

Abstract
The design and development of future molecular photonic/electronic systems pose the challenge of integrating functional molecular building blocks in a controlled, tunable, and reproducible manner. The modular nature and fidelity of the biosynthesis method provides a unique chemistry approach to one-pot synthesis of environmental factor-responsive chimeric proteins capable of energy conversion between the desired forms. In this work, facile tuning of dynamic thermal response in plasmonic nanoparticles was facilitated by genetic engineering of the structure, size, and self-assembly of the shell silk-elastin-like protein polymers (SELPs). Recombinant DNA techniques were implemented to synthesize a new family of SELPs, S4E8Gs, with amino acid repeats of [(GVGVP)4(GGGVP)(GVGVP)3(GAGAGS)4] and tunable molecular weight. The temperature-reversible conformational switching between the hydrophilic random coils and the hydrophobic β-turns in the elastin blocks were programmed to between 50 and 60 °C by site-specific glycine mutation, as confirmed by variable-temperature proton NMR and circular dichroism (CD) spectroscopy, to trigger the nanoparticle aggregation. The dynamic self-aggregation/disaggregation of the Au-SELPs nanoparticles was regulated in size and pattern by the β-sheet-forming, thermally stable silk blocks, as revealed by transmission electron microscopy (TEM) and dynamic light scattering (DLS). The thermally reversible, shell dimension dependent, interparticle plasmon coupling was investigated by both variable-temperature UV–vis spectroscopy and finite-difference time-domain (FDTD)-based simulations. Good agreement between the calculated and measured spectra sheds light on design and synthesis of responsive plasmonic nanostructures by independently tuning the refractive index and size of the SELPs through genetic engineering.
Introduction
The bottom-up approaches to molecular level devices and machines comprise a numerous set of synthesis and self-assembly methods for creating, accessing, and organizing a functional building block collection.1−4 Among the methods, genetically engineered synthesis offers a unique opportunity to exploit biologically inspired materials design and to introduce new, amino acid-based, supramolecular formulations for solving current pressing problems faced by nanodevice fabrication, e.g., highly efficient conversion of environmental stimuli into a desired form of energy, device reproducibility, and adaptive customization.2,3
Core–shell nanoparticles with self-regulating plasmonic,5−9 magnetic,10,11 luminescent,12,13 or catalytic functions14 have been intensively studied as service modules for controlled drug release and delivery,5,10,15,16 biochemical sensing and imaging,11,17,18 and biomedical optoelectronics.19 Gold nanoparticles have drawn extensive interest, as they often display intense color, arising from the collective oscillation of free conduction electrons induced by an interacting electromagnetic field, which is also known as surface plasmon resonance (SPR). The SPR of gold nanoparticles is size-dependent, which has been used to develop a variety of chemical and biosensors by modulating the gold nanoparticles self-assembly or aggregation status.20
In an aqueous or physiological environment, stimuli-responses of inorganic particle cores are often achieved by the dynamic covalent or reversible noncovalent interactions between polymeric shells.21 Conventional synthetic polymers, e.g., poly(N-isopropylacrylamide) (poly(NIPAM)), often adopt uniform conformations under certain conditions, lack structural tunability at the secondary structure level, and fall short in terms of controlling macroscopic material properties, such as mechanical and optical features. There is interest in developing rapid and versatile synthesis methods for tunable stimuli-responsive inorganic–polymer nanostructures with biocompatibility and biofunctionality. Recombinant modular protein polymers provide a novel chemistry approach to the design and one-pot synthesis of multifunctional polymeric materials. Bombyx mori silk-based materials, possessing optical transparency, remarkable mechanical strength, and stability,22−25 and mammalian elastin,26−30 possessing high resilience and stimuli-responsive features, have served as promising biomimetic targets.31
Recombinant silk-elastin-like protein polymers (SELPs) are a group of synthetic polypeptides consisting of tandemly repeated silk-inspired hexapeptide sequence GAGAGS and elastin-inspired pentapeptide sequence GVGVP (sometimes GXGVP where X represents a variable position and can be occupied by any amino acid residue other than proline).32,33 When heated, hydrogen bonds form between the valine residues in the main chain of the elastin-like pentapeptide repeats. The elastin-like blocks in SELPs therefore, undergo a transition from a hydrophilic random coil conformation to a putative hydrophobic β-spiral or β-turns, leading to aggregation of the SELPs.26−29 This thermally reversible conformational switching of SELP molecules, including chemically synthesized VPGVG pentapeptides and biosynthesized elastin-like polypeptides (ELPs), have been integrated into plasmonic gold nanostructures as shell materials to access a variety of optically responsive materials.7,34,35 Despite its temperature activation in plasmonic switching, elastin alone lacks kinetic control over phase separation as it tends to complete its phase separation and thermodynamically favors formation of a continuous elastin phase.36−38 Additional functional peptide module(s) is, therefore, necessary to gain control over secondary structures in the self-assembly and to facilitate tunable size and shape of the plasmonic aggregates.
Herein we report a new family of dynamic plasmonic gold nanoparticles with programmed thermoresponses, enabled by molecular-level engineering of the structure, size, and self-assembly behavior of the SELP shell. The β-structure-forming silk-like blocks were selected for enhancing the structural stability and for breaking the symmetry or pattern of the plasmonic assemblies. Characterization of the Au-SELP NPs is provided as an example to catalyze more extensive exploration of the hierarchical dynamic control for self-adaptive plasmonic nanodevices.
