

Abstract
Aim:
Retigabine, an activator of KCNQ2-5 channels, is currently used to treat partial-onset seizures. The aim of this study was to explore the possibility that structure modification of retigabine could lead to novel inhibitors of KCNQ2 channels, which were valuable tools for KCNQ channel studies.
Methods:
A series of retigabine derivatives was designed and synthesized. KCNQ2 channels were expressed in CHO cells. KCNQ2 currents were recorded using whole-cell voltage clamp technique. Test compound in extracellular solution was delivered to the recorded cell using an ALA 8 Channel Solution Exchange System.
Results:
A total of 23 retigabine derivatives (HN31-HN410) were synthesized and tested electrophysiologically. Among the compounds, HN38 was the most potent inhibitor of KCNQ2 channels (its IC50 value=0.10±0.05 μmol/L), and was 7-fold more potent than the classical KCNQ inhibitor XE991. Further analysis revealed that HN38 (3 μmol/L) had no detectable effect on channel activation, but accelerated deactivation at hyperpolarizing voltages. In contrast, XE991 (3 μmol/L) did not affect the kinetics of channel activation and deactivation.
Conclusion:
The retigabine derivative HN38 is a potent KCNQ2 inhibitor, which differs from XE991 in its influence on the channel kinetics. Our study provides a new strategy for the design and development of potent KCNQ2 channel inhibitors.
Keywords: KCNQ2 channel, retigabine, HN38, XE991, structure modification, CHO cell, whole-cell recording
Introduction
The KCNQ family consists of five members termed KCNQ1 to KCNQ5 (or Kv7.1 to Kv7.5). The subthreshold activating potassium currents mediated by KCNQ channels play inhibitory roles in controlling cellular excitability1,2,3. Expression of the five isoforms is tissue specific. KCNQ1 is most abundantly expressed in cardiac tissue, but its expression in non-cardiac tissue should not be ignored4. The rest four isoforms (KCNQ2 to KCNQ5) are expressed mainly in the central and peripheral nervous systems, for example, hippocampal and dorsal root ganglion5. The co-assembly of KCNQ2 with KCNQ3 is believed to be the major component of the M-channel5. Augmentation of the M-current dampens membrane excitability3. Recently there is an increasing interest in the discovery of small molecular modulators of Kv channels. Such compounds can be derived either from synthetic sources or from natural products6,7. Both activators and inhibitors are essential as tools for elucidating the mechanism of channel activity and for developing therapeutic agents. Activators capable of augmenting KCNQ2 function have proven effective in animal models of convulsion, pain and neuropsychopathy3,6,8. Retigabine, a KCNQ2-5 activator, was approved for adjunctive treatment of partial-onset seizures in adult patients in 2011. Linopirdine, an inhibitor of the KCNQ2-3 channels, was studied initially in Alzheimer's disease due to its enhancement of acetylcholine release in cholinergic nerve terminals in the brain. Subsequent clinical trials failed, as treatment with linopirdine did not improve memory performance in elderly subjects. However, linopirdine has become a valuable pharmacological tool compound for KCNQ channel studies9,10,11. Two analogs of linopirdine that exhibit higher potency are XE991 and DMP 543, shown in Figure 1.
Figure 1.

Modulators of KCNQ channels.
During our search for activators of KCNQ2, we found that certain chemical modifications on the N-2 and N-3 positions of retigabine gave rise to an inhibitor of KCNQ2 channels. This compound (hit 1, Figure 2) inhibits the current of the KCNQ2 channel by 70%. This encouraged us to explore this class of compounds further. The most potent derivative (HN38) inhibited the KCNQ2 current by 92% at 10 μmol/L. The IC50 of HN38 is 0.10±0.05 μmol/L, which is significantly more potent than either linopirdine or XE99112.
Figure 2.

KCNQ inhibitor hit 1.
Materials and methods
Chemistry
All the compounds other than HN311 were synthesized according to Scheme 1. The procedure for the synthesis of HN311 is described in the experimental section. Compound 6 was synthesized from commercially available compound 5 and 4-fluorobenzaldehyde via reductive amination reaction. Compound 6 was then hydrogenatedly catalyzed by Raney-Ni. Intermediate compound 7 was treated with Boc2O to give 8. Reaction of compound 8 with the corresponding reagents produced 9a–f. Deprotection of 9a–f, followed by further treatment with the appropriate alkyl chloroformate, gave the final compounds HN36-HN310 and HN41-HN410. Compound HN21 was synthesized in the same way. Reductive amination using HN21 gave compounds HN31-HN35.
