

Abstract
We recently developed “photo-unclick chemistry”, a novel chemical tool involving the cleavage of aminoacrylate by singlet oxygen, and demonstrated its application to visible light-activatable prodrugs. In this study, we prepared an advanced multifunctional prodrug, Pc-(L-CA4)2, composed of the fluorescent photosensitizer phthalocyanine (Pc), an SO-labile aminoacrylate linker (L), and a cytotoxic drug combretastatin A-4 (CA4). Pc-(L-CA4)2 had reduced dark toxicity compared with CA4. However, once illuminated, it showed improved toxicity similar to CA4 and displayed bystander effects in vitro. We monitored the time-dependent distribution of Pc-(L-CA4)2 using optical imaging with live mice. We also effectively ablated tumors by the illumination with far-red light to the mice, presumably through the combined effects of photodynamic therapy (PDT) and released chemotherapy drug, without any sign of acute systemic toxicity.
Introduction
The use of visible and near-infrared (NIR) light, penetrable to deep tissue, is an attractive method of spatiotemporally controlling drug release from various drug delivery forms, such as prodrugs, liposomes, polymers, and other nano- and macrodelivery systems.1 However, because of its lower energy, it is difficult to directly cleave a chemical bond using such light. Thus, novel mechanisms using lower energy light to trigger the release of biologically active compounds have been a major topic of interest.2 The photodynamic process and the unique chemistry of singlet oxygen (SO) were adopted to mediate lower energy light release of drugs.3−11 SO is formed during the photodynamic process and reacts with electron-rich olefins to form unstable dioxetanes. These dioxetanes decompose to release drugs.
Aminoacrylate was an ideal SO-cleavable linker.8 We named the cleavage of aminoacrylate by SO “photo-unclick chemistry”, and demonstrated the release of the anticancer drug combretastatin A-4 (CA4) from its prodrug using this method. We prepared CMP-L-CA4, a CA4 prodrug that can be activated by far-red light (690 nm), by combining a SO-labile aminoacrylate linker, a core-modified porphyrin (CMP) photosensitizer, and CA4.10 The prodrug released CA4 and enhanced cytotoxicity upon illumination. The released CA4 damaged cancer cells, demonstrating our system’s ability to generate bystander effects in vitro. We found that the antitumor effects of CMP-L-CA4 were superior to the effects of its pseudo-prodrug CMP-NCL-CA4 (NCL = noncleavable linker). The pseudo-prodrug cannot release CA4 even after illumination in vivo.
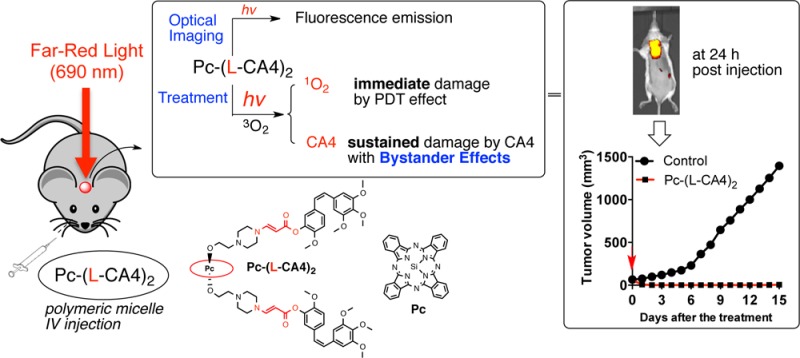
Optical imaging is a valuable tool for tracking fluorescent or luminescent molecules. The ability to optically image the light-activatable prodrug is useful for determining an illumination time when the prodrug is at its maximum concentration at the target site. It can be utilized in real time at a low cost. If the prodrug accumulates in the target sites, we can use optical imaging to detect the target areas, as well as to treat the disease. We expected that we could optically image our photounclickable prodrug in vivo using a fluorescent photosensitizer. Thus, we could track the prodrugs and then treat the tumor with the combined effects of photodynamic therapy (PDT) and local chemotherapy (Figure 1a). The released drug could damage the cancer cells that survived the initial PDT damage through bystander effects (Figure 1b).
Figure 1.
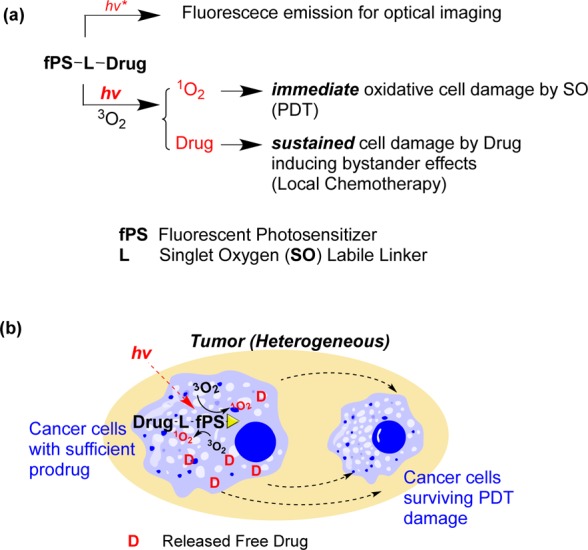
(a) Multifunctional prodrug for optical imaging and combined treatment with PDT and local chemotherapy. (b) Bystander effects from the released drugs can kill cancer cells that survive PDT damage. [The lifetime of SO is short (submicrosecond scale). Thus, direct cell damage by SO occurs only during illumination. The light dose used for imaging will be much lower than the light dose used for treatment. Thus, we do not expect any significant damage during optical imaging.]