Experimental Section
Construction of Expression Plasmids
DNA sequence was designed to encode the silk-elastin like sequence: [(GVGVP)4(GGGVP)(GVGVP)3(GAGAGS)4]. The monomer DNA sequence was purchased as synthetic gene that was cloned into EcoRV site of the vector pUC57 from GenScript. The BanII restriction sites were designed to flank the monomer DNA sequence. The monomer DNA sequence was liberated by digesting the pUC57 derivatives with BanII, isolated by preparative gel electrophoresis, and purified using the QIAquick Gel Extraction Kit. The purified monomer DNA was then self-ligated with T4 DNA ligase for 8 h at 16 °C to yield DNA multimers. Next, the BanII and alkaline phosphatase-treated pET-19b3 plasmid was added to the reaction mixture and incubated for an additional 16 h.32 The ligation mixture was then used to transform E. coli DH5α. The resulting transformants contained recombinant plasmids that carried repetitive genes of varying lengths. These expression plasmids were identified by restriction digest analysis with the enzymes NcoI and BamHI and confirmed by dideoxy sequencing with both forward and reverse primers based on the T7 promoter and terminator sequences (Tufts Core Facility).
Protein Expression and Purification
The screened plasmids were transformed to E. coli BL21(DE3)Star, respectively. Protein expression was conducted via standard methods employing chemical induction (isopropyl β-d-thiogalactopyranoside, IPTG, final concentration 1 mM) of cultures of the appropriate expression host [1 L, supplemented with ampicillin (100 μg/mL)]. Cells were harvested 4 h after induction via centrifugation (7500 rpm, 20 min), the supernatant was decanted, and the cell pellets were resuspended in denaturing lysis buffer (200 mL, 100 mM NaH2PO4, 10 mM Tris·HCl, 8 M urea, pH 8.0) overnight. The resuspension was centrifuged at 8000 rpm for 30 min and 4 °C. The supernatant was loaded onto nickel chelating resin that had been equilibrated with denaturing lysis buffer. The column was washed and eluted with buffers at pH 6.3 and 4.5, respectively. The purified proteins were dialyzed (MWCO 3.5 kDa) against deionized water for 5 days and stored at −20 °C. The purity of the proteins was monitored via SDS-PAGE. The molecular weight of the purified protein was confirmed via matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Tufts Core Facility).
Synthesis of Au-S4E8G NPs
Ni-NTA gold nanoparticles were synthesized according to previous reports.39−42 For the conjugation of SELP onto AuNPs surface, to 20 nM of Au-NTA-Ni2+ NPs dispersed in Tris-HCl buffer (10 mM, pH = 8.0) was added S4E8G to get a protein/Au ratio of 50/1, followed by a further 2 h of incubation at room temperature for TEM, DLS, and UV–vis spectroscopy studies.
Instrumentation
The molecular weights of the purified proteins were confirmed via matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry on an Applied Biosystems Voyager De-Pro MALDI (Applied Biosystems, Foster City, CA) at Tufts Core Facility, Boston, MA. The 1H NMR was recorded on a Bruker (Billerica, MA) Advance 500 MHz spectrometer with 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal standard.The CD measurements were performed using an Aviv model 62DS spectrophotometer equipped with a Peltier temperature controller (Aviv Biomedical, Lakewood, NJ). The proteins were dissolved in ultrapure water (0.2 mg/mL), equilibrated overnight at 4 °C, and measured in 1 mm path length quartz cuvettes at 25 °C. The CD spectra were obtained from 260 to 190 nm at a resolution of 0.5 nm and at a scanning speed of 50 nm/min. Each spectrum was taken after equilibrating at the desired temperature for 30 min. TEM images were taken with either a Zeiss Libra 120 TEM (Zeiss, Oberkochen, Germany) equipped with an in-column energy filter operated at 100 kV or an FEI Tecnai F20 TEM (FEI, Hillsboro, OR) operated at 120 kV. The air-dried samples were prepared through deposition of a drop of Au-S4E8G NP solution onto the TEM grid (copper, 400 mesh). Variable temperature UV–vis extinction spectra of the Au-S4E8G NPs were characterized by monitoring the absorption of 400–800 nm on an Aviv 14DS UV–vis spectrophotometer equipped with a Peltier temperature controller (Aviv Biomedical, Lakewood, NJ). Each spectrum was taken after equilibrating at the desired temperature for 10 min.
Simulation of UV–Vis Extinction Spectra
Computer simulations were performed using the commercial finite-difference time domain (FDTD) package CST Microwave Studio 2011 with the frequency-domain solver. The simulated silk gold nanoparticles (i.e., Au–S4E8G-18mer NP) had an overall size of 38 nm consisting of an Au core diameter of 12 nm and a uniform silk protein coating of 13 nm. The permittivity ε of the silk was 2.37 (ε = n2, where n = 1.54, obtained from experimentally measured refractive index for silk and the dielectric dispersion function of gold in the optical regime was fitted using a Drude model (epsilon infinity: 9.5; plasma frequency: 1.37 × 1016 rad/s; collision frequency: 1.05 × 1014 s–1). The simulated frequency dependent extinction cross section was obtained for the thermally reversible aggregation-disaggregation behaviors.
