

 A novel method for the parallel synthesis of peptide-biocargo conjugates was developed and applied to synthesize cell-penetrating peptide (CPP)–PNA705 conjugate libraries for screening in a splicing redirection assay.
A novel method for the parallel synthesis of peptide-biocargo conjugates was developed and applied to synthesize cell-penetrating peptide (CPP)–PNA705 conjugate libraries for screening in a splicing redirection assay.
Abstract
A novel method for the parallel synthesis of peptide-biocargo conjugates was developed that utilizes affinity purification for fast isolation of the conjugates in order to avoid time consuming HPLC purification. The methodology was applied to create two libraries of cell-penetrating peptide (CPP)–PNA705 conjugates from parallel-synthesized peptide libraries. The conjugates were tested for their ability to induce splicing redirection in HeLa pLuc705 cells. The results demonstrate how the novel methodology can be applied for screening purposes in order to find suitable CPP-biocargo combinations and further optimization of CPPs.
Introduction
Efficient cell delivery of bio-molecules, such as oligonucleotides and peptides, is a major hurdle in development of novel therapeutics. As a result, higher drug dosages are often required than would otherwise be needed, which increases costs and the possibility of off target effects. A promising method of enhancing cell delivery is by use of cell-penetrating peptides (CPPs) to facilitate cell-uptake.1 CPPs often contain positive charges and/or hydrophobic elements. Some are based on cell-permeable peptides obtained from larger proteins and are known as protein transduction domains (PTD), whereas others are synthetically derived. CPPs have been shown to deliver a range of different biologically active cargoes into cells and in vivo, including proteins, peptides, oligonucleotide analogues, siRNAs and small molecule drugs.2
In the case of oligonucleotide cargoes, CPPs can either be complexed with or conjugated covalently to the cargo. For example, CPP conjugates of charge neutral antisense phosphorodiamidate morpholino oligonucleotides (PMO) or peptide nucleic acids (PNA) have been shown to be very effective in induction of splicing redirection or exon skipping in cells and in vivo.3 However, there have been very many CPPs proposed, and individual research groups often utilise their own preferred peptide sequences. No single CPP has been found to be universally successful for the conjugate delivery of all cargoes into all cell types. Instead for a new drug target or application, success is often only achieved through painstaking conjugate synthesis on an individual basis and search for a suitable peptide sequence by trial and error using a cell or in vivo assay.
Only a small number of CPPs (commonly 2 or 3 known CPPs) can usually be tested as bio-cargo conjugates because of the difficulty and the time-consuming nature of their syntheses, even though cell-screening reporter assays, e.g. luciferase-based, are frequently available and adaptable for high-throughput. Significant demand exists therefore for a method of peptide conjugate library synthesis that is quick and efficient and is suitable for subsequent use of rapid screening assays.4 Following cell assay of an initial library of conjugates using peptides of widely ranging sequence, fine-tuning of a peptide candidate can then be accomplished either by further narrower library synthesis and cell re-assay, or by more conventional synthesis if further screening is to be carried out by an in vivo assay.
The most time-limiting step in synthesis of peptide-cargo conjugates is generally not the conjugation itself but the individual purifications required for peptide, cargo and conjugate. Recently, parallel multi-peptide synthesis machines have become available as well as new methods for rapid affinity-based purification of bio-molecules (e.g. histidine tags or biotin-streptavidin)4b that provide an opportunity both for rapid synthesis and for reaction workup and purification. In addition, recent developments in bio-orthogonal “click”-type ligation reactions provide a chemical basis for efficient conjugation of bio-molecules in aqueous solution.5 These new methodologies have now inspired us to develop a new strategy for the SELection of PEPtide CONjugates (SELPEPCON) of bio-cargoes that can act as an initial parallel synthesis approach useful for therapeutic screening.
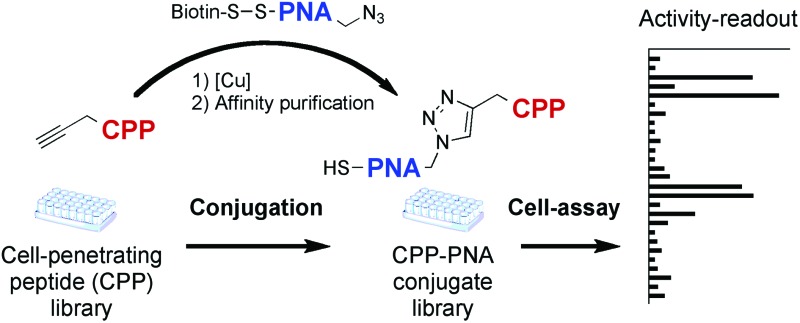
SELPEPCON allows for convenient and rapid parallel conjugation reactions and workup that avoid the need for HPLC purification (Fig. 1). The method utilizes a functionalized cargo linked through a cleavable linker to an affinity tag. The functionalized cargo is conjugated to the peptide after which it is purified by immobilization and isolated by release of the tag. Affinity purification is very rapid and is readily automated, making the overall strategy very suitable for high-throughput synthesis of conjugates for screening.
Fig. 1. Overview of conjugation and conjugate work-up strategy of SELPEPCON.