Scheme 1.

Synthesis of retigabine derivatives.
Reagents and conditions
a) 4-Fluorobenzaldehyde, cat. pTsOH, toluene, 120 °C, 77.2%; b) NaBH4, 1,4-dioxane:MeOH (4:1), rt, 94%; c) H2, Raney-Ni, MeOH, rt, 100%; d) Boc2O, NaHCO3, H2O:THF (1:2), rt, 85%; e) R2Br, DIPEA, 60 °C, DMF; f) (1) TFA, DCM, 0 °C; (2) Alkyl chloroformate, DIPEA, 1,4-dioxane, rt; g) Ethyl chloroformate, DIPEA, 1,4-dioxane, rt; h) Alkyl aldehyde, AcOH, then sodium triacetoxyborohydride, DCM, rt.
Electrophysiology experiment
Cell culture
Chinese hamster ovary (CHO) cells were grown in 50/50 DMEM/F-12 (Gibco™, Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS), and 2 mmol/L L-glutamine (Invitrogen™, Life Technologies, Carlsbad, CA, USA). To express KCNQ2 channels, cells were split for 24 h before transfection, plated in 60-mm dishes, and transfected with Lipofectamine 2000™ reagent (Invitrogen™, Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. At 24 h after transfection, cells were split and replated onto coverslips coated with poly-L-lysine (Sigma-aldrich, St Louis, MO, USA). A GFP cDNA (Amaxa, Gaithersburg, MD, USA) was cotransfected to identify the transfected cells by fluorescence microscopy.
Electrophysiological recording
Standard whole-cell recording was used to record KCNQ2 current in CHO cells. Pipettes were pulled from borosilicate glass capillaries (TW150-4, World Precision Instruments, Sarasota, FL, USA). When filled with the intracellular solution, the pipettes had resistances of 3–5 megaohms. During the recording, constant perfusion of extracellular solution was maintained using a BPS perfusion system (ALA Scientific Instruments). Pipette solution contained (in mmol/L): 145 KCl, 1 MgCl2, 5 EGTA,10 HEPES and 5 MgATP (pH 7.3); extracellular solution contained (in mmol/L): 140 NaCl, 3 KCl, 2 CaCl2, 1.5 MgCl2, 10 HEPES and 10 glucose (pH 7.4). Current and voltage were recorded using an Axopatch-200B amplifier, filtered at 2 kHz, and digitized using a DigiData 1440A with pClamp 10.2 software (Axon Instruments, Sunnyvale, CA, USA). Series resistance compensation was used and set to 60%–80%.
Compound application
XE991 and HN38 were dissolved in dimethylsulfoxide (DMSO) to prepare a 20 mmol/L stock solution, from which the appropriate volumes were added to the external solutions to produce the desired concentrations. DMSO (less than 0.1% in the final dilution) elicited no observable effect on the K+ currents. The testing concentration of each compound in electrophysiological recording was 10 μmol/L except otherwise stated. The external solution containing the drugs was delivered to the recorded cell using ALA 8 Channel Solution Exchange System (ALA Scientific Instruments Inc, Farmingdale, NY, USA).
Data analysis
Patch clamp data were processed using Clampfit 10.2 (Molecular Devices, Sunnyvale, CA, USA) and then analyzed in GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Voltage-dependent activation curves were fitted with the Boltzmann equation, G=Gmin+(Gmax−Gmin)/(1+exp(V−V1/2)/S), where Gmax is the maximum conductance, Gmin is the minimum conductance, V1/2 is the voltage for half-maximum conductance, and S is the slope factor. Dose-response curves were fitted with the Hill equation, E=Emax/(1−(EC50/C)P), where EC50 is the drug concentration producing half of the maximum response, and P is the Hill coefficient. The data are presented as the mean±SEM. Significance was estimated using Student's t test, where P<0.05 was considered significant.