Results and Discussion
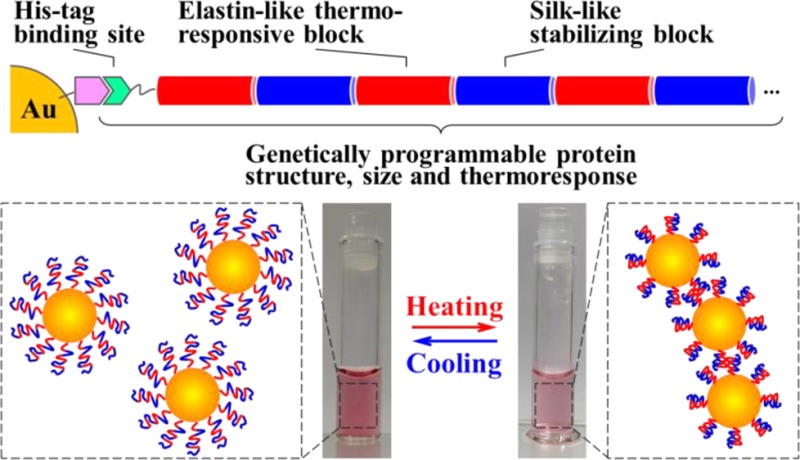
Figures 1 and 2 show the schematic diagram of the molecular construction of the plasmonic gold/SELP core/shell nanoparticles (Au-S4E8G NPs) investigated in this work and the targeted self-assembled structures. Construction of plasmids containing 3-, 8-, 13-, and 18-mer of [(GVGVP)4(GGGVP)(GVGVP)3(GAGAGS)4] (S4E8G) was achieved by a seamless cloning strategy, which avoided the introduction of extra amino acids residues at the junctions between monomers (please see details in the Experimental Section). The corresponding molecular weights of the proteins were 17.8, 42.1, 66.1, and 90.4 kDa, respectively. The identity and purity of the recombinant S4E8Gs were characterized by mass spectrometry (Figure S1) and SDS-PAGE (Figure 2c). Glycine was employed in the second amino acid position of the fifth elastin block in each monomer (underlined in Figure 2a). This neutral amino acid permitted the elastin block to display a conformational change between 50 and 60 °C determined from turbidity measurements.43 The desired temperature can be adjusted by introducing different amino acids.
Figure 1.

Schematic diagram of the NTA-Ni2+ functionalized Au NPs for surface recognition of the thermoresponsive S4E8G protein polymers, facilitated by an N-terminal polyhistidine tag.42 The responsive self-assembly characteristics of Au-S4E8Gs are based on the thermally reversible conformational switching in the elastin-like block between the hydrophilic random coil conformation and the hydrophobic β-spiral adopting repetitive type II β-turns.26−28,31
Figure 2.

(a) Molecular design and construction of the recombinant S4E8G protein polymers consisting of tandemly repeated elastin-like pentapeptide sequence GVGVP and silk-like hexapeptide sequence GAGAGS. The trigger temperatures for thermal response of the Au-S4E8G NPs were genetically engineered to between 50 and 60 °C (b). Identity (b) and purity of the recombinant S4E8Gs, containing four different numbers of silk-elastin-like repeats: 3-, 8-, 13-, and 18-mer, were characterized by mass spectrometry and (c) SDS-PAGE.
Conjugation and purification of the Au-S4E8Gs were facilitated by specific molecular recognition of a polyhistidine tag fused at the N-terminus of SELP on the nickel–chelate–nitrilotriacetate (NTA-Ni)-functionalized Au surface.39−41 The synthesis strategy following the method reported by Kitai et al.42 and is shown in Figure S2. The XPS characterization of the Au NPs at various steps of the reaction is summarized in Figure S3. By introducing Au NP into a specific site located at the end of the SELP chain, various functional amino acid structures can be modularly designed and genetically encoded into the family of plasmonic Au-S4E8G NPs.
The genetically programmed conformational transition of S4E8G (18-mer, 0.3 mg/mL in H2O) in response to temperature was verified by variable-temperature CD spectra (Figure 3a). Before heating, the S4E8Gs adopted a random coil-dominated conformation, as indicated by an intense negative band at 198 nm. As temperature increased, the 198 nm band decreased, and a positive ellipticity band evolved at 210 nm, characteristic of a type II β-turn conformation.44 The existence of an isodichroic point at ∼213 nm, during both heating and cooling, implies that the thermally induced conformational switching between random coil and type II β-turn occurred exclusively in the elastin-like domains.45 Hysteresis loops of ellipticities at 198 and 210 nm are plotted, respectively, in Figure 3b, showing that the elastin-based conformational switching in S4E8G was rapidly reversible (within minutes).
Figure 3.

Molecular origin of the reversible thermal response of the recombinant S4E8G protein polymers. A heating-induced conformational transition in the elastin-like domains of S4E8G-18-mer, from random coil to type II β-turn, was revealed by variable-temperature CD spectroscopy. (a) S4E8G-18-mers (0.3 mg/mL in H2O) at low temperatures were found to adopt random coil-dominated structures, containing small amounts of β-conformations. The inset shows reference CD spectra of regenerated B. mori silk (0.05 wt % in H2O), adopting 100% random coil at pH 10 and containing about 5% of β-sheets41 at pH 6. The evolution of a positive ellipticity band with a maximum at 210 nm during heating confirms the growth of type II β-turn.44 Note that an isodichroic point occurred at ∼213 nm, implying two-state solution behavior,45 and that the β-structures in the silk-like domains were not affected by temperature. (b) Thermally induced conversion between random coil and β-turn in the elastin-like domains was demonstrated to be rapidly reversible in the hysteresis loops.
As shown in the inset of Figure 3a, regenerated B. mori silk (0.05 wt % in H2O) adopted a completely unordered conformation at pH = 10. When the pH was adjusted to 6, about 5% β-sheet structure was induced in the regenerated silk,46 as indicated by the appearance of a dip at 218 nm. Therefore, Figure 3a reveals that the silk-like domains of S4E8G formed β-conformations in aqueous solution, and the small amount of β-structure remained unaffected by temperature. The possibility that Au-S4E8G aggregation was triggered merely by the silk blocks can be excluded.
