 156.2, 155.0, 142.1, 136.3, 132.3, 123.3, 122.1, 117.9, 105.1, 100.8, 98.8, 55.6, 55.5. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1541.
2-(2,4-Dimethoxyphenyl)-6-(2,5-dimethoxyphenyl)pyridine (4m)
Eluant: hexane–ethyl acetate (7:3). Yield 71%, 0.75 g. Solid, mp 76–77 °C. 1H NMR (CDCl3, 300 Hz): 7.93 (1H, d, J = 8.4 Hz), 7.79–7.66 (3H, m), 7.53 (1H, d, J = 2.7 Hz), 6.96–6.87 (2H, m), 6.62 (1H, dd, J = 2.4 and 8.7 Hz), 6.56 (1H, d, J = 2.1 Hz), 3.86 (3H, s), 3.85 (3H, s), 3.83 (3H, s), 3.80 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.2, 158.2, 155.1, 154.9, 153.9, 151.5, 153.3, 132.2, 130.5, 122.8, 122.5, 122.4, 116.5, 114.9, 113.1, 105.0, 98.8, 56.5, 55.8, 55.6, 55.4. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1560.
2-(2,3-Dimethoxyphenyl)-6-(2,4-dimethoxyphenyl)pyridine (4n)
Eluant: hexane–ethyl acetate (9:1). Yield 90%, 0.95 g. Solid, mp 79–80 °C. 1H NMR (CDCl3, 300 Hz): 7.96 (1H, d, J = 8.4 Hz), 7.80 (1H, dd, J = 2.1 and 7.2 Hz), 7.76–7.68 (2H, m), 7.50 (1H, dd, J = 1.5 and 7.8 Hz), 7.16 (1H, t, J = 8.1 Hz), 6.96 (1H, dd, J = 1.5 and 7.8 Hz), 6.63 (1H, dd, J = 2.4 and 8.4 Hz), 6.56 (1H, d, J = 2.4 Hz), 3.91 (3H, s), 3.87 (3H, s), 3.85 (3H, s), 3.71 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.2, 158.2, 155.1, 155.0, 153.0, 147.2, 153.6, 134.9, 132.2, 124.1, 123.0, 122.9, 122.2, 112.3, 105.1, 98.8, 61.0, 55.9, 55.5, 55.4. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1557.
2-(2,4-Dimethoxyphenyl)-6-(2,6-dimethoxyphenyl)pyridine (4o)
Eluant: hexane–ethyl acetate (8:2). Yield 96%, 1.0 g. Solid, mp 155–156 °C. 1H NMR (CDCl3, 300 Hz): 7.79 (1H, d, J = 8.4 Hz), 7.74–7.67 (2H, m), 7.29 (1H, t, J = 7.8 Hz), 7.18 (1H, dd, J = 2.1 and 6.9 Hz), 6.64 (2H, d, J = 8.4 Hz), 6.58 (1H, dd, J = 2.4 and 8.4 Hz), 6.54 (1H, d, J = 2.1 Hz), 3.85 (3H, s), 3.83 (3H, s), 3.74 (6H, s). 13C NMR (CDCl3, 75 Hz): 160.9, 158.3, 158.0, 155.2, 153.8, 135.2, 132.6, 129.4, 123.6, 122.8, 122.7, 119.8, 105.0, 104.4, 98.6, 56.0, 55.5, 55.4. MS-EI: 351 (M+). HRMS (ESI(+)) calcd for C21H22NO4 (M + H): 352.1549; found 352.1536.
2-(2,4-Dimethoxyphenyl)-6-(3-methoxyphenyl)pyridine (4p)
Eluant: hexane–ethyl acetate (9:1). Yield 68%, 0.66 g. Solid, mp 75–76 °C. 1H NMR (CDCl3, 300 Hz): 8.03 (1H, d, J = 8.7 Hz), 7.82 (1H, dd, J = 0.6 and 7.8 Hz), 7.75–7.69 (2H, m), 7.65 (1H, d, J = 7.8 Hz), 7.59 (1H, dd, J = 0.6 and 7.5 Hz), 7.38 (1H, t, J = 7.8 Hz), 6.95 (1H, dd, J = 1.8 and 8.1 Hz), 6.66 (1H, dd, J = 2.4 and 8.7 Hz), 6.56 (1H, d, J = 2.1 Hz), 3.89 (3H, s), 3.87 (6H, s). 13C NMR (CDCl3, 75 Hz): 161.3, 159.9, 158.4, 156.3, 155.1, 141.4, 136.3, 132.3, 129.5, 123.2, 122.1, 119.4, 117.9, 114.3, 112.4, 105.1, 98.8, 55.6, 55.4, 55.3. MS-EI: 321 (M+). HRMS (ESI(+)) calcd for C20H20NO3 (M + H): 322.1443; found 322.1452.