To demonstrate the utility of this SELPEPCON methodology, two case studies are shown. In the first example, a small conjugate library is synthesized to investigate the roles of individual or groups of amino acids in a pre-selected CPP attached to a cargo. The second shows a larger CPP screen of a variety of quite different peptide sequences to find CPP candidates suitable for delivery of a novel cargo into a cell. Both studies utilize CPP conjugates of a PNA705 cargo, a well-known 16-mer splice-redirecting oligonucleotide analogue cargo that has been used many times for CPP development in the past.6 A HeLa pLuc705 cell assay was employed to assess the construct for their ability to enter cells. In this assay, PNA705, upon entering the cell, causes splicing redirection of an aberrant β-globin intron, which leads to the up-regulation of firefly luciferase.7 The assay is convenient, has a high dynamic range and is very suitable for testing of a large number of conjugates in parallel.
Conjugations are carried out using copper catalyzed Huisgen “click” reactions8 between alkyne-functionalized CPPs and an azide-functionalized PNA705 containing a disulphide-linked biotin-tag. The disulphide-linked biotin tag allows for solid-phase immobilization purification of the resultant conjugates after which the conjugates can be isolated by reduction of the disulphide releasing it from the solid support. The results demonstrate how SELPEPCON can be utilized to find active CPPs for a cargo such as PNA705 in a rapid synthesis, isolation and screening procedure.
Results and discussion
Cargo synthesis
The SELPEPCON method of synthesis of a bifunctionalized PNA705 cargo suitable for attachment of peptides and subsequent immobilization is shown in Scheme 1. Because a known side product of the click reaction is alkyne homocoupling, we decided to locate the azide functionality on the PNA cargo and the alkyne functionality on the peptide component. In this setup any alkyne-homocoupling side reaction will lead to a product that is easily removed by washing of the streptavidin solid support. Thus, 5-azidopentanoic acid was used for N-terminal modification of the PNA by manual solid phase amide coupling following machine-aided assembly of the PNA by solid-phase synthesis.9
Scheme 1. Synthesis of N3-PNA705-S-S-biotin cargo for conjugation to CPP-libraries.

The PNA contains a C-terminal S-trityl-protected Cys residue, which was introduced at the first step of solid-phase synthesis. After deprotection and cleavage from the solid support, a biotin group was introduced on to the N3-PNA705 by disulfide bridge formation via reaction of the C-terminal Cys residue with commercially available N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (EZ-link™ HPDP Biotin). This straight forward reaction provided the N3-PNA(705)-S-S-biotin after HPLC purification in 80% yield.
Pip5h based CPP-library (LB1)
In order to demonstrate the methodology, a small test library of 16 peptides was synthesized on Tentagel using an Intavis Parallel Peptide Synthesizer (Table 1). The test library was narrowly based around a known relatively short (17 amino acids) CPP, Pip5h, which we have previously evaluated in exon skipping as a conjugate with a PMO targeting the pre-mRNA of dystrophin,10 but which was not chosen for further development. Peptide variants included modifications in the central 5-amino acid core (LB1_1–8) or on one of the Arg-rich flanking regions (LB1_9–16) of the sequences. In each case an N-terminal alkyne was introduced using 4-pentynoic acid through standard amide bond formation as the final coupling step (Scheme 2).
Table 1. Library of Pip5h analogues containing an N-terminal alkyne linker, peptide and conjugate synthesis yield.
| Peptide | Sequence (N to C – term) a | Net charge | Number of Arg's | Yield peptide b (μmol (%)) | Yield conjugate c (nmol (%)) |
| LB1_1 | RXRRXRILFQYRXRRXR | +8 | 8 | 2.7 (54) | 21.0 (70) |
| LB1_2 | RXRRXRYQFLIRXRRXR | +8 | 8 | 3.0 (59) | 21.0 (70) |
| LB1_3 | RXRRXRFLQIYRXRRXR | +8 | 8 | 2.6 (51) | 21.4 (71) |
| LB1_4 | RXRRXRAAAAARXRRXR | +8 | 8 | 3.0 (59) | 17.1 (57) |
| LB1_5 | RXRRXRIKFQYRXRRXR | +9 | 8 | 2.9 (58) | 19.6 (65) |
| LB1_6 | RXRRXRIEFQYRXRRXR | +7 | 8 | 3.7 (73) | 18.3 (61) |
| LB1_7 | RXRRXRIWFQYRXRRXR | +8 | 8 | 2.6 (51) | 20.9 (70) |
| LB1_8 | RXRRXRIPFQYRXRRXR | +8 | 8 | 4.0 (79) | 17.4 (58) |
| LB1_9 | KXRKXRILFQYKXRKXR | +8 | 4 | 3.4 (68) | 18.4 (61) |
| LB1_10 | HXRHXRILFQYHXRHXR | +4 | 4 | 1.7 (35) | 19.5 (65) |
| LB1_11 | SXRSXRILFQYSXRSXR | +4 | 4 | 3.2 (64) | 17.3 (58) |
| LB1_12 | EXREXRILFQYEXREXR | 0 | 4 | 2.0 (40) | 18.4 (61) |
| LB1_13 | RMKWKKILFQYRXRRXR | +8 | 5 | 1.7 (34) | 18.8 (63) |
| LB1_14 | RXRRXRILFQYRMKWKK | +8 | 5 | 2.8 (56) | 18.0 (60) |
| LB1_15 | KKWKMRILFQYRXRRXR | +8 | 5 | 1.5 (31) | 18.2 (61) |
| LB1_16 | RXRRXRILFQYKKWKMR | +8 | 5 | 2.8 (56) | 19.2 (64) |
aAll peptides contain a C-terminal amide and a 4-pentynoic acid group coupled to the N-terminus.
bCrude yield from a 5 μmol synthesis after Oasis HLB cartridge workup. Based on crude weight and corrected for TFA salts.
cYield of the LB1-PNA705 conjugate obtained via SELPEPCON (Schemes 1–3) from a 30 nmol scale synthesis based on N3-PNA705-S-S-biotin, determined by UV absorption at 260 nm.