We generated Pc-(L-CA4)2, an advanced multifunctioning CA4 prodrug, for both fluorescence optical imaging and combination therapy with PDT and released CA4. We chose phthalocyanine (Pc) because Pc is a fluorescent photosensitizer that can generate both fluorescence and singlet oxygen.12−14 Although fluorescence emission and SO generation are competing processes, Pc has uniquely balanced yields of both functionality (i.e., Si-Pc: Φ1O2 = 0.22 and Φf = 0.4) with a high molar extinction coefficient (ε).15,16 Its brightness (BT) is greater than that of CMP (e.g., ε = 150,000 M–1 cm–1 at 675 nm, BT = 6000 M–1 cm–1 for Pc vs ε = 5000 at 690 nm, BT = 50 M–1 cm–1 for CMP).17,18 We prepared Pc-(NCL-CA4)2 as its pseudo-prodrug. This pseudo-prodrug is similar to Pc-(L-CA4)2 in structure, but cannot release CA4 upon illumination. It will mimic the PDT effects of Pc-(L-CA4)2, but cannot induce damage from released CA4. We evaluated the cytotoxic effects of these two prodrugs with and without illumination, the inhibition of tubulin polymerization, the in vitro bystander effects, tumor localization using optical imaging, and the antitumor effects.
Results and Discussion
Synthesis
We developed a synthetic scheme using high-yield reactions, such as esterification, nucleophilic substitution, and the yne-amine reaction, to make the process easily adaptable to other alcohol-containing drugs (Scheme 1). CA4 was esterified at room temperature to yield compound 1. Alkylation of CA4 gave compound 2. A nucleophilic substitution reaction of silicon phthalocyanine dichloride (Pc-Cl2) yielded compound 3. Pc-(L-CA4)2 was synthesized through a click (yne-amine) reaction of compounds 1 and 3. Under the basic condition, Pc-(NCL-CA4)2 was synthesized by N-alkylation of compounds 2 and 3. Overall, the synthesis was straightforward and all steps gave high yields (>73% each step).
Scheme 1. Synthesis of Pc-(L-CA4)2 and Pc-(NCL-CA4)2.

Reagents and conditions: (i) propynoic acid, DCC, DMAP, CH2Cl2, room temp, 24 h; (ii) 1,3-dibromopropane, anhydrous K2CO3, acetone, reflux, 12 h; (iii) 2-(piperazin-1-yl)ethanol, pyridine, toluene, reflux, 4 h; (iv) 1, anhydrous THF, 30 min; (v) 2, anhydrous K2CO3, acetone, reflux, 12 h.
Formulation in PEG–PLA Polymeric Micelle
We formulated the prodrugs using PEG–PLA [poly(ethylene glycol)-poly(d,l-lactide)] copolymer micelles to take advantage of the enhanced permeability and retention (EPR) effect to enhance the delivery to tumor.19 The nanosized PEG–PLA polymer micelle was expected to provide three advantages: (1) passive targeting to tumors via the EPR effect,20,21 (2) prolonged circulation in the plasma, and (3) solubilization of the nonpolar prodrug. The biodegradable and nontoxic PEG–PLA micelle of paclitaxel (PCX) was approved by the FDA.22,23 PEG–PLA polymer micelles of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 were readily prepared. The zeta potentials and mean diameters of the micelles of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 were determined by dynamic light scattering (DLS) (zeta potential = 11.64 ± 1.38 mV, 16.81 ± 1.67 mV and mean diameter = 71.96 ± 1.34 nm, 75.07 ± 1.45 nm, respectively). To visualize the formation of the polymeric micelles, we used transmission electron micrographs (TEM). TEM images of the micelles showed consistent particle sizes (61–78 nm for Pc-(L-CA4)2 and 65–80 nm for Pc-(NCL-CA4)2 micelles). The prodrug concentrations in the micelles were 211 and 210 μM, respectively. The stability of the micelles was monitored by their particle sizes and zeta potentials at 4 °C under dark conditions. These values remained within 95% of the initial values for up to 21 days.
Figure 2.

(a) Particle size distribution and TEM images (inset) of micelles of (a) Pc-(L-CA4)2 and (b) Pc-(NLC-CA4)2.
Effects of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 on Tubulin Polymerization
CA4 is known to inhibit tubulin polymerization by binding to the colchicine binding pocket of tubulin.24,25 Because the bulky groups (Pc-L and Pc-NCL) are attached to CA4, we expected lower inhibitory activity of tubulin polymerization. We determined the effects of these prodrugs using the tubulin polymerization assay, in which fluorescence emission increases as tubulins polymerize (Figure 3a). The polymerization enhancer PCX and polymerization inhibitor CA4 were used as positive controls. Consistent with our data on the previous CA4 prodrug CMP-L-CA4,10 both Pc-(L-CA4)2 and Pc-(NCL-CA4)2 had significantly (p < 0.02) lower inhibitory activity (23% and 17%, respectively) than the parent drug CA4 (100%, Figure 3b).
Figure 3.
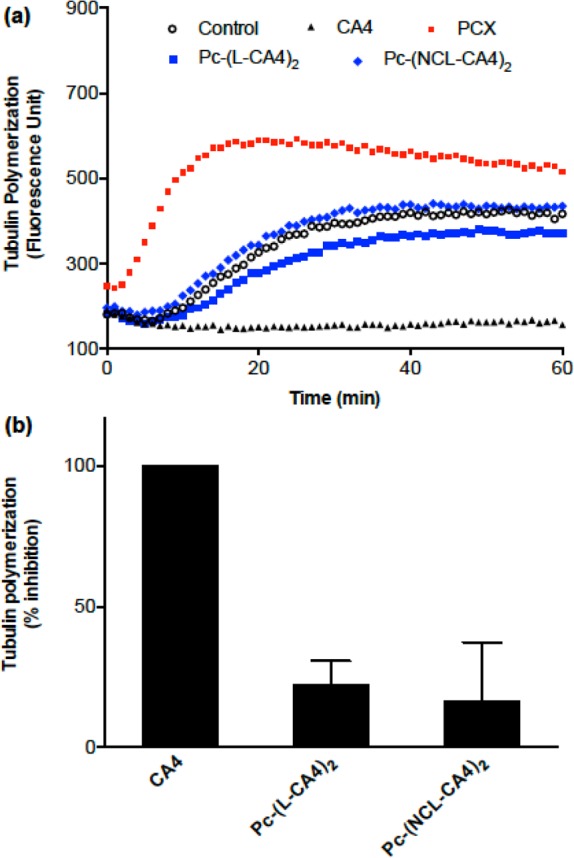
Effects of 3 μM each of PCX, CA4, Pc-(L-CA4)2, and Pc-(NCL-CA4)2 on tubulin polymerization: (a) one data set of representative kinetic traces (data from two more experiments are found in Figure S1, Supporting Information) and (b) inhibition of tubulin polymerization by CA4, Pc-(L-CA4)2, and Pc-(NCL-CA4)2 after 1 h incubation (±SD of three experiments).