The heat-induced hydrogen-bond formation of β-turns in the elastin-like domains of S4E8G (18-mer, 1 mg/mL in D2O) was also investigated by variable-temperature 1H NMR spectroscopy (Figure 4a). During heating, the amide proton NMR resonances of the elastin-originated glycine and valine residues showed distinct downfield shifts, while the silk-originated glycine residues moved slightly downfield and the alanine residues remained almost unchanged (as calibrated with an external standard HMDS). It was previously reported that an elastin-like pentapeptide, poly(AVGVP), forming intra- and interchain hydrogen bonds above its transition temperature, showed downfield shifts of amide protons.47,48 Regenerated silk, when forming β-sheet hydrogen bonding, showed upfield shifts of amide protons.49,50 Therefore, the observed chemical shifts of S4E8G were consistent with the CD results that the conformation adopted by the silk-like domains in aqueous solution was unaffected by the thermal switch of conformation occurred in the elastin-like domains.
Figure 4.
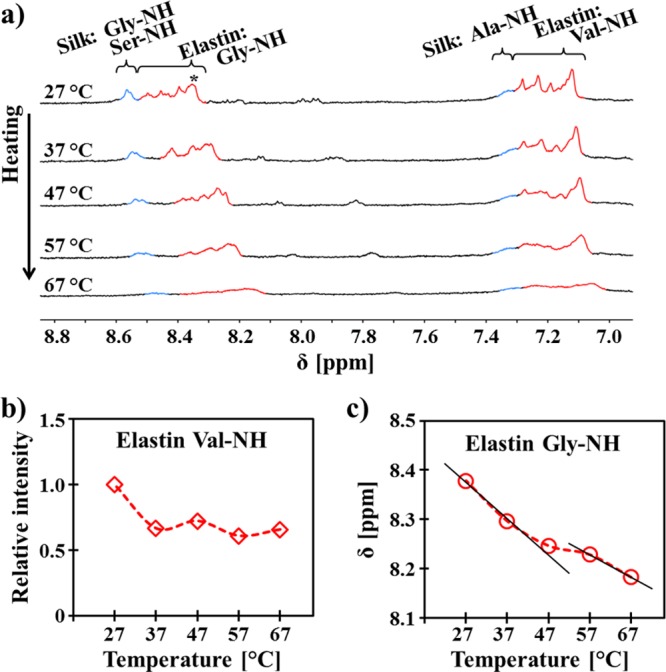
Heating-induced conformational switching in S4E8G-18-mer was further studied by variable-temperature 1H NMR spectroscopy. (a, b) Heating-induced changes (broadening, intensity decrease, and downfield shifts) in the amide proton NMR signals verified the decrease of chain mobility in S4E8G-18-mers (1 mg/mL in D2O, containing the external standard HMDS) at higher temperatures.47,48 (c) Thermal activation of the proton exchange, for example, between the elastin glycine NH and H2O, determined from the slope of the temperature dependence of NH chemical shifts, was found weaker above the transition temperature, suggesting that the formation of hydrogen bonds in the elastin-like domains.
Meanwhile, when the temperature was increased, the intensities of all amide proton signals decreased relative to the CH proton signal at the same temperature. The temperature dependence of the normalized relative intensity of the elastin Gly-NH was plotted, as an example, in Figure 4b, suggesting a decrease of chain mobility and aggregation of S4E8G. Since no sedimentation took place under the experimental conditions, the aqueous solution underwent microphase separation. The thermal activation of the chemical exchange, for example, of the elastin glycine NH protons (Figure 4c), was determined from the slope of the temperature dependence of chemical shifts. The weaker thermal activation of the proton exchange between NH and H2O above the transition temperature was strong evidence of the formation of hydrogen bonds in the elastin-like domains.
The complex index of refraction and all other optical properties of a protein polymer in solution is determined by its sequence and structure. The size of the polymer phase with desired optical properties, on the other hand, was programmed by the molecular weight. The family of the S4E8Gs in this contribution, genetically synthesized with various numbers of silk-elastin-like repeats, was found to exhibit similar temperature-induced conformational behaviors. The modular design approach, therefore, uniquely enables independent tuning of the properties and dimensions of the polymer phases.
Chelation of Ni2+-NTA with oligo-histidines was selected as a universal chemical strategy to integrate the SELPs into the plasmonic gold nanostructures. TEM micrographs revealed the existence of S4E8G shells on the surfaces of 12 nm diameter Au cores (inset of Figure 5a). The Au-S4E8G NPs (1.2 nM) were well dispersed in 10 mM PBS (pH 7.4) at 25 °C and aggregated and formed clusters at 60 °C (Figure 5b).
Figure 5.
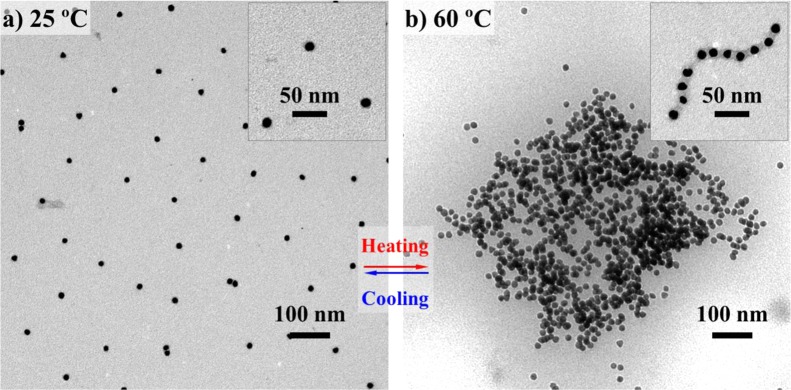
Thermally reversible assembly of plasmonic NPs programmed by genetically engineered S4E8G proteins. (a) TEM images of Au-S4E8G-18mer NPs (1.2 nM in 10 mM PBS at pH 7.4) in individual form at 25 °C and (b) in aggregated form when heated to 60 °C. The square-like shape of (b) the Au-S4E8G NP aggregate was an indication for the presence of β-strands/β-sheets in the silk blocks of S4E8G, as also confirmed by CD. The thermal stability of the silk blocks, as a resistance mechanism opposing the temperature-induced coacervation, functionalized to regulate the aggregate size.