Scheme 2. Synthesis of alkyne functionalized peptides for conjugation to N3-PNA705-S-S-biotin.

The peptides were deprotected and cleaved from the solid support by standard TFA treatment for 4 h with gentle mixing and rapidly isolated by solid phase extraction (SPE) using Oasis HLB cartridges. Good yields were achieved using this synthesis and purification method (Table 1). MALDI-TOF mass-spectrometry of the products showed in all cases a major signal corresponding to the expected peptide and only minor amounts of peptide impurities (ESI Table S1 and Fig. S1, S2 and S13, S14†). None of the observed shorter peptide impurities should form conjugates in the subsequent conjugation step because the alkyne linker is introduced in the final step of the synthesis and truncated peptides are capped after each coupling step during solid phase synthesis. Thus, time-consuming HPLC purification of each synthesized peptide is avoided. The peptide purification method used is fast, such that cleavage, deprotection and purification of 16 peptides can be carried out within one day, thus allowing for easy parallelization.
LB1–PNA705 conjugate library synthesis
Peptide-PNA conjugates were synthesized, purified and isolated in parallel using the procedure shown in Scheme 3. Copper(ii) sulphate in combination with sodium ascorbate proved to be the most promising catalyst system. Procedures involving copper(i) salts, such as copper(i) bromide or copper(i) iodide, were also tested but provided less reproducible conversions. Diisopropylethylamine was used as base to quench TFA salts of the cargo and peptides, and 2,6-lutidine and tris(benzyltriazolylmethyl)amine (TBTA) were utilized to stabilize the active copper(i) species. After 1 h, reactions were quenched by the addition of EDTA, diluted and immobilized on a streptavidin-functionalized Sepharose™-support held in SpinTrap™ centrifuge tubes. Following several washing steps, the conjugates were released from the support by addition of a TBS buffer solution containing 10 mM tris(2-carboxyethyl)phosphine (TCEP) followed by centrifugation, leaving the biotin tag on the support. The use of organic solvent (e.g. 40% acetonitrile) for dilution of the reaction mixtures, washing and release proved to be essential for good recovery of the conjugates.
Scheme 3. Outline of conjugation and purification strategy to obtain CPP–PNA705 conjugate libraries.

The resultant conjugates still contained buffer and TCEP, which were removed by SPE using Oasis HLB cartridges. The conjugates were obtained in good isolated yields (50–70%, Table 1). In all cases, analysis by MALDI-TOF mass-spectrometry showed the expected signal for the conjugation product (ESI Table S1 and Fig. S15–S17†). In addition, a signal corresponding to the unconjugated PNA705 was observed. Quantitative analysis by RP-HPLC revealed <10% of the unconjugated cargo. As the cargo by itself is unable to enter cells efficiently and has been demonstrated to be inactive in the HeLa pLuc705 cell splicing redirection assay (data not shown), the conjugates containing small amounts of unconjugated PNA705 were used without further purification.
Compared to standard methods of preparing CPP–PNA conjugates, SELPEPCON is very fast and convenient. For a library of 16 conjugates, parallel peptide synthesis and purification took 3 days and conjugation and purification took only two days including lyophilisation time. Thus, conjugates were available within a week and enough peptide remains for many more conjugation reactions. By contrast 16 individual conjugate syntheses, RP-HPLC purifications for each peptide, and subsequent conjugations as well as conjugate desalting would take a minimum of 48 hours each, excluding method optimization.
Splicing redirection activity of LB1–PNA705 conjugate library
The PNA705 conjugates of LB1 were assessed for their ability to enter HeLa pLuc705 cells and redirect splicing, resulting in up-regulation of luciferase expression. Attempts were made to use 96-well plates, instead of the commonly used 24-well plates, to allow for faster screening. However, the obtained results were hard to interpret because of large inconsistencies in luminescence values. More reliable and reproducible results were obtained with 48-well plates, which were used for all screening assays shown in this work. Several conjugates were purified by RP-HPLC and assayed, which provided similar results compared to the crude conjugates obtained by SELPEPCON (ESI Fig. S30†).
The full LB1–PNA705 conjugate library was assayed at a single-concentration (5 μM) and the results analysed as the fold increase in luminescence compared to buffer only (Fig. 2). Several of the LB1–PNA705 constructs were strongly active, as was expected for Arg-rich peptides. Substantial reduction in the number of Arg residues (e.g. multiple replacements by His, Ser or Glu) led to significantly lower activity (LB1_10–12). When the Arg residues were replaced by Lys, retaining the overall positive charge, some splicing redirection activity was retained (LB1_9 and 13–16). LB1–PNA705 conjugates differ slightly from CPP–PNA constructs previously synthesized by us.6,10 Thus, LB1–PNA conjugates contain an additional free Cys residue and the peptide to PNA conjugation is N- to N-terminus rather than C- to N-terminal conjugation used in previous work. However, LB1_1 containing the original Pip5h sequence showed similar splicing redirection activity to that of previously synthesised Pip5h-PNA705 (data not shown).