Dark and Phototoxicity
Due to the dramatic reduction of the inhibition of tubulin polymerization, it was expected that Pc-(L-CA4)2 and Pc-(NCL-CA4)2 would have lower dark toxicity (cytotoxicity without illumination) than CA4. Using MCF-7 cells, we found that the dark toxicity of the prodrugs decreased by 19- and 101-fold [IC50D values = 9, 173, and 916 nM for CA4, Pc-(L-CA4)2 and Pc-(NCL-CA4)2, respectively]. Pc-L and Pc-NCL reduced the cytotoxicity, presumably by interfering with its binding to tubulin (Figure 3). However, illumination enhanced the toxicity of both the prodrug and the pseudo-prodrug [IC50P = 6 nM and 34 nM for Pc-(L-CA4)2 and Pc-(NCL-CA4)2, respectively].
Figure 4.
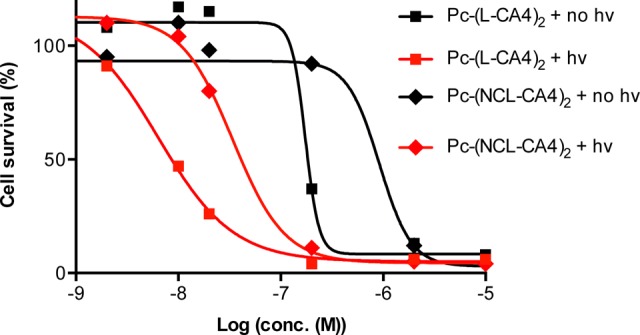
Dark and phototoxicity of Pc-L-(CA4)2 and Pc-NCL-(CA4)2.
Bystander Effects by Pc-(L-CA4)2
The difference in the magnitudes of phototoxicity and dark toxicity effects may be the result of each prodrug using a different mechanism. While Pc-(NCL-CA4)2 can only kill cells via the PDT effects of SO, Pc-(L-CA4)2 can potentially kill cells by both PDT effects and released CA4. To prove this mechanistic difference, we tested the bystander effects after the illumination. After treating the cells in wells of 24-well plates with the prodrug or pseudo-prodrug, one-half of each well was exposed to light. At 48 h post-illumination, live cells were stained with Calcein AM, and the center of each well was imaged using a fluorescence microscope.
Because the lifetime of SO in aqueous medium/biological systems is very short (∼40 ns),26 SO generated in the illuminated half of the well cannot kill the cells in the unilluminated half. SO should decay before reaching the other half of the well (diffusion distance of SO = ∼20–200 nm).27,28 However, the released CA4 can induce bystander effects because it can diffuse to the unilluminated half of the well. As we expected, bystander effects were found in the Pc-(L-CA4)2-treated wells: cells in the nonilluminated side were damaged as much as cells in the illuminated side (Figure 5b). However, wells treated with Pc-(NCL-CA4)2 had cell damage only in the illuminated halves of the wells (Figure 5c). This clearly supported our hypothesis that illuminated Pc-(L-CA4)2 releases CA4 that can induce bystander effects, which is consistent with data from our previous study with CMP-L-CA4.10
Figure 5.
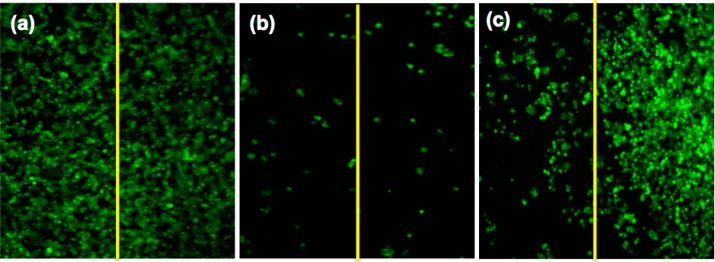
Fluorescence live cell images of the center of each well treated with (a) vehicle (diluting solution without a prodrug), (b) 25 nM Pc-(L-CA4)2, and (c) 25 nM Pc-(NCL-CA4)2. The left half of each well was illuminated with a 690 nm diode laser (11 mW/cm2 for 15 min). At these concentrations, Pc-(L-CA4)2 and Pc-(NCL-CA4)2 did not produce any significant dark toxicity.
Preclinical Optical Imaging in Live Mice
One of our major goals was to make Pc-(L-CA4)2 detectable in tissues using fluorescence optical imaging. We expected that the fluorescent photosensitizer Pc would provide sufficient fluorescence emission for this goal. To deliver the prodrugs to tumors by EPR effects, we prepared PEG–PLA polymer micelles of Pc-(L-CA4)2 or Pc-(NCL-CA4)2. As a control formulation, we also made solutions of these prodrugs in 5% Cremophor EL, which does not produce EPR effects. Balb/c mice with SC tumors (colon-26 cells, 4–6 mm in length) were injected retro-orbitally with 2 μmol/kg of the prodrug. Then, the mice were imaged at various postinjection time points (Figure 6 and Figure S4). As anticipated, we could clearly see the fluorescence emissions from the two prodrugs in live mice.
Figure 6.

Fluorescence optical images of the mice after retro-orbital injection of 2 μmol/kg of prodrug: (a) Pc-(L-CA4)2 in polymer micelles, (b) Pc-(NCL-CA4)2 in polymer micelles, (c) Pc-(L-CA4)2 in 5% Cremophor solution, and (d) Pc-(NCL-CA4)2 in 5% Cremophor solution. BG: background image before prodrug injection. Scale bar unit: fluorescence arbitrary unit.
The imaging data revealed two important findings. First, the images from the mice that received the polymer micelles of both prodrugs showed “hot spots” in tumors, with a peak at around 24 h postinjection, presumably due to the EPR effects of the nanosized polymer micelles. These hot spots were more evident in the mice who received injections of Pc-(L-CA4)2. Second, the “hot spots” resulting from formulations with the polymer micelles persisted longer than those “hot spots” resulting from formulations with Cremophor solutions. The polymer micelles appeared to delay the clearance of the prodrugs from the system.