While the elastin blocks serve as commander to trigger the temperature responses of Au-S4E8G NPs, the β-sheet structure-forming silk blocks play a significant role in the dimension control over the coacervation transition by stabilizing the packing structure of the aggregates. The presence of β-strands/β-sheets in the S4E8Gs and the thermal stability over the range of 10–80 °C (as revealed by CD in Figure 3a) functioned as a counter-aggregation mechanism, preventing the overall phase separation36−38 and mediating the self-regulation of aggregate size. Morphological studies by TEM provided additional evidence for the existence of and the contribution from the silk domains. In contrast to the round-shaped ELP aggregates dictated by surface tension,51,52 the Au-S4E8G NPs aggregated into square-like shapes (Figure 5b), similar to the β-pleated-sheet crystal dominated silk-like proteins.53,54 When the local concentration was low, the Au-S4E8G NPs arranged into linear chains after heating, in a similar way as the β-sheet-rich fibrils from silk.55−58 The inset of Figure 5b shows a representative example of a chain of Au-S4E8G-18mer composed of 11 NPs with a 8–13 nm SELP spacing between the gold cores.
The thermally reversible aggregation–disaggregation behavior of the Au-S4E8G NPs were investigated in situ in solution (0.1 nM in 10 mM PBS, pH 7.4) using DLS (Figure 6). An increase in the apparent size of the Au-S4E8G NPs with increasing Mw of the shell proteins was observed. The average diameters of the Au-S4E8G-3-mer, -8-mer, -13-mer, and -18mer NPs, in the individual form at 25 °C, were ∼24, ∼29, ∼32, and ∼38 nm, respectively (the blue bars in Figure 6). After activating the thermal trigger, longer protein chains with more elastin-like repeats led to a larger population of NPs of bigger sizes. A 2-fold increase in size was identified for Au-S4E8G-3-mer aggregates (∼60 nm), a 3-fold increase for both Au-S4E8G-8-mer (∼80 nm) and -13-mer (∼100 nm), and a 4-fold increase for Au-S4E8G-18-mer (∼160 nm) (the red bars in Figure 6). When cooled back to 25 °C, all of the four Au-S4E8Gs disaggregated and reversed to ∼25, ∼28, ∼32, and ∼41 nm, respectively. The increase in the aggregation size was well correlated with the increase in the polymer chain length from 3-mer to 18-mer. Since the full S4E8G macromer sequence was repeated, the experimental evidence collected was insufficient to conclude the role played by individual silk-like or elastin-like blocks.
Figure 6.

DLS studies on the reversible, temperature dependent aggregation behavior of the four Au-S4E8G NPs (all at 0.1 nM) in PBS buffer (10 mM, pH 7.4).
The on/off thermal switching between the individual and collective plasmon resonances in the Au-S4E8Gs was monitored by the variable-temperature UV–vis extinction spectra (Figure 7a). For example, the solution of Au-S4E8G-18-mer NPs as prepared at room temperature was ruby red with peak extinction measured at approximately 522 nm. As the temperature rose to 60 °C, the color turned to pink-red reflected by a broad absorption peak centered at 545 nm. Modeling of the extinction spectra (Figure 7b) was performed with a finite-difference time-domain (FDTD) method using size parameters determined by DLS and optical parameters estimated from regenerated silk.59 The simulated extinction profile of the well-dispersed Au-S4E8G-18-mers peaked at ∼521 nm, and the peak red-shifted to ∼545 nm for the plasmonic aggregates, which match well with the experimental results. As predicted, the Au-S4E8Gs with thinner protein shell, such as Au-S4E8G-3-mer, shows a reduced red-shift (to ∼534 nm) during the heating-induced aggregation. The experimentally characterized absorption peaks were consistently broader than the calculated values, partially due to a nonuniform distribution of the spacing between Au-S4E8G NPs in the cluster, but the absorption peak shift between the two operation modes was well captured in the simulated spectra.
Figure 7.

On/off thermal switching between the individual and collective plasmon resonances in the Au-S4E8G NPs. (a) Variable-temperature UV–vis extinction spectra of the Au-S4E8G-18-mer NPs (1.0 nM in 10 mM PBS at pH 7.4) revealed a peak extinction at ∼522 nm for the ruby red solution as-prepared at room temperature. When heated to 60 °C, the absorption peak red-shifted to ∼545 nm, and the color turned to pink-red (see the inset). (b) Simulated extinction spectra were in good agreement with the experimental results. (c) Thermal cycles of the Au-S4E8G-3-mer and -18-mer NPs between 25 and 60 °C were monitored by UV–vis spectroscopy showing good reversibility.
Thermal cycles of the Au-S4E8G-3-mer and -18-mer NPs between 25 and 60 °C were monitored by UV–vis spectroscopy (Figure 7c). The extinction peak shifts were completely reversible within two or three cycles without agitation. After three cycles, pipetting was required to help resuspend the dense nanoparticles in the aqueous phase. When stored at 4 °C, the suspension of Au-S4E8G NPs, either with or without thermal history, was stable. Precipitation was minimal for at least a month.