Fig. 2. Fold increase in luminescence caused by PNA705 conjugates of LB1-peptides compared to a buffer blank induced by the conversion of Beetle luciferin to oxyluciferin by expressed luciferase via splicing redirection in HeLa pLuc705 cells at 5 μM conjugate concentration.

Amongst the more active conjugates, variations in splicing redirection activity observed in the HeLa cells were not identical to those that might have been expected based on knowledge of exon skipping effects in mouse mdx muscle cells for which the Pip5 and the later Pip6 series were designed.10,11 Scrambling of the hydrophobic core (LB1_3), replacement by Ala residues (LB1_4), substitution of Leu by Pro (LB1_8) or a negatively charged Glu (LB1_6) all appeared to be detrimental to the activity (Fig. 2). Good splicing redirection activity was retained by reversal of the core sequence (LB1_2) or by replacement of a Leu by a Trp (LB1_7). The latter gives an IWFQ sequence also found in Penetratin, from which the hydrophobic core sequence of the Pip series was originally derived. Interestingly, the replacement of same Leu by a Lys residue significantly increased activity (LB1_5).
The results of CPP–PNA705 conjugate library based on LB1 demonstrate how SELPEPCON can be effective for study and optimization of the amino acid sequence of a CPP for delivery of a cargo into a particular cell system providing that a cellular assay is available.
Parallel synthesis and purification of LB2 CPP-library
We now wished to apply the SELPEPCON methodology to a significantly larger library in order to demonstrate its potential for selection of a CPP from a large range of sequence-dissimilar peptides, which would be necessary in the case of a new cargo candidate where relatively little was known about likely requirements for intracellular activity. Thus we designed peptide library LB2 consisting of 78 peptides that included many well-known CPPs, some sequences obtained from searching scientific literature, and some newly designed (Table S2, ESI†). The peptides in this library consisted of both hydrophobic and hydrophilic peptides, ranged in length from 6–28 amino acids and carried a net charge ranging from –1 to +13. The library consisted of standard l-amino acids with the addition of β-alanine (B) and ε-aminocaproic acid (X), which are commonly found in CPPs.2b,c
Since it was found that free thiol groups inhibited the copper-catalysed cycloaddition reaction, Cys residues could not be introduced using Cys(Trt), which is the standard protecting group used in peptide synthesis. Instead an alternative procedure was developed using S-tert-butylthio (StBu) protected l-cysteine, which contains a disulfide-protecting group that is stable to TFA cleavage but which is released in the reducing step of the conjugate workup procedure.
The library of CPPs (LB2) was synthesised in parallel using the procedure described above for LB1. Only standard synthesis protocols were used for the parallel synthesizer and a single method for purification and isolation was employed without any peptide specific optimization. Using these standard conditions, all but 5 of the 78 peptides (>90%) were successfully obtained, where the expected masses were seen as main signals in mass spectrometry (ESI Tables S2, S3 and Fig. S3–S12†). Because of the type of work-up procedure, small amounts of impurities were seen in most of the mass-spectra obtained for the successfully synthesised peptides. In 4 peptide syntheses a significant amount of shorter acetyl-capped peptide sequences was observed, which is the result of incomplete coupling reactions (Fig. S11 and S12†). Overall yields for the 73 successful syntheses were good (±70% average) and provided plenty of peptide material in principle for many different conjugation reactions on nanomole scale for this CPP library.
Since 20 peptides could be purified easily in parallel, the library was obtained in a total of only 9 days, including time for automated peptide assembly. Further parallelization to allow for the purification of more peptides simultaneously should lead to further decreases in this time.
LB2–PNA705 conjugate library synthesis
The LB2 peptides were conjugated to PNA705 using the procedure described previously for LB1 conjugations. Most conjugates were obtained in good yield (>60% average, ESI Tables S2 and S3†). As in the case of LB1, a small amount of unconjugated PNA was observed by MALDI-TOF mass-spectroscopy and HPLC for all conjugates obtained (ESI Fig. S18–S29†). Out of the 73 peptides where there was a peptide product available for conjugation, 10 conjugates were either not obtained or showed only small amounts of conjugate product compared to unconjugated PNA following affinity purification and desalting. The failed conjugations were practically all for the longer peptides that are likely to be more prone to form secondary structures, which may sterically inhibit the conjugation reactions.
In the cases of all 4 peptides (vide supra) that showed significant amounts of acetyl-capped peptide impurities, good quality conjugate products were nevertheless obtained. This demonstrates the effectiveness of the introduction of the alkyne linker in the final step of assembly in combination with acetic anhydride capping after each synthesis cycle during peptide synthesis, which allows for the removal of these peptide-impurities during affinity purification of the conjugates.
The four LB2–PNA705 conjugates that contained Cys residues (see Table S3, ESI†) showed by mass-spectrometry impurities that resulted from a statistical mixture of StBu modifications. Some modification was even observed of the Cys residue that remains attached to PNA705 after release from the affinity support, which was evidenced by the fact that conjugate masses were found corresponding to two StBu groups for conjugates that contained only one Cys residue in the peptide part. This may be because residual free StBu that is not effectively removed by the Oasis HLB cartridge workup of the conjugates can slowly allow StBu disulfides to form with available Cys residues in the conjugates. The StBu disulfide protecting groups are clearly cleaved off during the reduction step in SELPEPCON, since the alternative use of centrifugal filter units (Amicon Ultra 3 K 0.5 mL, Millipore Ireland Ltd) to remove salts, TCEP reducing agent and StBu did provide the main product conjugates without StBu modifications. However, this procedure resulted in low yields, especially for hydrophobic conjugates.