Because the illumination for optical imaging could theoretically generate SO and thus release CA4 from Pc-(L-CA4)2, we monitored the body weight change and tumor growth pattern of the mice imaged with Pc-(L-CA4)2. We did not see any significant impact on mouse body weight or tumor growth since the light dose used for imaging was negligible [675 filter (660–690 nm) at ∼1.5 μW/cm2 for 2 s (3.0 × 10–6 J/cm2)]. The treatment used a (1.2 × 108)-fold higher light dose than the dose used for imaging.
Antitumor Efficacy
We expected that the new prodrug Pc-(L-CA4)2 would show better antitumor effects than our previous prodrug CMP-L-CA4, because Pc has superior light absorption properties and the prodrug releases two CA4 instead of one. We used BALB/c mice with SC tumors to evaluate the antitumor effects of the prodrugs with illumination. Twenty-four hours post-retro-orbital injection of 1 or 2 μmol/kg of the prodrug, the tumor was illuminated by a 690 nm diode laser for 30 min at 100 mW/cm2 (180 J/cm2) or 200 mW/cm2 (360 J/cm2). These treatment conditions were chosen based on data from pilot studies. Six groups were used to assess the antitumor effects of the prodrug and pseudo-prodrug: G1, negative control; G2, [CA4 (1 μmol/kg) + no hv]; G3, [Pc-(NCL-CA4)2 (1 μmol/kg) + hv (100 mW/cm2)]; G4, [Pc-(L-CA4)2 (1 μmol/kg) + hv (100 mW/cm2)]; G5, [Pc-(L-CA4)2 (2 μmol/kg) + hv (100 mW/cm2)]; and G6, [Pc-(L-CA4)2 (2 μmol/kg) + hv (200 mW/cm2)]. Antitumor effects were monitored by measuring tumor volume (Figure 7a and b). [The tumor growth curves of G5 and G6 were nearly identical to G4 until day 15. So, these curves were omitted from Figure 7a for clarity (Figure S2).]
Figure 7.
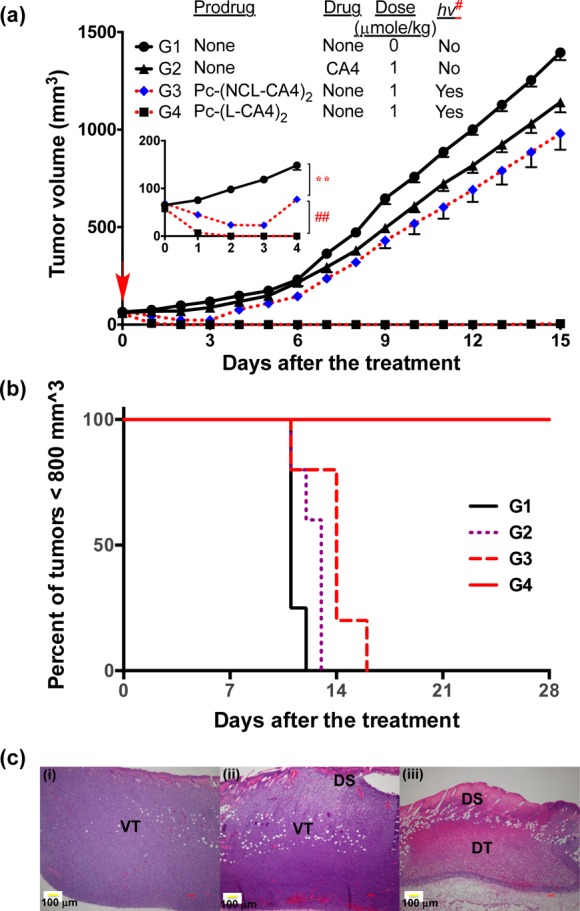
Antitumor effects. (a) Tumor growth curves, drug IV administration: once a day on day −1, illumination 24 h postdrug administration [hv#: 100 mW/cm2 for 30 min (180 J/cm2) or 200 mW/cm2 for 30 min (360 J/cm2)], 5 mice per group except the control group (4 mice). Error bars represent SE. In inset: the order of tumor size was G1 > G3 > G4 during day 1 to day 4 [** p < 0.01 (G1 vs G3, from day 1 to day 4) and ## p < 0.01 (G3 vs G4, from day 1 to day 4)]. (b) Kaplan–Meier plot of response to treatment. (c) H&E staining of tumors 24 h post-illumination from (i) control mice or mice treated with (ii) Pc-(NCL-CA4)2, or (iii) Pc-(L-CA4)2. Prodrug administration and illumination conditions were same as for those in (a). VT, viable tumor; DT, damaged tumor; and DS, damaged skin.
We found outstanding antitumor effects in the mice treated with Pc-(L-CA4)2, G4–G6. After 24 h illumination, all tumors shrank to a nonmeasurable size and remained so for almost 15 days (Figure 7a). Mice in group G4 experienced tumor growth only after day 16 (Figure S2). All mice in G4–G6 lived until day 30, while tumor size of all 4 mice in the control group (G1) reached >800 mm3 in 12 days (Figure 7b). The PDT effects resulting from Pc-(NCL-CA4)2 treatment (G3) had a significant impact on tumor size until day 3 (p < 0.05). However, after day 3, the tumors grew back at a rate similar to the tumors in the control group G1. Throughout the observation period, Pc-(L-CA4)2 treatment yielded significantly better antitumor effects (p < 0.01), than Pc-(NCL-CA4)2 treatment. Thus, it seemed that the PDT effects alone might not be sufficient to produce such a robust antitumor effect as seen in group G4. We hypothesized that our findings stemmed from the contribution of the released CA4 in addition to PDT effects. Interestingly, no mice in G1–G6 experienced a significant decrease in body weight (Figure S3).