Results
Structure and activity relationship of retigabine derivatives
The electrophysiology experiment was conducted using the whole-cell patch clamp technique and the effects on the amplitude of outward current (I/I0) were analyzed. I0 is the amplitude of outward current in the absence of a compound. I is the amplitude of outward current in the presence of a compound. Compounds leading to I/I0>1 were defined as activators, while compounds giving with I/I0<1 were defined as inhibitors. Compound concentration in the assay was 10 μmol/L and testing voltage was −10 mV, unless otherwise stated. We first evaluated the effect of substitutions on the N-2 and N-3 positions of retigabine. Both compounds HN21 and HN22 (Figure 3) lost inhibitory activities (data not shown). These results indicate that both N-2 and N-3 groups are important for inhibition. Other compounds and their effects on the KCNQ2 channel are shown in Table 1. The effects of substitutions at N-3 are shown by compounds HN31–HN310. Alkyl chains of moderate lengths appear to be preferred, as shown in HN33 and HN34, which are nearly equipotent as hit 1. Increasing or decreasing the length of the chain had a detrimental effect on activity (HN31 vs HN33, HN35 vs HN33). Incorporation of a methyl group at N-3 gave a weak agonist (HN31). Although saturated alkyl chains did not improve activity, allylic substitutions gave some promising results (HN36-HN311). The effect of branching on the allylic group was thus studied. Substitution with 3-methyl allylic (HN36) and 3,3-dimethyl allylic (HN37) groups afforded weak inhibitors, while introduction of a 2-methyl allylic (HN38) side chain caused 92% inhibition. This compares favorably with 70% inhibition shown by hit 1. A derivative with increased length of the side chain on the allylic group (HN39) was devoid of activity. A polar branching group was also investigated, giving rise to a compound with moderate