Once again the synthesis and purification procedures were very fast and effective. In addition to the good yields of the large majority of the conjugates, the conjugations and purifications involved in the whole library could be carried out within 8 days.
Splicing redirection activity of LB2–PNA705 conjugate library
The LB2–PNA705 conjugate library was screened at 5 μM concentration for their ability to enter HeLa pLuc705 cells and redirect splicing, resulting in up-regulation of luciferase expression (Fig. 3). A significant number of active conjugates were identified from this initial screen, showing 20- to 200-fold increases in luminescence values. In some cases, high error-bars were observed for conjugates that showed over a 20-fold increase, because conjugates could not all be tested in a single 48-well plate experiment and samples with high luminescence readouts show large variability between different experiments. However, the main purpose of the luciferase readout is to distinguish active from inactive conjugates.
Fig. 3. Fold increase in luminescence caused by PNA705 conjugates of LB2-peptides compared to a buffer blank induced by the conversion of Beetle luciferin into oxyluciferin by expressed luciferase via splicing redirection in HeLa pLuc705 cells at 5 μM conjugate concentration.

As a check on the reliability of purifications of conjugates several conjugates were purified by HPLC and tested for their ability to up-regulate luciferase expression (Fig. 4). None of the tested low-activity crude conjugates showed significantly higher activity after purification, revealing good reliability of the crude conjugates in this assay. The mildly active PNA705 conjugate of LB2_21 does show improved activity after purification. This can be explained because the crude conjugates contain a small amount (±10%) of unconjugated PNA, lowering the effective concentration of the LB2_21–PNA705. This highlights once again that the assay results using this crude library should only be used to find suitable active CPP-candidates for further investigation and active conjugates should not be directly compared.
Fig. 4. Fold increase in luminescence caused by PNA705 conjugates of LB2-peptides obtained by SELPEPCON (Crude) and RP-HPLC purified (Purified) compared to a buffer blank induced by the conversion of Beetle luciferin into oxyluciferin by expressed luciferase via splicing redirection in HeLa pLuc705 cells at 5 μM conjugate concentration.

Some interesting observations can be inferred from the results of the luciferase expression readouts. First, strong splicing redirection for the PNA705 cargo in this HeLa-cell assay is clearly directly related to the numbers of positive charges, particularly Arg residues, in the peptide part of the conjugates (Fig. 5a and 5b). A charge of at least +8 and at least 7–8 arginines are revealed as good CPPs properties. Association between the number of Arg residues and cell penetrating ability for CPP conjugates of PNA and PMO cargoes is well known from previous literature,12 but never before has such a large number and variety of CPPs of varying Arg content been tested in parallel. Interestingly several standard CPP sequences previously proposed for conjugation to PNA/PMO cargoes were found to be ineffective in this assay (for example, two containing Tat sequences LB2_30 and 52, and two containing Penetratin sequences LB2_28 and 35). These results show that effective cell penetration is cargo and cell dependent and demonstrates how SELPEPCON can be a valuable tool in the selection of a good CPP candidate for a particular application. An additional experiment was carried out in which all conjugates were evaluated in a single point assay to demonstrate how a first round of selection can be performed after a minimal screening effort (ESI Fig. S31†). Although several active conjugates give very high activity spikes, the same trend for active conjugates is observed as for the multiple data-point screening shown in Fig. 3.
Fig. 5. Fold increase in luminescence as a function of (a) the net charge and (b) the number of arginines of the CPPs in the LB2–PNA705 conjugates compared to a buffer blank induced by the conversion of Beetle luciferin into oxyluciferin by expressed luciferase via splicing redirection in HeLa pLuc705 cells at 5 μM conjugate concentration. The dots represent the data points and the line represents the average.

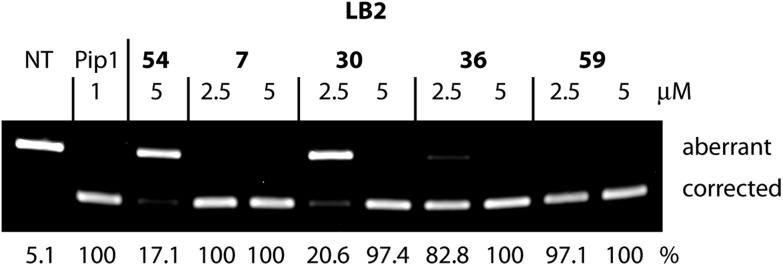
A follow-up to screening can provide further information on selected candidate conjugates. For example, initial “hits” can be confirmed by evaluation of splicing redirection at the RNA level.6 This assay is more reliable compared to the luciferase readout but is less suitable in a high-throughput mode because of the need for gel electrophoresis to separate and quantify DNA strands. An example is shown in Fig. 6 in which several LB2–PNA705 conjugates were assessed by extraction of RNA from the treated HeLa pLuc705 cells followed by RT-PCR. Previously reported Pip-1-PNA,6 prepared by conventional conjugation through a C- to N-terminal disulfide linkage, was used as a positive control. As expected, the splice corrected RNA levels were low when cells were exposed to LB_54–PNA705, which also shows very low activity in the luciferase readout. LB2_7 contains the same Pip1 sequence as the positive control. The PNA705 conjugate was N- to N-terminally linked as obtained through SELPEPCON and showed high splice correction activity at both 2.5 and 5 μM. At 5 μM fully spliced RNA levels are high for the PNA705 conjugates of LB2_30–PNA705, 36 and 59, while LB2_30 showed significantly lower luciferase activity. The saturation of the splicing redirection observed at 5 μM can explain the large variation in the luciferase activity assay. At 2.5 μM these conjugates show the same activity trend by RT-PCR as seen in the luciferase readout obtained at 5 μM, highlighting the difference in sensitivity between the two methods.