The histological data were consistent with the antitumor effects (Figure 7c). After 24 h of treatment, tumors were collected and stained with H&E to visualize the tissue damage. While mice treated with Pc-(NCL-CA4)2 experienced tissue damage only on the skin, mice receiving Pc-(L-CA4)2 also experienced direct tumor damage. In fact, the volume of tumors treated with Pc-(L-CA4)2 shrank to about 1/8 of the volume of the tumors in the control group.
Conclusion
We successfully demonstrated a multifunctional photounclickable prodrug that can be visualized by optical imaging, and ablates tumors with a combination of PDT and local chemotherapy. The prodrug Pc-(L-CA4)2 and its pseudo-prodrug Pc-(NCL-CA4)2 were prepared in high yields through a facile and flexible scheme. The cytotoxicity of these prodrugs was lower than that of the parent drug CA4, but both prodrugs showed enhanced cytotoxicity upon illumination.
The mechanisms of cell damage of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 combined with illumination should be different. While the use of Pc-(NCL-CA4)2 and illumination killed cancer cells through PDT effects, the use of Pc-(L-CA4)2 and illumination combined these PDT effects with local chemotherapy through the released CA4. This was supported by the bystander effects demonstrated in vitro. Through the use of optical imaging, we found that both prodrugs were detected at the therapeutic dose within tumors. Optical imaging also provided the information about the PK profiles of the prodrugs so that we could find the optimal time point for illumination. As expected, the antitumor effects in mice treated with Pc-(L-CA4)2 were dramatically better than in mice treated with Pc-(NCL-CA4)2. Treatment with either Pc-(NCL-CA4)2 or CA4 produced minimal antitumor effects, suggesting that the outstanding antitumor effects of Pc-(L-CA4)2 may have been a result of the synergistic effects of PDT and chemotherapy.
In addition to confirming that our current SO-activatable prodrug strategy provides improved antitumor efficacy, we demonstrated the innovative use of a fluorescent photosensitizer within the prodrug. Using optical imaging, we were able to noninvasively generate PK information about the prodrug without causing any observable acute toxicity to mice. Our multifunctional prodrug strategy includes (1) activation by the clinical translatable far-red light (or NIR), (2) the unique combination of PDT and local chemotherapy, and (3) the dual function of optical imaging and treatment with one prodrug. We anticipate that this strategy will be applicable to various drug delivery forms, clinically approved drugs, and advanced drug delivery systems targeted at tumors.
Experimental Section
Synthesis
CA4,29 compound 1,9 and compound 2(10) were synthesized as reported previously. The purity of the biologically evaluated compounds Pc-(L-CA4)2 and Pc-(NCL-CA4)2 was confirmed to be >95% by high-performance liquid chromatography (HPLC) (Figures S9 and S10).
Compound 3
1-(2-Hydroxyethyl)-piperazine (1.91 g, 14.70 mmol) and pyridine (2.5 mL) were added to a solution of silicon(IV) phthalocyanine dichloride (Pc-Cl2, 1 g, 1.63 mmol) in 50 mL toluene. The reaction mixture was refluxed for 12 h. The solvent was then removed under reduced pressure. The residue was dissolved in CH2Cl2 and washed with water. The solvent of the combined organic layers was removed by evaporation, and the crude was recrystallized with CHCl3/n-hexane (1:4 v/v) to give a blue solid compound 3 (1.22 g, 94%). 1H NMR (300 MHz, CD2Cl2) δ −1.19 (t, J = 5.8 Hz, 4H), −0.82 (t, J = 5.8 Hz, 1H), 0.28 (m, 8H), 1.75 (m, 8H), 8.30–8.40 (m, 8H, Pc-Hβ), 9.55–9.69 (m, 8H, Pc-Hα); 13C NMR (300 MHz, CD2Cl2) δ 123.4, 131.0, 135.9, 149.2; HRMS ESI (m/z): [M + H]+ calculated for C44H43N12O2Si, 799.3401; found, 799.3395.
Pc-(L-CA4)2
Compound 1 (46 mg, 0.13 mmol) and compound 3 (50 mg, 0.06 mmol) were dissolved in 20 mL dry THF, and the solution was stirred at room temperature for 15 min. The solvent was removed under reduced pressure to yield the crude product, which was then recrystallized from CHCl3/n-hexane (1:5 v/v) to give Pc-(L-CA4)2 (83 mg, 87%). 1H NMR (300 MHz, CD2Cl2)) δ −1.96 (t, J = 5.6 Hz, 4H), −0.55 (t, J = 5.0 Hz, 4H), 0.29 (br s, 8H), 2.20 (br s, 8H), 3.67 (s, 12H), 3.72 (s, 6H), 3.77 (s, 6H), 4.34 (d, J = 12.8 Hz, 2H), 6.45 (d, J = 4.9 Hz, 4H), 6.62 (s, 4H), 6.84 (d, J = 8.1 Hz, 2H), 6.96 (s, 2H), 7.00 (s, 2H), 7.09 (d, J = 12.8 Hz, 2H), 8.38–8.43 (m, 8H, Pc-Hβ), 9.65–9.70 (m, 8H, Pc-Hα); 13C NMR (300 MHz, CD2Cl2) δ 55.8, 56.6, 60.4, 81.8, 105.9, 111.8, 123.5, 123.9, 126.6, 128.7, 129.1, 129.8, 131.2, 132.5, 135.8, 137.2, 140.3, 149.3, 151.1, 152.2, 153.0, 167.2; HRMS ESI (m/z): [M + H]+ calculated for C86H83N12O14Si, 1535.5921; found, 1535.5914.