2-(2,4-Dimethoxyphenyl)-6-(2,4,6-trimethoxyphenyl)pyridine (4q)
Eluant: hexane–ethyl acetate (7:3). Yield 57%, 0.65 g. Solid, mp 144–145 °C. 1H NMR (CDCl3, 300 Hz): 7.79 (1H, d, J = 8.7 Hz), 7.72–7.65 (2H, m), 7.16 (1H, dd, J = 2.1 and 6.6 Hz), 6.58 (1H, dd, J = 2.1 and 8.4 Hz), 6.53 (1H, d, J = 2.1 Hz), 6.22 (2H, s), 3.85 (6H, s), 3.83 (3H, s), 3.73 (6H, s). 13C NMR (CDCl3, 75 Hz): 161.1, 160.9, 159.0, 158.0, 155.1, 153.8, 135.1, 132.6, 123.9, 122.8, 122.5, 113.1, 105.0, 98.6, 91.1, 56.0, 55.5, 55.4. MS-EI: 381 (M+). HRMS (ESI(+)) calcd for C22H24NO5 (M + H): 382.1654; found 382.1662.
2-(2,4-Dimethoxyphenyl)-6-(2-methoxyphenyl)pyridine (4r)
Eluant: hexane–ethyl acetate (9:1). Yield 89%, 0.86 g. Solid, mp 95–96 °C. 1H NMR (CDCl3, 300 Hz): 7.94 (1H, d, J = 8.7 Hz), 7.92 (1H, dd, J = 1.8 and 7.8 Hz), 7.77–7.65 (3H, m), 7.36 (1H, m), 7.08 (1H, td, J = 1.2 and 7.5 Hz), 7.00 (1H, d, J = 8.1 Hz), 6.62 (1H, dd, J = 2.4 and 8.7 Hz), 6.56 (1H, d, J = 2.4 Hz), 3.87 (3H, s), 3.86 (3H, s), 3.85 (3H, s). 13C NMR (CDCl3, 75 Hz): 161.1, 158.2, 157.1, 155.2, 155.1, 135.2, 132.3, 131.5, 129.7, 129.6, 122.7, 122.6, 122.5, 121.0, 111.4, 105.1, 98.8, 55.64, 55.61, 55.4. MS-EI: 321 (M+). HRMS (ESI(+)) calcd for C20H20NO3 (M + H): 322.1443; found 322.1435.
5-(6-(2,4-Dimethoxyphenyl)pyridine-2-yl)-2-methoxyphenol (4s)
Eluant: hexane–ethyl acetate (7:3). Yield 83%, 0.84 g. Solid, mp 142–144 °C. 1H NMR (DMSO-d6, 300 Hz): 9.16 (1H, s), 7.89 (1H, d, J = 9.3 Hz), 7.78–7.66 (4H, m), 7.53 (1H, dd, J = 1.8 and 8.4 Hz), 7.01 (1H, d, J = 8.4 Hz), 6.70–6.69 (2H, m), 3.86 (3H, s), 3.83 (3H, s), 3.82 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 161.1, 158.2, 155.4, 154.3, 148.7, 146.7, 136.8, 132.0, 131.7, 122.2, 121.2, 117.7, 116.9, 113.7, 112.1, 105.7, 98.7, 55.72, 55.66, 55.4. MS-EI: 337 (M+). HRMS (ESI(+)) calcd for C20H20NO4 (M + H): 338.1392; found 338.1386.
2-Methoxy-5-(6-(3,4,5-trimethoxyphenyl)pyridine-2-yl)phenol (4t)
Eluant: hexane–ethyl acetate (7:3). Yield 66%, 0.72 g. Solid, mp 129–130 °C. 1H NMR (DMSO-d6, 300 Hz): 9.24 (1H, s), 7.87 (2H, d, J = 4.2 Hz), 7.76 (1H, m), 7.71 (1H, d, J = 2.1 Hz), 7.60 (1H, dd, J = 2.1 and 8.4 Hz), 7.48 (2H, s), 7.03 (1H, d, J = 8.4 Hz), 3.91 (6H, s), 3.83 (3H, s), 3.73 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 155.4, 155.1, 153.2, 148.9, 146.7, 138.6, 138.0, 134.5, 131.5, 118.1, 117.9, 113.7, 112.2, 104.1, 60.2, 56.1, 55.7. MS-EI: 367 (M+). HRMS (ESI(+)) calcd for C21H22NO5 (M + H): 368.1498; found 368.1490.