Conclusion
In summary, we have demonstrated a novel strategy for design and synthesis of responsive plasmonic nanostructures by genetic engineering of SELPs. Thermal responses of gold nanoparticles in aqueous solution were programmed at the molecular level by tuning the ratio of silk to elastin and site-specific mutation of the shell SELPs. Reversible self-assembly of Au-SELPs was studied at the secondary structure level; variable-temperature 1H NMR and CD spectra revealed, in the elastin-like blocks, the formation of hydrogen bonds upon heating and the thermally reversible conformational transition between random coil and type II β-turns; in the silk-like blocks, the presence of thermally stable β-sheet conformations; TEM and DLS investigated the resulting dynamic self-aggregation/disaggregation of the Au-SELP NPs and confirmed the stabilizing effects of the silk-like blocks on the regulation of aggregate size and pattern. The thermally reversible interparticle plasmon coupling was observed by variable-temperature UV–vis spectroscopy. The refractive index of the SELP was determined by the amino acid sequence and polymer chain conformation/structures, and the size/dimension of the SELP phase was tuned independently by the molecular weight. By showing good agreement between the experiments and the FDTD-based simulations, this contribution represents a promising approach to a new class of self-adaptive gold/protein core/shell nanoparticles.
Acknowledgments
This work was supported by the NIH (EB002520, AR005593), the AFOSR (FA9550-10-1-0172), and DARPA (N68001-12-C-4194). The authors thank Professor Barbara Brodsky for valuable comments concerning the interpretation of the CD results. The authors also thank Ke Shang, Yajing Zhu, and Yucheng He for their help with protein purification.
Supporting Information Available
MALDI-TOF mass spectra of the recombinant SELPs; surface functionalization of gold nanoparticles for molecular recognition of the histidine-tagged S4E8Gs; X-ray photoelectron spectroscopy (XPS) characterizations of the Au-NTA-Ni2+ and Au-NTA-Ni2+-SELP NPs deposited on silicon wafers. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Y.L., X.X., and M.W. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fan J. A.; Wu C.; Bao K.; Bao J.; Bardhan R.; Halas N. J.; Manoharan V. N.; Nordlander P.; Shvets G.; Capasso F. Self-assembled plasmonic nanoparticle clusters. Science 2010, 328, 1135–1138. [DOI] [PubMed] [Google Scholar]
- Chen Y. S.; Hong M. Y.; Huang G. S. A protein transistor made of an antibody molecule and two gold nanoparticles. Nat. Nanotechnol. 2012, 7, 197–203. [DOI] [PubMed] [Google Scholar]
- Gao B.; Arya G.; Tao A. R. Self-orienting nanocubes for the assembly of plasmonic nanojunctions. Nat. Nanotechnol. 2012, 7, 433–437. [DOI] [PubMed] [Google Scholar]
- Shafiei F.; Wu C.; Wu Y.; Khanikaev A. B.; Putzke P.; Singh A.; Li X.; Shvets S. Plasmonic nano-protractor based on polarization spectro-tomography. Nat. Photonics 2013, 7, 367–372. [Google Scholar]
- Cobley C. M.; Chen J. Y.; Cho E. C.; Wang L. V.; Xia Y. N. Gold nanostructures: a class of multifunctional materials for biomedical applications. Chem. Soc. Rev. 2011, 40, 44–56. [DOI] [PubMed] [Google Scholar]
- Hu M.; Chen J. Y.; Li Z. Y.; Au L.; Hartland G. V.; Li X. D.; Marquez M.; Xia Y. N. Gold nanostructures: engineering their plasmonic properties for biomedical applications. Chem. Soc. Rev. 2006, 35, 1084–1094. [DOI] [PubMed] [Google Scholar]
- Lemieux V.; Hans P.; Adams H. M.; van Hest J. C. M. Elastin-based stimuli-responsive gold nanoparticles. Chem. Commun. 2010, 46, 3071–3073. [DOI] [PubMed] [Google Scholar]
- Liu L.; Michelsen K.; Kitova E. N.; Schnier P. D.; Klassen J. S. Energetics of lipid binding in a hydrophobic protein cavity. J. Am. Chem. Soc. 2012, 134, 3054–3060. [DOI] [PubMed] [Google Scholar]
- Xia Y. N.; Xiong Y. J.; Lim B.; Skrabalak S. E. Shape-controlled synthesis of metal nanocrystals: Simple chemistry meets complex physics?. Angew. Chem., Int. Ed. 2009, 48, 60–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham S.; Chang P. E. J.; Rumpel H.; Kee I. H. C.; Ng R. T. H.; Chow P. K. H.; Tan C. K.; Ramanujan R. V. Thermoresponsive core-shell magnetic nanoparticles for combined modalities of cancer therapy. Nanotechnology 2009, 20, 305101. [DOI] [PubMed] [Google Scholar]
- Li Q.; Zhang L. F.; Bai L. J.; Zhang Z. B.; Zhu J.; Zhou N. C.; Cheng Z. P.; Zhu X. L. Multistimuli-responsive hybrid nanoparticles with magnetic core and thermoresponsive fluorescence-labeled shell via surface-initiated RAFT polymerization. Soft Matter 2011, 7, 6958–6966. [Google Scholar]
- Lao U. L.; Mulchandani A.; Chen W. Simple conjugation and purification of quantum dot-antibody complexes using a thermally responsive elastin-protein L scaffold as immunofluorescent agents. J. Am. Chem. Soc. 2006, 128, 14756–14757. [DOI] [PubMed] [Google Scholar]
- Son J. S.; Hyeon T. Large-scale soft colloidal template synthesis of 1.4 nm-thick CdSe nanosheets. Angew. Chem., Int. Ed. 2009, 48, 6861–6864. [DOI] [PubMed] [Google Scholar]