Pc-(NCL-CA4)2
Anhydrous K2CO3 (68 mg, 0.500 mmol) and compound 3 (200 mg, 0.25 mmol) were added to a solution of compound 2 (218 mg, 0.50 mmol) in 10 mL dry DMF. The reaction mixture was stirred at room temperature for 12 h. The K2CO3 was removed by suction filtration and the solvent was removed under reduced pressure. The crude product was then recrystallized from CHCl3/n-hexane (1:5 v/v) to give Pc-(NCL-CA4)2 (302 mg, 80%). 1H NMR (300 MHz, CD2Cl2) δ −1.96 (t, J = 5.6 Hz, 4H), −0.78 (t, J = 5.8 Hz, 4H), 0.41 (br s, 8H), 1.50 (m, 2H), 1.90 (m, 2H), 2.18 (m, 2H), 2.49 (m, 2H), 3.32 (s, 4H), 3.60 (br s, 8H), 3.65 (s, 12H), 3.75 (s, 6H), 3.80 (s, 6H), 6.46 (m, 4H), 6.51 (m, 4H), 6.74 (m, 2H), 6.81 (m, 2H), 6.92 (m, 2H), 8.38 (m, 8H, Pc-Hβ), 9.66 (m, 8H, Pc-Hα); 13C NMR (300 MHz, CD2Cl2) δ 55.8, 60.4, 105.1, 111.4, 113.6, 115.2, 117.5, 123.4, 126.7, 126.8, 126.9, 130.9, 132.8, 135.9, 149.1; HRMS ESI (m/z): [M + H]+ calculated for C86H91N12O12Si, 1511.6649; found, 1511.6612.
Preparation of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 Micelles
Briefly, 3 mg of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 was dissolved in 1.3 mL of THF. Ten mg of mPEG–PLA (cat #AK09, vendor: Polyscitech) was dissolved in 1 mL of THF. 600 μL of the 3 mg of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 dissolved in 1.3 mL THF was added to the mPEG-PLA-THF mixture. The volume of the resulting mixture was reduced to 300 μL under reduced pressure. The 300 μL of the mixture of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 and mPEG–PLA was added to 3 mL of distilled water dropwise while stirring. The mixture was stirred for 3 h, after which the organic solvent was evaporated under reduced pressure at 40 °C. The resultant solution was filtered through a 0.2 μm filter. The concentration of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 micelles was determined by diluting the micelles in THF: the absorbance was measured by absorbance of Pc group. The concentration was calculated from the molar extinction coefficient (EC) of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 at 675 nm in THF (EC of Pc-(L-CA4)2 = 205,000; Pc-(NCL-CA4)2 = 206,110 M–1 cm–1) using the Beer–Lambert law. The concentrations of Pc-(L-CA4)2 or Pc-(NCL-CA4)2 micelles were determined to be 211 and 210 μM, respectively. Freshly prepared Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelles were used for all the experiments.
Cremophor Solution
2.0 mM stock solutions of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 prepared in THF were further diluted with 5% Cremophor EL in PBS to achieve appropriate concentrations.
Characterization of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 Micelles
The Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelles in an aqueous solution were characterized by measuring their hydrodynamic diameter and zeta potential via dynamic light scattering (DLS). The size measurement was carried out at a concentration of 1.0 mg/mL of Pc-(L-CA4)2 or Pc-(NCL-CA4)2. Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelles were also imaged by transmission electron microscopy (TEM) at 20,000× operating at 80 kV. TEM samples were prepared by depositing 20 μL of diluted Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelle solution on a 300 mesh copper TEM grid with a carbon film. The sample grid was air-dried before the measurements were taken.
Tubulin Polymerization Assay
A fluorescence-based tubulin polymerization assay was conducted using a kit supplied by Cytoskeleton, Inc. (cat # BK011P). The basic principle is that an increase in fluorescence will occur as a fluorescence reporter is incorporated into microtubules during the course of polymerization. The assay was performed following the experimental procedure described in version 2.1 of the tubulin polymerization assay kit manual. Briefly, a drug in DMSO stock solution was added to a mixture of tubulin and GTP in a buffer solution, to give a final concentration of 3 μM. The reaction mixture was incubated at 37 °C. Fluorescence was monitored (excitation = 360 nm and emission = 450 nm) every 2 min for 1 h. PCX and CA4 were included in the assay as positive controls, as well as a vehicle-only negative control.
Dark and Phototoxicity
The cytotoxicity of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 was determined with and without illumination. MCF-7 cells were maintained in minimum essential medium (α-MEM) supplemented with 10% bovine growth serum, 2 mM l-glutamine, 50 units/mL penicillin G, 50 μg/mL streptomycin, and 1.0 μg/mL fungizone. MCF-7 cells (5000 cells/well) were seeded on 96-well plates in the medium and incubated for 24 h at 37 °C in 5% CO2. Stock Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelle solutions (200 μM) were prepared in distilled water. The stock solutions were further diluted with medium to obtain the necessary final concentrations. The diluted solution (10 μL) was then added to each well (190 μL). The plates were incubated for 24 h and then removed from the incubator. For the phototoxicity study: The uncovered plate was illuminated for 30 min using a diode laser (690 nm, 5.6 mW/cm2). To ensure uniformity of the light during the illumination, each plate was shaken gently on an orbital shaker (Lab-line, Barnstead International). For the dark toxicity study: Plates were kept in the dark for 30 min and then returned to the incubator. After 3 days, cell viability was determined using an MTT assay. Briefly, a 10 μL solution of MTT (10 mg in 1 mL PBS buffer) was added to each well and the plate was incubated for 4 h at room temperature or 37 °C. Then, the MTT solution was removed and the cells were dissolved in 200 μL of DMSO. The absorbance of each well was measured at 570 nm with background subtraction at 650 nm. Cell viability was quantified by measuring the absorbance of the treated wells compared to that of the untreated control wells, and expressed as a percentage.
Bystander Effect
Colon-26 cells were seeded at 5000 cells/well on 24-well plates and incubated for 24 h. Stock Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelle solutions (200 μM) were prepared in distilled water. 200 μM of the stock solutions were added to wells (1 mL) to obtain appropriate final concentrations of both Pc-(L-CA4)2 and Pc-(NCL-CA4)2 at 25 nM. After 24 h incubation, the plates were illuminated from the bottom with a 690 nm diode laser at 11 mW/cm2 for 15 min. During illumination, half of each well was blocked with black masking tape (cat# T743-1.0, vendor: Thorlabs). The illuminated plates were incubated for an additional 48 h. Then, a Calcein AM live cell staining assay (cat # 4892-010-K, vendor: Molecular, Probes Tervigen) was performed. The cells were washed once with 1 mL Calcein AM wash buffer, then 250 μL fresh wash buffer and 250 μL working reagent were added to the wells. The cells were incubated for 30 min. Fluorescent images were obtained with an Olympus IX51 inverted microscope with a green fluorescence channel to visualize live cells. All images were taken at 10× magnification.