2-(2,4-Dimethoxyphenyl)-6-(3-hydroxy-4-methoxyphenyl)pyridin-1-ium Chloride (5)
To the solution of 4s (0.3 g, 0.88 mmol) in THF (5 mL) was added HCl solution in ether (0.44 mL, 0.88 mmol, 2 M). The resultant mixture was stirred at rt overnight. The precipitate was filtered and washed with dichloromethane to afford 5 (0.32 g) in yield of 97%. Solid, mp 140 °C (d). 1H NMR (DMSO-d6, 300 Hz): 8.28 (1H, t, J = 7.8 Hz), 7.96 (1H, d, J = 7.8 Hz), 7.89 (1H, d, J = 7.8 Hz), 7.75 (1H, d, J = 8.4 Hz), 7.45–7.42 (2H, m), 7.13 (1H, d, J = 8.7 Hz), 3.92 (3H, s), 3.861 (3H, s), 3.855 (3H, s). 13C NMR (DMSO-d6, 75 Hz): 163.1, 158.6, 152.8, 151.6, 150.6, 147.1, 143.1, 132.5, 126.4, 124.1, 121.0, 119.9, 115.7, 114.8, 112.5, 106.4, 99.0, 56.3, 56.0, 55.9. MS-ESI: 338 (M+-Cl). HRMS (ESI(+)) calcd for C20H20NO4 (M – Cl): 338.1392; found 338.1387.
Antiproliferative Assays
Human breast adenocarcinoma (MDA-MB-231), human nonsmall cell lung carcinoma (A549), and human cervical carcinoma (HeLa) cells were grown in DMEM medium supplemented with 115 units/mL of penicillin G, 115 μg/mL of streptomycin, and 10% fetal bovine serum (all from Life Technologies, Grand Island, NY). Cells were seeded in 96-well plates (5 × 103 cells/well) containing 50 μL of growth medium for 24 h. After medium removal, 100 μL of fresh medium containing individual analogue compounds at different concentrations was added to each well and incubated at 37 °C for 72 h. After 24 h of culture, the cells were supplemented with 50 μL of analogue compounds dissolved in DMSO (less than 0.25% in each preparation). After 72 h of incubation, 20 μL of resazurin was added for 2 h before recording fluorescence at 560 nm (excitation) and 590 nm (emission) using a Victor microtiter plate fluorimeter (PerkinElmer, USA). The IC50 was defined as the compound concentration required to inhibit cell proliferation by 50% in comparison with cells treated with the maximum amount of DMSO (0.25%) and considered as 100% viability.
Immunofluorescence
MDA-MB-231 and HeLa cells were grown on a Laboratory-Tek chamber slide (VWR International, Radnor, PA) and treated with vehicle (DMSO) or 1 μM CA-4 or 4h for 24 h to 4 days. At the completion of treatment, cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100 in PBS for 4 min. The cells were first incubated for 1 h in a solution of PBS containing 1% BSA and calf serum to block nonspecific antibody binding. The cells were then incubated with the mouse antitubulin antibody (1:200) (Life Technologies, Grand Island, NY), washed three times in PBS containing 1% BSA, and incubated for 2 h at room temperature with secondary goat antimouse antibody Alexa 488 labeled (1:200) for tubulin staining. The chamber slides were examined and photographed using a Nikon ES800 fluorescence microscope with a digital camera.
Tubulin Polymerization Assays
A tubulin polymerization kit (Cytoskeleton, Denver, CO) was used to evaluate effect of the pyridine-linked CA-4 analogues on tubulin assembly in vitro.24,25 It is based on the principal that light is scattered by microtubules to an extent that is proportional to the concentration of the microtubule polymer. Compounds that interact with tubulin will alter the polymerization of tubulin, and this can be detected using a spectrophotometer. The absorbance at 340 nm at 37 °C is monitored. The experimental procedure of the assay was performed as described in version 8.2 of the tubulin polymerization assay kit manual. Varying concentrations of compounds were preincubated with 10 μM bovine brain tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After the addition of 0.4 mM GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C. Tubulin assembly was monitored by measuring the optical density at 340 nm using a BioTek Synergy 4 multifunction microplate spectrophotometer.