activity (HN310). One-carbon homologation of HN39 gave HN311, a compound with attenuated activity. Compounds HN41-HN45 and HN46-HN410 enabled investigation of SAR for the N-1 and N-2 positions. The trends observed were similar to those seen in the two N-3 substituent series. The nature of the functional groups at N-1 and N-2 was very important. A critical difference was observed between the activity of methyl (HN41, HN46) and that of ethyl (HN38, hit 1) analogues, with HN41 and HN46 behaving as activators while HN38 and hit 1 as inhibitors. Increasing the length of the chain led to decreased activity (HN42). Reducing flexibility at these positions did not improve activity (HN43). Branching at α- or β-position of the substituents led to attenutated (HN44) or agonistic (HN45) activity.
Figure 3.

Chemical structures of compounds HN21-HN22.
Table 1. Structures of retigabine derivatives and effects on KCNQ2 channels.
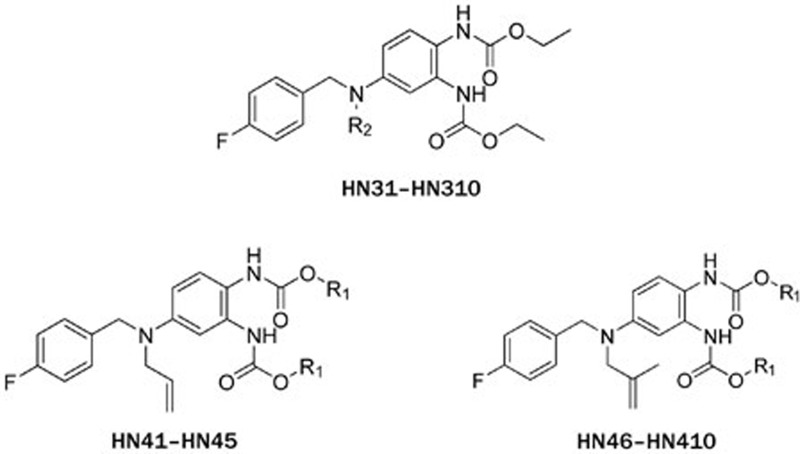
| Compound | R2 | I/I0[a] | Compound | R1 | I/I0[a] |
|---|---|---|---|---|---|
| HN31 | Me | 1.55±0.20 | HN41 | Me | 1.43±0.05 |
| HN32 | Et | 1.17±0.09 | HN42 | nPr | 0.16±0.04 |
| HN33 | nPr | 0.28±0.10 | HN43 | Allyl | 0.24±0.10 |
| HN34 | nPen | 0.36±0.12 | HN44 | iBu | 0.50±0.14 |
| HN35 | nBu | 1.01±0.05 | HN45 | tBu | 1.24±0.10 |
| HN36 | 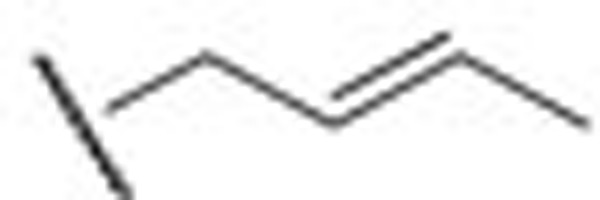 |
0.87±0.06 | HN46 | Me | 1.14±0.20 |
| HN37 | 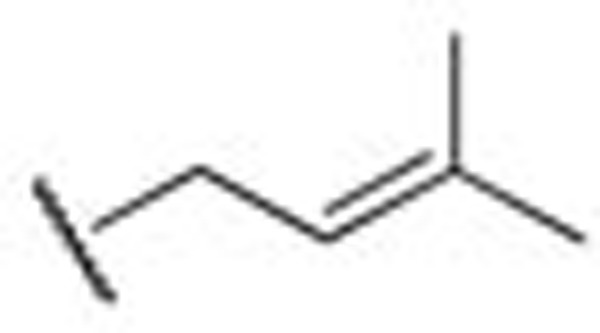 |
0.66±0.16 | hit 1 | Et | 0.30±0.01 |
| HN38 | 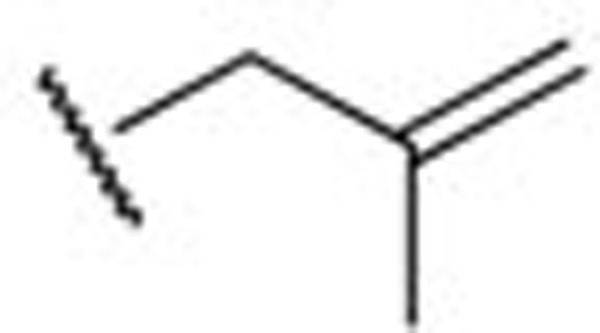 |
0.08±0.02 | HN47 | nPr | 0.27±0.12 |
| HN39 | 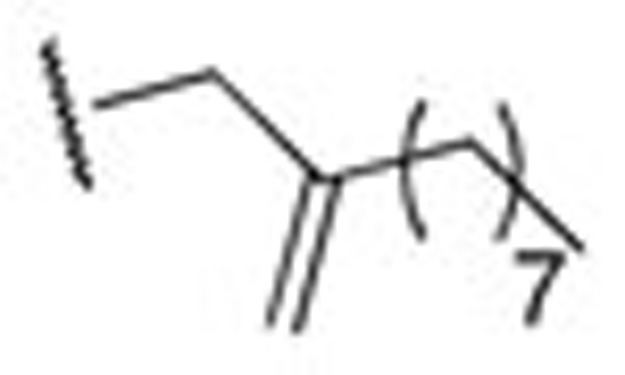 |
1.18±0.05 | HN48 | Allyl | 0.17±0.07 |
| HN310 | 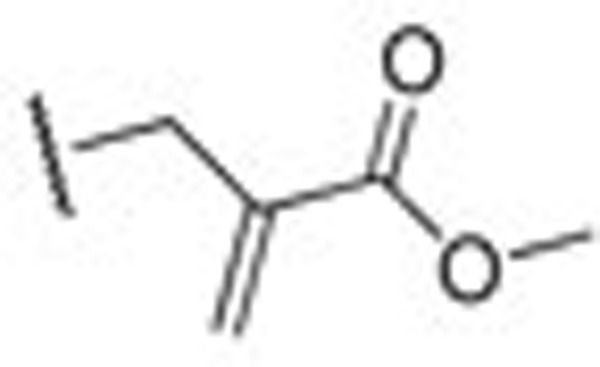 |
0.24±0.02 | HN49 | iPr | 0.24±0.10 |
| HN311 | 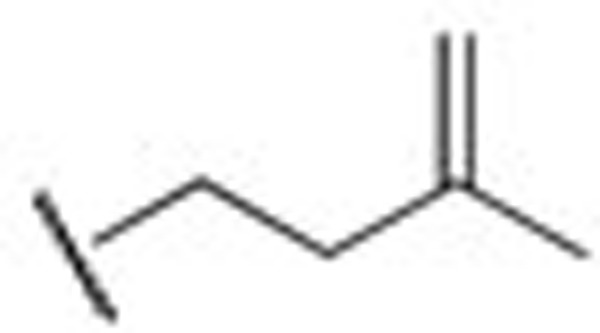 |
0.29±0.10 | HN410 | iBu | 1.08±0.03 |
[a] The testing concentration was 10 μmol/L. Each compound was tested in more than three cells.