In Vivo Optical Imaging
The concentrations of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelles in aqueous solution were determined by diluting 10 μL of the formulation stock in 1 mL of THF and measuring the absorbance of phthalocyanine. The concentration was calculated from the EC of Pc-(L-CA4)2 at 672 nm in THF (EC of Pc-(L-CA4)2 = 205,000; Pc-(NCL-CA4)2 = 206,110 M–1 cm–1) using the Beer–Lambert law.
We used four- to six-week-old BALB/c mice to investigate the biodistribution and tumor targeting ability of the polymeric micelles. The mice were shaved before the imaging experiments, and were imaged using the IVIS Imaging system. The mice were injected with Pc-(L-CA4)2 and Pc-(NCL-CA4)2 in the micelle formulation (2 μmol/kg, i.v.). As a comparison, the prodrugs were also evaluated in the Cremophor solution. Fluorescence images were taken 0, 3, 6, 12, 24, 48, and 72 h after retro-orbital injection. Before taking the images, the mice were anesthetized in an acrylic chamber with a 2.5% isoflurane/air mixture. The following parameters were used to acquire images with Living Image software: fluorescence mode, exposure time: 2 s, binning: medium, F/Stop: 2, excitation: 675 filter (660–690 nm), and emission: 720 filter (710–730 nm). During post processing, image counts were adjusted to 3 × 104 as minimum and 6.0 × 104 a.u. as maximum color scale.
Antitumor Efficacy Study
Four- to six-week-old BALB/c mice (18–20 g) were used for the murine tumor model. The mice were implanted SC with 2 × 106 colon 26 cells in PBS (100 μL) on the lower back of the neck. Tumor growth was monitored using digital calipers. The longest axis of the tumor (1) and the axis perpendicular to l (w) were used to calculate tumor volume (lw2/2). Mice with tumors 5–6 mm in diameter were used for the experiments.
We used stock solutions of Pc-(L-CA4)2 and Pc-(NCL-CA4)2 micelles in aqueous solution and further dilutions to achieve final doses as follows: [CA4, (1 μmol/kg each)], [Pc-(NCL=CA4)2, (1 μmol/kg each) ], [Pc-(L-CA4)2, (1 μmol/kg each)], and [Pc-(L-CA4)2, (2 μmol/kg each)]. To each mouse, 200 μL of sample was injected via IV once on day −1. Twenty-four hours later mice were anesthetized by IP injection of 80 mg/kg ketamine and 6 mg/kg xylazine. Tumors were illuminated with a 690 nm diode laser at 100 mW/cm2 or 200 mW/cm2 for 30 min −180 or 360 J/cm2, respectively. Tumor size was measured every day after the treatment.
Histology Study (H&E Staining)
To evaluate antitumor effect, mice from various groups were euthanized 24 h after laser illumination and tumors were collected. The specimens were fixed in 10% buffered formalin, embedded in paraffin, and ∼4 mm diameter tissue sections were stained with hematoxylin and eosin following a standard procedure at the tissue pathology core facility at OUHSC. The sections were viewed and photographed by bright-field microscopy at 4× magnification.
Statistical analysis
Student’s t test was used for statistical analysis. P values less than 0.05 were considered significant.
Acknowledgments
The helpful discussion with Drs. Michael Ihnat and Sukyung Woo are appreciated. We thank the Peggy and Charles Stephenson Cancer Center at the University of Oklahoma Health Sciences Center, Oklahoma City, OK, and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103639 for the use of the Molecular Imaging Core facility that provided services for the optical in vivo imaging and Cancer Tissue Pathology Core facility that provided services for the H&E staining. We also thank the Oklahoma Medical Research Foundation imaging core facility that provided the services for the transmission electron micrographs. This research was supported by the DoD [Breast Cancer Research Program] under Award Number W81XWH-09-1-0071. Views and opinions of and endorsements by the authors do not reflect those of the U.S. Army or the DoD.
Glossary
Abbreviations
- CA4
combretastatin A-4
- DCC
N,N′-dicyclohexylcarbidiimide
- DMAP
4-dimethylaminopyridine
- DS
damaged skin
- DT
damaged tumor
- Pc
silicon (iv) phthalocyanine
- L
aminoacrylate linker
- NCL
noncleavable linker
- PDT
photodynamic therapy
- PEG
poly(ethylene glycol)
- PLA
ploy(d,l-lactide)
- ROS
reactive oxygen species
- SO
singlet oxygen
- VT
viable tumor
Supporting Information Available
Supporting Information includes detailed descriptions of general experimental conditions, additional tubulin polymerization data, extended tumor growth curves, body weight changes during antitumor efficacy study, an additional set of the fluorescence optical images, and 1H NMR spectra and HPLC chromatograms. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
M. B. and Y. Y. designed the project and wrote the manuscript. M.B. and G.N. prepared the prodrugs, and M.B. and P.R. carried out the biological evaluations of the compounds.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Alvarez-Lorenzo C.; Bromberg L.; Concheiro A. Light-sensitive intelligent drug delivery systems. Photochem. Photobiol. 2009, 85, 848–860. [DOI] [PubMed] [Google Scholar]
- Bio M.; You Y. Emerging strategies for controlling drug release by visible/near IR light. Med. Chem. 2013, 3, 192–198. [Google Scholar]
- Ruebner A.; Yang Z.; Leung D.; Breslow R. A cyclodextrin dimer with a photocleavable linker as a possible carrier for the photosensitizer in photodynamic tumor therapy. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14692–14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M. Y.; Dolphin D. Site-specific prodrug release using visible light. J. Am. Chem. Soc. 2008, 130, 4236–4237. [DOI] [PubMed] [Google Scholar]