Fig. 6. RT-PCR analysis to measure aberrant and redirected RNA levels in HeLa pLuc705 cells after exposure to CPP–PNA705 conjugates at different concentrations. The activity is presented as a percentage of corrected RT-PCR product obtained from splicing redirection.
SELPEPCON assessment and application prospects
The SELPEPCON methodology described above has been exemplified by use of libraries of CPPs conjugated to a PNA705 cargo where a reliable cell assay is available to gauge the effectiveness of the peptides in delivering the cargo into the nucleus of HeLa cells. Clearly the methodology is in principle applicable to any bio-cargo capable of being functionalized by an azido group, such as PMO or other oligonucleotide types, siRNA, peptides or other biomolecules where there is a need to search for peptides that when conjugated to a cargo may enhance its delivery into cells or for cell or tissue targeting. For example, we are currently applying the SELPEPCON technology to cell delivery of a potential anti-cancer peptide known to poorly enter cells itself. The only limitation is that the constructs need to be compatible with the conjugation step. For example, a conjugation can fail for longer peptide sequences that contain strong secondary structures, as shown for several peptides in the LB2 library. This might be overcome by further optimizing the conjugation conditions, such as use of a denaturing agent in the conjugation step.
We chose to conjugate the N-terminus of peptides to the N-terminus of the cargo, since N-terminal peptide functionalization is technically easier and in addition capping of unreacted chains during synthesis reduces the possibility of shorter chain peptide impurities becoming conjugated to the cargo. Thus, 4 less pure peptides could nevertheless be conjugated to the PNA cargo and still lead to an acceptable conjugate product. However, the method is not limited to N-terminal conjugation via a N-terminal alkyne. To demonstrate this we synthesized two peptides based on LB1_1 containing a C-terminal alkyne (LB1_17 and LB1_18, Tables S4 and S5†). The alkyne was placed on the C-terminus by using N-Fmoc-l-bishomopropargylglycine-OH (Fmoc-Bpg-OH, Chiralix, Nijmegen, The Netherlands) during automated synthesis. Both peptides were obtained in good yield. The double glycine spacer of LB1_18 did not appear to be necessary to improve the conversion of the conjugation reaction, as both conjugates were readily obtained in good yield and purity. Note that it should also be possible to achieve conjugation at the C-terminus of PNA by use of an azido side-chain derivative of an Fmoc amino acid (such as Fmoc-l-azido-lysine) at the C-terminus and a Cys attached at the N-terminus of the PNA.
Following initial selection and verification of hits by the SELPEPCON methodology using a wide CPP library, such as LB2 described above, lead optimization would likely involve further rounds of SELPEPCON screening using narrower peptide libraries, as exemplified in LB1. Because the peptides in LB1 are relatively similar, conjugation efficiency and thus effective concentration and purity of the conjugates obtained by SELPEPCON will be less variable compared to a broad library such as LB2.
The resultant libraries are also suitable for other screening purposes such as comparisons of serum stabilities of the conjugates, if in vivo studies are to be contemplated. Once one or more candidate peptides have been chosen for the desired cargo, thereafter drug development might consist of investigation of alternative conjugation methods to vary the type of linkage between peptide and cargo, as well in some cases their respective orientations to each other. Commonly such studies are best carried out in an in vivo model of a disease state using conjugates prepared more conventionally on larger scale,6,9,12a,13 since pharmacological properties of conjugates may vary substantially depending on cargo type as well as on peptide sequence. It is possible that SELPEPCON could be used for preparing conjugates for assay by intramuscular injection (e.g. into a mouse mdx model of Duchenne muscular dystrophy8) where only μg quantities are needed. However, use of SELPEPCON should reduce considerably the time and effort required to obtain potential leads that are demonstrated to be capable of the required biological effect in a cell model, hitherto a major bottleneck.
Experimental
General
MALDI-TOF mass spectrometry was carried out using a Voyager DE Pro BioSpectrometry workstation. A stock solution of 10 mg mL–1 of α-cyano-4-hydroxycinnamic acid in 60% acetonitrile in water was used as matrix. Error bars are ±0.1%. Reversed phase HPLC purifications and analyses were carried out using a Varian 940-LC. A Nanodrop 2000 UV analyser (Thermo Scientific) was used for the quantification of PNA concentrations using the absorption at 260 nm with the following values to calculate absorption coefficients; C 6.6 cm–1 M–1, T 8.6 cm–1 M–1, A 13.7 cm–1 M–1, G 11.7 cm–1 M–1. TBTA (tris-(benzyltriazolylmethyl)amine) was synthesised according to a literature procedure.14 The synthesis of the Pip-1-PNA705 conjugate had been reported previously.6,13b
N3-PNA705-SH synthesis
The PNA705 sequence containing a C-terminal Cys and two flanking Lys residues (Lys-CCTCTTACCTCAGTTACA-Lys-Cys) was synthesised on a 50 μmol scale using a modified Liberty Peptide Synthesizer (CEM) according to a published procedure[10] using a Chem-Matrix solid support and Fmoc amino acid monomers (Novabiochem) or Fmoc (Bhoc) PNA monomers (PolyOrg Inc.). After the final deprotection, the support was removed from the synthesizer and 5-azidopentanoic acid was manually double coupled using a standard PyBop/NMM coupling reaction at room temperature for 2 × 15 min. The PNA was cleaved from the support and deprotected using TFA (1.5 mL) containing 10% triisopropylsilane/2.5% water and 1% phenol. The mixture was agitated for 2 h after which it was filtered and concentrated. The crude PNA was isolated by cold diethyl ether precipitation, dissolved in water, filtered and purified by HPLC. A Phenomenex Prep C18 Jupiter column (250 × 21.2 mm, 10 micron) was used with the following gradient (A: 0.1% TFA, B: 90% acetonitrile, 0.1% TFA) 0–2 min 7.5% B 2–20 min 7.5%–25% B 20–25 min 25%–90% B (retention time: 20.4 min). The fractions containing the desired PNA were combined and freeze-dried giving a fluffy white solid (yield 10–40% based on support loading). Mass, expected: m/z 5250.0 found: m/z 5256.9.