Cytofluorimetric Analysis of Cell Cycle Distribution
Cells treated with tested compounds for 24 h were washed once in PBS and resuspended in 1 mL of 70% ice-cold ethanol and stored at –20 °C. Fixed cells were washed twice in PBS and then treated with 1 mL of 0.1 mg/mL of RNase A solution at 37 °C for 1 h. DNA was then stained with a PBS solution containing 0.1 mg/mL propidium iodide for 30 min at room temperature in the dark. Cell cycle analysis was determined with Accuri C6 (BD Biosciences, Mountain View, CA).
Chick Embryo Chorioallantoic Membrane (CAM) Assay
Fertilized embryos were obtained from Charles River Laboratories and incubated at 37.5 °C for 3 days, removed from their shell using a Dremel tool and placed into a covered weighboat for 10 further days of incubation. Solidified 30 μL onplants containing 2.1 mg/mL rat tail collagen (BD Biosciences, Bedford, MA) and 10 ng/plug bFGF and 30 ng/plug VEGF in the presence or absence of Ca-4 and 4s were placed on the embryo chorioallantoic membrane (CAM) over two pieces of nylon mesh approximately 0.5 cm2. Four collagen onplants were added per egg on at least three separate eggs. After 4 days of incubation, images were taken of each plug on surviving embryos using a mini-Vid camera (LW Scientific, Lawrenceville, GA) and quantified in a masked fashion on a scale from 0 to 3 with 0 representing no angiogenesis and 3 representing extreme angiogenesis. Data from one scorer (confirmed by a second scorer) are presented as the means ± standard errors of the mean. Statistical significances were determined by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test (GraphPad Prism, La Jolla, CA).
Molecular Modeling
All the docking studies were carried out using Sybyl-X 1.3 on a linux workstation. The initial coordinates for tubulin was taken from the crystal structure of tubulin in complex with colchicine (PDB: 1SA0.pdb). After removing all the ions and substructures, such as GDP and GTP present in the crystal structure, hydrogen atoms were added to the protein system using Amber99 force field. Four representative CA-4 analogues, including the parent compound CA-4, were selected for the docking studies. The 3D structures of these selected compounds were first built using Sybyl-X 1.3 sketch followed by energy minimization using the MMFF94 force field and Gasteiger–Marsili charges. We employed Powell’s method for optimizing the geometry with a distance dependent dielectric constant and a termination energy gradient of 0.05 kcal/mol. Structural resemblance of combretastatin to colchicine indicates that combretastatin binds to tubulin at the same site as colchicine and presumably has a similar mode of action. Hence we used colchicine binding site for docking our selected compounds to tubulin. We used the Surflex docking program to automatically dock all the selected compounds into the binding pocket of tubulin. Surflex is a fully automatic flexible molecular docking algorithm that combines Hammerhead’s empirical scoring function with a molecular similarity method to generate putative poses of ligand fragments. Docking process was guided by the protomol, which is a computational representation of the intended binding site. We used the position of the native ligand, colchicine, in the crystal structure to generate the protomol. In all the cases, we used the default screening protocol of Surflex that employs Surflex’s ligand preminimization and postdocking all-atom minimization. Standard parameters were used to estimate the binding affinity characterized by the Surflex-Dock scores. In each docking experiment, 20 poses were saved for each compound based on their total docking score values. A higher score represents stronger binding affinity. The optimal binding pose of the docked compounds was selected based on the Surflex scores and visual inspection of the docked complexes. The binding free energies of the complexes were calculated using the MM/GBSA method. OPLS-AA force field and GB/SA continuum solvent model were used to calculate the necessary energies of the complex.
Competitive Binding Assay Using LC-MS/MS
A Thermo TSQ Advantage instrument was used to determine the competitive binding affinity of the pyridine-bridged CA-4 analogues by measuring the concentration of colchicine displaced by the analogues. Briefly, colchicine (1.2 μM) was incubated with tubulin (1.3 mg/mL) in the incubation buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, pH 6.9) at 37 °C for 1 h. Varying concentrations (0.1–125 μM) of 4h, 4s, and 4t were used to compete with colchicine originally bound to tubulin. After incubation, the filtrate was obtained as previously described.26 The ability of the analogue to inhibit the binding of colchicine was expressed as a percentage of control binding in the absence of any competitor.