- Murthy R. S.; Bio M.; You Y. J. Low energy light-triggered oxidative cleavage of olefins. Tetrahedron Lett. 2009, 50, 1041–1044. [Google Scholar]
- Zamadar M.; Ghosh G.; Mahendran A.; Minnis M.; Kruft B. I.; Ghogare A.; Aebisher D.; Greer A. Photosensitizer drug delivery via an optical fiber. J. Am. Chem. Soc. 2011, 133, 7882–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahendran A.; Kopkalli Y.; Ghosh G.; Ghogare A.; Minnis M.; Kruft B. I.; Zamadar M.; Aebisher D.; Davenport L.; Greer A. A hand-held fiber-optic implement for the site-specific delivery of photosensitizer and singlet oxygen. Photochem. Photobiol. 2011, 87, 1330–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio M.; Nkepang G.; You Y. Click and photo-unclick chemistry of aminoacrylate for visible light-triggered drug release. Chem. Commun. 2012, 48, 6517–6519. [DOI] [PubMed] [Google Scholar]
- Hossion A. M. L.; Bio M.; Nkepang G.; Awuah S. G.; You Y. Visible light controlled release of anticancer drug through double activation of prodrug. ACS Med. Chem. Lett. 2013, 4, 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio M.; Rajaputra P.; Nkepang G.; Awuah S. G.; Hossion A. M.; You Y. Site-specific and far-red-light-activatable prodrug of combretastatin A-4 using photo-unclick chemistry. J. Med. Chem. 2013, 56, 3936–3942. [DOI] [PubMed] [Google Scholar]
- Lee J.; Park J.; Singha K.; Kim W. J. Mesoporous silica nanoparticle facilitated drug release through cascade photosensitizer activation and cleavage of singlet oxygen sensitive linker. Chem. Commun. 2013, 49, 1545–1547. [DOI] [PubMed] [Google Scholar]
- Jiang X. J.; Yeung S. L.; Lo P. C.; Fong W. P.; Ng D. K. Phthalocyanine-polyamine conjugates as highly efficient photosensitizers for photodynamic therapy. J. Med. Chem. 2011, 54, 320–330. [DOI] [PubMed] [Google Scholar]
- Jiang X. J.; Lo P. C.; Yeung S. L.; Fong W. P.; Ng D. K. A pH-responsive fluorescence probe and photosensitiser based on a tetraamino silicon(IV) phthalocyanine. Chem. Commun. 2010, 46, 3188–3190. [DOI] [PubMed] [Google Scholar]
- Urano Y.; Asanuma D.; Hama Y.; Koyama Y.; Barrett T.; Kamiya M.; Nagano T.; Watanabe T.; Hasegawa A.; Choyke P. L.; Kobayashi H. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat. Med. 2009, 15, 104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. E.; Zhang P.; Azizuddin K.; Delos Santos G. B.; Chiu S. M.; Xue L. Y.; Berlin J. C.; Peng X.; Wu H.; Lam M.; Nieminen A. L.; Kenney M. E.; Oleinick N. L. Structural factors and mechanisms underlying the improved photodynamic cell killing with silicon phthalocyanine photosensitizers directed to lysosomes versus mitochondria. Photochem. Photobiol. 2009, 85, 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Larkin H. E.; Li Y. S.; Rihter D.; Zaidi S. I.; Rodgers M. A.; Mukhtar H.; Kenney M. E.; Oleinick N. L. The synthesis, photophysical and photobiological properties and in vitro structure-activity relationships of a set of silicon phthalocyanine PDT photosensitizers. Photochem. Photobiol. 1997, 65, 581–586. [DOI] [PubMed] [Google Scholar]
- Sekkat N.; van den Bergh H.; Nyokong T.; Lange N. Like a bolt from the blue: phthalocyanines in biomedical optics. Molecules 2012, 17, 98–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y. J.; Gibson S. L.; Hilf R.; Davies S. R.; Oseroff A. R.; Roy I.; Ohulchanskyy T. Y.; Bergey E. J.; Detty M. R. Water soluble, core-modified porphyrins. 3. Synthesis, photophysical properties, and in vitro studies of photosensitization, uptake, and localization with carboxylic acid-substituted derivatives. J. Med. Chem. 2003, 46, 3734–3747. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug. Delivery Rev. 2013, 65, 71–79. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Controlled Release 2000, 65, 271–284. [DOI] [PubMed] [Google Scholar]
- Lim W. T.; Tan E. H.; Toh C. K.; Hee S. W.; Leong S. S.; Ang P. C.; Wong N. S.; Chowbay B. Phase I pharmacokinetic study of a weekly liposomal paclitaxel formulation (Genexol-PM) in patients with solid tumors. Ann. Oncol. 2010, 21, 382–388. [DOI] [PubMed] [Google Scholar]
- Kim T. Y.; Kim D. W.; Chung J. Y.; Shin S. G.; Kim S. C.; Heo D. S.; Kim N. K.; Bang Y. J. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [DOI] [PubMed] [Google Scholar]
- Kong Y.; Grembecka J.; Edler M. C.; Hamel E.; Mooberry S. L.; Sabat M.; Rieger J.; Brown M. L. Structure-based discovery of a boronic acid bioisostere of combretastatin A-4. Chem. Biol. 2005, 12, 1007–1014. [DOI] [PubMed] [Google Scholar]
- Jin Y. H.; Qi P.; Wang Z. W.; Shen Q. R.; Wang J.; Zhang W. G.; Song H. R. 3D-QSAR study of combretastatin A-4 analogs based on molecular docking. Molecules 2011, 16, 6684–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovsen E.; Snyder J. W.; Lambert J. D. C.; Ogilby P. R. Lifetime and diffusion of singlet oxygen in a cell. J. Phys. Chem. B 2005, 109, 8570–8573. [DOI] [PubMed] [Google Scholar]
- Niedre M.; Patterson M. S.; Wilson B. C. Direct near-infrared luminescence detection of singlet oxygen generated by photodynamic therapy in cells in vitro and tissues in vivo. Photochem. Photobiol. 2002, 75, 382–391. [DOI] [PubMed] [Google Scholar]
- Moan J. On the diffusion length of singlet oxygen in cells and tissues. J. Photochem. Photobiol., B 1990, 6, 343–347. [Google Scholar]
- Gaukroger K.; Hadfield J. A.; Hepworth L. A.; Lawrence N. J.; McGown A. T. Novel syntheses of cis and trans isomers of combretastatin A-4. J. Org. Chem. 2001, 66, 8135–8138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.