N3-PNA705-S-S-biotin construct
To a solution of 4 μmol PNA705 in 3 mL water, was added 8 μmol EZ-link HPDP-Biotin (Thermo Scientific) from a 3.7 μmol mL–1 stock in DMSO. To this solution 600 μl 2 M sodium acetate pH 7 was added and the resulting solution shaken for two hours. The reaction was quenched by the addition of 15 mL 0.1% TFA and 150 μl TFA in order to solubilise the precipitate formed. This solution was filtered and then purified by HPLC using a Phenomenex Prep C18 Jupiter reversed phase column (250 × 21.20 mm, 10 micron) with a the following gradient (A: 0.1% TFA, B: 90% MeCN 0.1% TFA) 0–2 min 12% B 2–25 min 12%–25% B 25–30 min 25%–90% (retention time: 25.0 min). The purified N3-PNA-705-S-S-biotin was obtained as a fluffy white powder in a typical yield of 60–80%. Mass, expected: m/z 5678.4 found: m/z 5687.1.
Peptide synthesis
Peptide library synthesis was carried out on a 5 μmol scale using an Intavis Parallel Peptide Synthesizer, applying standard Fmoc chemistry and following manufacturer's recommendations. The solid support was as supplied by Intavis (Tentagel, 0.2 mmol g–1). Double coupling steps were used with a PyBop/NMM coupling mixture followed by acetic anhydride capping after each coupling step. Terminal alkyne functionalization involved a standard coupling procedure using 4-pentynoic acid in the final step of the synthesis. The peptides were cleaved from the support and deprotected by addition of TFA (1.5 mL) containing 5% triisopropylsilane/2.5% water and 1% phenol with shaking for 4 h. The support suspension was then concentrated to a volume of ±500 μl using a flow of nitrogen and diluted with 5 mL water. After mixing, the resulting mixture was loaded on a 20 cc Oasis HLB cartridge (Waters), which was previously washed with acetonitrile (10 mL) and equilibrated with 0.1% TFA (3 × 10 mL). After loading, the cartridge was washed with 0.1% TFA (2 × 10 mL) and 5% acetonitrile in 0.1% TFA (2 × 10 mL). The peptide was eluted with 40% acetonitrile (10 mL). The solution obtained was freeze-dried and the yield was calculated using the weight obtained (Table 1), corrected for the amount of TFA salts based on the number of positive charges present in the peptide. For mass spectroscopy data see Tables S1 and S2.†
Conjugate synthesis
A mixture was prepared containing 30 nmol PNA from a stock solution in water (±2 mM), 150 nmol peptide from a stock solution in NMP (±10 mM), 0.2 μl 2,6-lutidine (1.7 μmol) and 1 μl diisopropylethylamine (5.7 μmol). 7.5 μl of 20 mM CuSO4–TBTA solution (150 nmol) premixed in a 1 : 1 mixture of DMSO/water and 10 μl of a 20 mM solution of sodium ascorbate (200 nmol) were added to this mixture. The mixture was left for one hour and quenched with 100 μl 0.2 M EDTA 40% acetonitrile and 800 μl TBS 40% acetonitrile. The solution obtained was vortexed and loaded in two batches of 500 μl on a streptavidin HP SpinTrap (GE Healthcare UK Limited) and each batch was incubated for 20 minutes while being mixed by inversion. The column was washed with 400 μl 0.1 M EDTA in TBS 40% acetonitrile and with 5 × 400 μl TBS 40% MeCN. The conjugate was released from the resin by 2 × 20 minutes reactions with 2 × 400 μl 10 mM TCEP in TBS 40% acetonitrile and the solid support washed with 200 μl 40% acetonitrile in TBS. The solutions collected were combined and freeze-dried. The solid was dissolved in 500 μl 20% acetonitrile and loaded on an equilibrated 1 cc Oasis HLB cartridge (Waters) together with 500 μl 0.1% TFA. The column was washed with 3 × 1 mL 0.1% TFA, 3 × 1 mL 5% acetonitrile 0.1% TFA and 1 × 1 mL 10% acetonitrile 0.1% TFA. These extensive washes are required to remove the TCEP from the cartridge. The conjugates are released using 500 μl 60% acetonitrile and diluted with 500 μl 0.1% TFA. The resulting solution was freeze-dried and dissolved in 500 μl water. The concentration was determined and the solution was stored in the freezer for use in cellular assays.