- Li G. L.; Tai C. A.; Neoh K. G.; Kang E. T.; Yang X. L. Hybrid nanorattles of metal core and stimuli-responsive polymer shell for confined catalytic reactions. Polym. Chem. 2011, 2, 1368–1374. [Google Scholar]
- Savla R.; Taratula O.; Garbuzenko O.; Minko T. Tumor targeted quantum dot-mucin 1 aptamer-doxorubicin conjugate for imaging and treatment of cancer. J. Controlled Release 2011, 153, 16–22. [DOI] [PubMed] [Google Scholar]
- Sershen S. R.; Westcott S. L.; Halas N. J.; West J. L. Temperature-sensitive polymer-nanoshell composites for photothermally modulated drug delivery. J. Biomed. Mater. Res. 2000, 51, 293–298. [DOI] [PubMed] [Google Scholar]
- Lee J. E.; Chung K.; Jang Y. H.; Jang Y. J.; Kochuveedu S. T.; Li D.; Kim D. H. Bimetallic multifunctional core@shell plasmonic nanoparticles for localized surface plasmon resonance based sensing and electrocatalysis. Anal. Chem. 2012, 84, 6494–6500. [DOI] [PubMed] [Google Scholar]
- Anker J. N.; Hall W. P.; Lyandres O.; Shah N. C.; Zhao J.; Van Duyne R. P. Biosensing with plasmonic nanosensors. Nat. Mater. 2008, 7, 442–453. [DOI] [PubMed] [Google Scholar]
- Qu D.; Liu F.; Yu J. F.; Xie W. L.; Xu Q.; Li X. D.; Huang Y. D. Plasmonic core-shell gold nanoparticle enhanced optical absorption in photovoltaic devices. Appl. Phys. Lett. 2011, 98, 113119. [Google Scholar]
- Giljohann D. A.; Seferos D. S.; Daniel W. L.; Massich M. D.; Patel P. C.; Mirkin C. A. Gold nanoparticles for biology and medicine. Angew. Chem., Int. Ed. 2010, 49, 3280–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtecki R. J.; Meador M. A.; Rowan S. J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [DOI] [PubMed] [Google Scholar]
- Jiang C. Y.; Wang X. Y.; Gunawidjaja R.; Lin Y. H.; Gupta M. K.; Kaplan D. L.; Naik R. R.; Tsukruk V. V. Mechanical properties of robust ultrathin silk fibroin films. Adv. Funct. Mater. 2007, 17, 2229–2237. [Google Scholar]
- Mandal B. B.; Grinberg A.; Gil E. S.; Panilaitis B.; Kaplan D. L. High-strength silk protein scaffolds for bone repair. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7699–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenetto F. G.; Kaplan D. L. New opportunities for an ancient material. Science 2010, 329, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood D. N.; Preda R. C.; Yucel T.; Wang X. Q.; Lovett M. L.; Kaplan D. L. Materials fabrication from Bombyx mori silk fibroin. Nat. Protoc. 2011, 6, 1612–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Z.; Scaffidi J. P.; Asher S. A. Circular dichroism and UV-resonance Raman investigation of the temperature dependence of the conformations of linear and cyclic elastin. Biopolymers 2009, 91, 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook W. J.; Einspahr H.; Trapane T. L.; Urry D. W.; Bugg C. E. Crystal-structure and conformation of the cyclic trimer of a repeat pentapeptide of elastin, cyclo-(L-valyl-L-prolylglycyl-L-valylglycyl)3. J. Am. Chem. Soc. 1980, 102, 5502–5505. [Google Scholar]
- Urry D. W.; Cunningh W. D.; Ohnishi T. Studies on conformation and interactions of elastin - proton magnetic-resonance of repeating pentapeptide. Biochemistry 1974, 13, 609–616. [DOI] [PubMed] [Google Scholar]
- Urry D. W.; Mitchell L. W.; Ohnishi T. Studies on conformation and interactions of elastin secondary structure of synthetic repeat hexapeptides. Biochim. Biophys. Acta 1975, 393, 296–306. [DOI] [PubMed] [Google Scholar]
- Urry D. W.; Ohnishi T.; Long M. M.; Mitchell L. W. Studies on conformation and interactions of elastin - nuclear magnetic-resonance of polyhexapeptide. J. Pept. Protein Res. 1975, 7, 367–378. [DOI] [PubMed] [Google Scholar]
- Kushner A. M.; Guan Z. B. Modular design in natural and biomimetic soft materials. Angew. Chem., Int. Ed. 2011, 50, 9026–9057. [DOI] [PubMed] [Google Scholar]
- Xia X. X.; Xu Q. B.; Hu X.; Qin G. K.; Kaplan D. L. Tunable self-assembly of genetically engineered silk-elastin-like protein polymers. Biomacromolecules 2011, 12, 3844–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiram M.; Quiroz F. G.; Callahan D. J.; Chilkoti A. A highly parallel method for synthesizing DNA repeats enables the discovery of ‘smart’ protein polymers. Nat. Mater. 2011, 10, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. C.; Koria P.; Parker S. M.; Selby L.; Megeed Z.; Rege K. Optically responsive gold nanorod-polypeptide assemblies. Langmuir 2008, 24, 14139–14144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N.; Chilkoti A. Interfacial phase transition of an environmentally responsive elastin biopolymer adsorbed on functionalized gold nanoparticles studied by colloidal surface plasmon resonance. J. Am. Chem. Soc. 2001, 123, 8197–8202. [DOI] [PubMed] [Google Scholar]
- Yeo G. C.; Keeley F. W.; Weiss A. S. Coacervation of tropoelastin. Adv. Colloid Interface Sci. 2011, 167, 94–103. [DOI] [PubMed] [Google Scholar]