Pharmacokinetic Study in Mice
Female C57BL/6 mice were used for the pharmacokinetic study of selected CA-4 analogues. Mice were given oral gavage containing PBS and ethanol-dissolved CA-4, 4h, 4s, and 4t, at a single dose of 5 mg/kg/mouse. After oral administration, blood samples were collected from the orbital sinus of the mice at 1, 3, 6, and 24 h, with each group of mice subjected to only one sampling. All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by Xavier University Animal Care and Use Committee. The facilities and laboratory animals program of Xavier University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Acknowledgments
This work was supported by NIH-NIMHD through grant 8G12MD007595 and NIH-NIGMS grant 8P20GM103424, and in part by Louisiana Cancer Research Consortium. We thank Thomas Vu, Bria Carmichael, Breanna Jarrett, Eric Stewart, and Sydnie Turner for technical assistance in the CAM assays.
Glossary
Abbreviations Used
- CA-4
combretastatin-A4
- G0/G1
Gap0/Gap1 phase
- G2/M
Gap2/mitosis phase
- DMSO
dimethyl sulfoxide
- CAM
chorioallantoic membrane
- PBS
phosphate buffered saline
- bFGF
basic fibroblast growth factor
- VEGF
vascular endothelial growth factor
- BV
bFGF + VEGF
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Jordan A.; Hadfield J. A.; Lawrence N. J.; Mcgown A. T. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med. Res. Rev. 1998, 18, 259–296. [DOI] [PubMed] [Google Scholar]
- Giannakakou P.; Sackett D.; Fojo T. Tubulin/microtubules: still a promising target for new chemotherapeutic agents. J. Natl. Cancer Inst. 2000, 92, 182–183. [DOI] [PubMed] [Google Scholar]
- Checchi P. M.; Nettles J. H.; Zhou J.; Snyder J. P.; Joshi H. C. Microtubule-interacting drugs for cancer treatment. Trends Pharmacol. Sci. 2003, 24, 361–365. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Cragg G. M.; Herald D. L.; Schmidt J. M.; Lohavanijaya P. Isolation and structure of combretastatin. Can. J. Chem. 1982, 60, 1374–1376. [Google Scholar]
- Pettit G. R.; Temple J. R. C.; Narayanan V.; Varma R.; Simpson M. J.; Boyd M. R.; Rener G. A.; Bansal N. Antineoplastic agents 322. synthesis of combretastatin A-4 prodrugs. Anti-Cancer Drug Des. 1995, 10, 299–309. [PubMed] [Google Scholar]
- Patterson D.; Rustin G. Vascular damaging agents. Clin. Oncol. 2007, 19, 443–456. [DOI] [PubMed] [Google Scholar]
- Dowlati A.; Robertson K.; Cooney M.; Petros W. P.; Stratford M.; Jesberger J.; Rafie N.; Overmoyer B.; Makkar V.; Stambler B.; Taylor A.; Waas J.; Lewin J. S.; McCrae K. R.; Remick S. C. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin A-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002, 62, 3408–3416. [PubMed] [Google Scholar]
- Rustin G. J.; Galbraith S. M.; Anderson H.; Stratford M.; Folkes L. K.; Sena L.; Gumbrell L.; Price P. M. Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. J. Clin. Oncol. 2003, 21, 2815–2822. [DOI] [PubMed] [Google Scholar]
- Stevenson J. P.; Rosen M.; Sun W.; Gallagher M.; Haller D. G.; Vaughn D.; Giantonio B.; Zimmer R.; Petros W. P.; Stratford M.; Chaplin D.; Young S. L.; Schnall M.; O’Dwyer P. J. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J. Clin. Oncol. 2003, 21, 4428–4438. [DOI] [PubMed] [Google Scholar]
- Mooney C. J.; Nagaiah G.; Fu P.; Wasman J. K.; Cooney M. M.; Savvides P. S.; Bokar J. A.; Dowlati A.; Wang D.; Agarwala S. S.; Flick S. M.; Hartman P. H.; Ortiz J. D.; Lavertu P. N.; Remick S. C. A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid 2009, 19, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [DOI] [PubMed] [Google Scholar]
- Kong Y.; Grembecka J.; Edler M. C.; Hamel E.; Mooberry S. L.; Sabat M.; Rieger J.; Brown M. L. Structure-based discovery of a boronic acid bioisostere of combretastatin A-4. Chem. Biol. 2005, 12, 1007–1014. [DOI] [PubMed] [Google Scholar]