Splice-redirection assay
The splicing redirection assay was carried out similarly to a previously reported procedure with minor changes.6 The assay was performed using 48-well plates with cells seeded the previous day (7.5 × 104 cells per well) and a total volume 100 μl per well for the conjugates’ solutions in OptiMEM. Note that we found that 96-well plates tended to give too high a scatter probably because of the smaller volumes in pipetting. RT PCR experiments were carried out as reported in previous publications.12c All experiments were performed in triplicates, unless specifically stated as designed one-point experiments.
Conclusion
We have developed a novel rapid parallel synthesis methodology (SELPEPCON) for the synthesis of libraries of conjugates of peptides with a PNA cargo and assayed the conjugates in a splicing redirection assay in HeLa pLuc705 cells. Alkyne and azide functional groups, which are known to be compatible with the most commonly used bio-molecules, are readily introduced on peptide and cargo moieties respectively. The methodology is capable of robotization and is applicable to large libraries of peptide conjugates of biomolecules, such as oligonucleotides and their analogues, peptides, siRNA etc., and in amounts suitable for in vitro or cell screening. We expect the SELPEPCON method to have wide applicability in drug discovery.
Acknowledgments
P.D. acknowledges a fellowship grant from the MRC-Technology Development Gap Fund. The work was supported by the Medical Research Council (MRC programme number U105178803).
Footnotes
References
- (a) Sebbage V. Biosci. Horizons. 2009;2:64. [Google Scholar]; (b) Madani F., Lindberg S., Langel U., Futaki S., Graslund A. J. Biophys. 2011;2011:414729. doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Heitz F., Morris M. C., Divita G. Br. J. Pharmacol. 2009;157:195. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Simeoni F., Morris M. C., Heitz F., Divita G. Nucleic Acids Res. 2003;31:2717. doi: 10.1093/nar/gkg385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bechara C., Sagan S. FEBS Lett. 2013;587:1693. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]; (c) Langel U., Cell-Penetrating Peptides. Methods and Protocols, Humana Press, New York, 2011 [Google Scholar]
- (a) Lebleu B., Moulton H. M., Abes R., Ivanova G. D., Abes S., Stein D. A., Iversen P. L., Arzumanov A., Gait M. J. Adv. Drug Delivery Rev. 2008;60:517. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hassane F. S., Saleh A. F., Abes R., Gait M. J., Lebleu B. Cell. Mol. Life Sci. 2010;67:715. doi: 10.1007/s00018-009-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Singh Y., Murat P., Defrancq E. Chem. Soc. Rev. 2010;39:2054. doi: 10.1039/b911431a. [DOI] [PubMed] [Google Scholar]; (b) Craik D. J., Fairlie D. P., Liras S., Price D. Chem. Biol. Drug Des. 2013;81:136. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- (a) Stephanopoulos N., Francis M. B. Nat. Chem. Biol. 2011;7:876. doi: 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]; (b) Hermanson G. T., Bioconjugate techniques, Elsevier Inc., 2nd edn, 2008 [Google Scholar]
- Ivanova G. D., Arzumanov A., Abes R., Yin H., Wood M. J., Lebleu B., Gait M. J. Nucleic Acids Res. 2008;36:6418. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. H., Cho M. J., Kole R. Biochemistry. 1998;37:6235. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- (a) Tornøe C. W., Christensen C., Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]; (b) Kolb H. C., Finn M. G., Sharpless K. B. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Fabani M. M., Abreu-Goodger C., Williams D., Lyons P. A., Torres A. G., Smith K. G., Enright A. J., Gait M. J., Vigorito E. Nucleic Acids Res. 2010;38:4466. doi: 10.1093/nar/gkq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Saleh A. F., Betts C., Camelliti P., Seow Y., Ashraf S., Arzumanov A., Hammond S., Merritt T., Gait M. J., Wood M. J. Mol. Ther. 2011;19:1295. doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts C., Saleh A. F., Arzumanov A. A., Hammond S. M., Godfrey C., Coursindel T., Gait M. J., Wood M. J. Mol. Ther. Nucleic Acids. 2012;1:e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Abes S., Turner J. J., Ivanova G. D., Owen D., Williams D., Arzumanov A., Clair P., Gait M. J., Lebleu B. Nucleic Acids Res. 2007;35:4495. doi: 10.1093/nar/gkm418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Abes R., Arzumanov A., Moulton H., Abes S., Ivanova G., Gait M. J., Iversen P., Lebleu B. J. Pept. Sci. 2008;14:455. doi: 10.1002/psc.979. [DOI] [PubMed] [Google Scholar]; (c) Saleh A. F., Arzumanov A., Abes R., Owen D., Lebleu B., Gait M. J. Bioconjugate Chem. 2010;21:1902. doi: 10.1021/bc100275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Gogoi K., Mane M. V., Kunte S. S., Kumar V. A. Nucleic Acids Res. 2077;35:e139. doi: 10.1093/nar/gkm935. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Turner J. J., Williams D., Owen D. and Gait M. J., Curr. Protoc. Nucleic Acid Chem., 2006, 4.28.1 [DOI] [PubMed]; (c) Joshi R., Jha D., Su W., Engelmann J. J. Pept. Sci. 2011;17:8. doi: 10.1002/psc.1305. [DOI] [PubMed] [Google Scholar]
- Chan T. R., Hilgraf R., Sharpless K. B., Fokin V. V. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.