- Vrhovski B.; Jensen S.; Weiss A. S. Coacervation characteristics of recombinant human tropoelastin. Eur. J. Biochem. 1997, 250, 92–98. [DOI] [PubMed] [Google Scholar]
- Muiznieks L. D.; Keeley F. W. Proline periodicity modulates the self-assembly properties of elastin-like polypeptides. Biophys. J. 2011, 100, 199–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinas R. P.; Hu M. H.; Qian L. P.; Lymar E. S.; Hainfeld J. F. Gold nanoparticle size controlled by polymeric Au(I) thiolate precursor size. J. Am. Chem. Soc. 2008, 130, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. H. K.; Jiang S. Y.; Pun S. H. Localized cell uptake of his-tagged polyplexes immobilized on NTA self-assembled monolayers. Langmuir 2010, 26, 15445–15452. [DOI] [PubMed] [Google Scholar]
- Wegner G. J.; Lee N. J.; Marriott G.; Corn R. M. Fabrication of histidine-tagged fusion protein arrays for surface plasmon resonance imaging studies of protein-protein and protein-DNA interactions. Anal. Chem. 2003, 75, 4740–4746. [DOI] [PubMed] [Google Scholar]
- Kitai T.; Watanabe Y.; Toyoshima Y. Y.; Kobayashi T.; Murayama T.; Sakaue H.; Suzuki H.; Takahagi T. Simple method of synthesizing nickel-nitrilotriacetic acid gold nanoparticles with a narrow size distribution for protein labeling. Jpn. J. Appl. Phys. 2011, 50, 095002. [Google Scholar]
- Urry D. W.; Urry K. D.; Szaflarski W.; Nowicki M. Elastic-contractile model proteins: physical chemistry, protein function and drug design and delivery. Adv. Drug Delivery Rev. 2010, 62, 1404–1455. [DOI] [PubMed] [Google Scholar]
- Kim W.; Thevenot J.; Ibarboure E.; Lecommandoux S.; Chaikof E. L. Self-assembly of thermally responsive amphiphilic diblock copolypeptides into spherical micellar nanoparticles. Angew. Chem., Int. Ed. 2010, 49, 4257–4260. [DOI] [PubMed] [Google Scholar]
- Rosenblatt M. M.; Wang J. Y.; Suslick K. S. De novo designed cyclic-peptide heme complexes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13140–13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G. Y.; Zhou P.; Shao Z. Z.; Xie X.; Chen X.; Wang H. H.; Chunyu L. J.; Yu T. Y. The natural silk spinning process - A nucleation-dependent aggregation mechanism?. Eur. J. Biochem. 2001, 268, 6600–6606. [DOI] [PubMed] [Google Scholar]
- Dybal J.; Schmidt P.; Kurkova D.; Kriz J.; Rodriguez-Cabello J. C.; Alonso M. Temperature induced conformational transitions of elastin-like polypentapeptides studied by Raman and NMR spectroscopy. Spectroscopy 2002, 16, 251–255. [Google Scholar]
- Kurkova D.; Kriz J.; Schmidt P.; Dybal J.; Rodriguez-Cabello J. C.; Alonso M. Structure and dynamics of two elastin-like polypentapeptides studied by NMR spectroscopy. Biomacromolecules 2003, 4, 589–601. [DOI] [PubMed] [Google Scholar]
- Zainuddin; Le T. T.; Park Y.; Chirila T. V.; Halley P. J.; Whittaker A. K. The behavior of aged regenerated Bombyx mori silk fibroin solutions studied by H-1 NMR and rheology. Biomaterials 2008, 29, 4268–4274. [DOI] [PubMed] [Google Scholar]
- Gotoh Y.; Niimi S.; Hayakawa T.; Miyashita T. Preparation of lactose-silk fibroin conjugates and their application as a scaffold for hepatocyte attachment. Biomaterials 2004, 25, 1131–1140. [DOI] [PubMed] [Google Scholar]
- Osborne J. L.; Farmer R.; Woodhouse K. A. Self-assembled elastin-like polypeptide particles. Acta Biomater. 2008, 4, 49–57. [DOI] [PubMed] [Google Scholar]
- Koria P.; Yagi H.; Kitagawa Y.; Megeed Z.; Nahmias Y.; Sheridan R.; Yarmush M. L. Self-assembling elastin-like peptides growth factor chimeric nanoparticles for the treatment of chronic wounds. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valluzzi R.; Gido S. P.; Zhang W. P.; Muller W. S.; Kaplan D. L. Trigonal crystal structure of Bombyx mori silk incorporating a threefold helical chain conformation found at the air-water interface. Macromolecules 1996, 29, 8606–8614. [Google Scholar]
- He S. J.; Valluzzi R.; Gido S. P. Silk I structure in Bombyx mori silk foams. Int. J. Biol. Macromol. 1999, 24, 187–195. [DOI] [PubMed] [Google Scholar]
- Anderson J. P.; Cappello J.; Martin D. C. Morphology and primary crystal-structure of a silk-like protein polymer synthesized by genetically-engineered Escherichia-coli bacteria. Biopolymers 1994, 34, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Anderson J. P. Morphology and crystal structure of a recombinant silk-like molecule, SLP4. Biopolymers 1998, 45, 307–321. [Google Scholar]
- Zhang J. M.; Hao R. W.; Huang L.; Yao J. R.; Chen X.; Shao Z. Z. Self-assembly of a peptide amphiphile based on hydrolysed Bombyx mori silk fibroin. Chem. Commun. 2011, 47, 10296–10298. [DOI] [PubMed] [Google Scholar]
- Gong Z. G.; Huang L.; Yang Y. H.; Chen X.; Shao Z. Z. Two distinct beta-sheet fibrils from silk protein. Chem. Commun. 2009, 7506–7508. [DOI] [PubMed] [Google Scholar]
- Lawrence B. D.; Cronin-Golomb M.; Georgakoudi I.; Kaplan D. L.; Omenetto F. G. Bioactive silk protein biomaterial systems for optical devices. Biomacromolecules 2008, 9, 1214–1220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.