- Maya A. B. S.; Rey B. D.; Pelaez Lamamie De Clairac R.; Caballero E.; Barasoain I.; Andreu J. M.; Medarde M. Design, synthesis and cytotoxic activities of naphthyl analogues of combretastatin A-4. Bioorg. Med. Chem. Lett. 2000, 10, 2549–2551. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Minardi M. D.; Rosenberg H. J.; Hamel E.; Bibby M. C.; Martin S. W.; Jung M. K.; Pettit R. K.; Cuthbertson T. J.; Chapuis J. C. Antineoplastic agents. 509. Synthesis of fluorcombstatin phosphate and related 3-halostilbenes. J. Nat. Prod. 2005, 68, 1450–1458. [DOI] [PubMed] [Google Scholar]
- Gaukroger K.; Hadfield J. A.; Lawrence N. J.; Nolan S.; Mcgown A. T. Structural requirements for the interaction of combretastatins with tubulin: how important is the trimethoxy unit?. Org. Biomol. Chem. 2003, 1, 3033–3037. [DOI] [PubMed] [Google Scholar]
- Ohsumi K.; Hatanaka T.; Fujita K.; Nakagawa R.; Fukuda Y.; Nihei Y.; Suga Y.; Morinaga Y.; Akiyama Y.; Tsuji T. Syntheses and antitumor activity of cis-restricted combretastatins: 5-membered heterocyclic analogues. Bioorg. Med. Chem. Lett. 1998, 8, 3153–3158. [DOI] [PubMed] [Google Scholar]
- Bailly C.; Bal C.; Barbier P.; Combes S.; Finet J. P.; Hildebrand M. P.; Peyrot V. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. J. Med. Chem. 2003, 46, 5437–5444. [DOI] [PubMed] [Google Scholar]
- Pati H. N.; Wicks M.; Holt H. L. Jr.; LeBlanc R.; Weisbruch P.; Forrest L.; Lee M. Synthesis and biological evaluation of cis-combretastatin analogs and their novel 1,2,3-triazole derivatives. Heterocycl. Commun. 2005, 11, 117–120. [Google Scholar]
- Zaninetti R.; Cortese S. V.; Aprile S.; Massarotti A.; Canonico P. L.; Sorba G.; Grosa G.; Genazzani A. A.; Pirali T. A concise synthesis of pyrazole analogues of combretastatin a1 as potent anti-tubulin agents. ChemMedChem 2013, 8, 633–643. [DOI] [PubMed] [Google Scholar]
- Chen H.; Li Y.; Sheng C.; Lv Z.; Dong G.; Wang T.; Liu J.; Zhang M.; Li L.; Zhang T.; Geng D.; Niu C.; Li K. Design and synthesis of cyclopropylamide analogues of combretastatin-A4 as novel microtubule-stabilizing agents. J. Med. Chem. 2013, 56, 685–699. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Toki B.; Herald D. L.; Verdier-Pinard P.; Boyd M. R.; Hamel E.; Pettit R. K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41, 1688–1695. [DOI] [PubMed] [Google Scholar]
- Ducki S.; Forrest R.; Hadfield J. A.; Kendall A.; Lawrence N. J.; McGown A. T.; Rennison D. Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorg. Med. Chem. Lett. 1998, 8, 1051–1056. [DOI] [PubMed] [Google Scholar]
- Simoni D.; Grisolia G.; Giannini G.; Roberti M.; Rondanin R.; Piccagli L.; Baruchello R.; Rossi M.; Romagnoli R.; Invidiata F. P.; Grimaudo S.; Jung M. K.; Hamel E.; Gebbia N.; Crosta L.; Abbadessa V.; Di Cristina A.; Dusonchet L.; Meli M.; Tolomeo M. Heterocyclic and phenyl double-bond-locked combretastatin analogues possessing potent apoptosis-inducing activity in HL60 and in MDR cell lines. J. Med. Chem. 2005, 48, 723–736. [DOI] [PubMed] [Google Scholar]
- Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Salvador M. K.; Preti D.; Aghazadeh Tabrizi M.; Brancale A.; Fu X. H.; Li J.; Zhang S. Z.; Hamel E.; Bortolozzi R.; Porcù E.; Basso G.; Viola G. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl) thiazoles as novel anticancer agents. J. Med. Chem. 2013, 55, 5433–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. M.; Lu Y.; Ahn S.; Narayanan R.; Miller D. D.; Dalton J. T. Competitive mass spectrometry binding assay for characterization of three binding sites of tubulin. J. Mass Spectrom. 2010, 45, 1160–1166. [DOI] [PubMed] [Google Scholar]