Electrophysiological results
Inhibitory effects on KCNQ2 channels by HN38
Compound HN38 was selected to be tested further based on its potent inhibition of the KCNQ2 channel. Figure 4A shows that the inhibition of KCNQ2 current by HN38 is concentration-dependent. Analysis of the dose-response curves revealed an IC50 value of 0.10±0.05 μmol/L (Figure 4B). This value shows that HN38 is 7-fold more potent than XE991 and 40-fold more potent than linopirdine12.
Figure 4.

Dose-response relationship of HN38 on KCNQ2 channels. (A) Representative current traces of KCNQ2 in the absence or presence of HN38 (1, 3, or 10 μmol/L, as indicated). To elicit the currents, holding potential was set at −80 mV followed by 1500 ms depolarization step to −10 mV. A hyperpolarization step to −120 mV was used to record the tail current. (B) Dose-response curve of HN38 on KCNQ2 channels.
Comparison of HN38 with XE991
We next compared the effects of HN38 and XE991 on the KCNQ2 channel. Homomeric KCNQ2 mediates a characteristic outward current with slow activation and deactivation (Figures 5A and 5B). In the presence of 3 μmol/L HN38 or XE991, steady-state outward currents were significantly inhibited at all applied depolarizing voltages. Figure 5C shows the current-voltage relationships of the KCNQ2 channel in the absence and presence of 3 μmol/L HN38 or 3 μmol/L XE991. Then the effects of HN38 and XE991 on the activation and deactivation kinetics of the KCNQ2 channel were analyzed. HN38 at 3 μmol/L had no detectable effect on channel activation (Figures 5D and 5F), while it accelerated deactivation in hyperpolarizing voltages (Figures 5E and 5G). After application of HN38, the time constant of deactivation was accelerated from 38.3±6.6 ms to 17.2±4.2 ms (n=3, P<0.05). In contrast, XE991 did not affect the kinetics of either channel activation or deactivation (Figures 5D–5G).
Figure 5.

Effects of HN38 and XE991 on KCNQ2 channels. Representative traces of KCNQ2, before and after application of 3 μmol/L HN38 (A) or 3 μmol/L XE991 (B). The holding potential was −80 mV, followed by a series of 1500 ms depolarization steps from −70 to +50 mV in 10 mV increments. A 1000 ms hypopolarization step to −120 mV was then applied to record the tail current. (C) I–V curves in the absence (open circle) and presence (filled circle) of 3 μmol/L HN38 or presence (filled triangle) of 3 μmol/L XE991 as indicated. (D) Normalized activation phase from full traces in the absence and presence of 3 μmol/L HN38 (left) or 3 μmol/L XE991 (right). The testing depolarization was −10 mV. (E) Normalized deactivation phase from full traces in the absence and presence of 3 μmol/L HN38 (left) or 3 μmol/L XE991 (right). The testing depolarization was −120 mV. (F) Histogram summarized the effects of 3 μmol/L HN38 and 3 μmol/L XE991 on activation of KCNQ2 channels. (G) Histogram summarized the effects of 3 μmol/L HN38 or 3 μmol/L XE991 on deactivation of KCNQ2 channels (n>3, bP<0.05 vs control).
Discussion
XE991 and linopirdine are two classical KCNQ inhibitors and exhibit potent inhibition of both recombinant and native M-currents5. DMP 543 is a derivative of linopirdine. Although these three compounds have shown potential in pre-clinical models of memory enhancement, the results of the clinical trials for Alzheimer's disease were equivocal. This result may reflect the suboptimal pharmacokinetics and distribution properties of these drugs11. UCL2077 and ML252 are KCNQ2 inhibitors that have been reported recently13,14. Of all the KCNQ2 inhibitors reported to date, only ML252, a synthetic compound discovered from a high-throughput screen of 300 000 compounds, shows potency comparable to HN38. The IC50 of ML252 against KCNQ2 is approximately 70 nmol/L14. Detailed kinetics studies suggest the compound HN38 might belong to a different class of inhibitor of KCNQ2 channels from XE991. It accelerated the deactivation phase of KCNQ2 channels significantly while XE991 exhibited negligible effects on the deactivation. As a derivative of retigabine, an approved anti-epileptic drug, compound HN38 with its potent efficiency might be a good leading compound for clinical development of KCNQ inhibitors.
Conclusion
In summary, we have discovered a series of KCNQ2 inhibitors based on SAR studies of retigabine, an approved anti-epileptic drug (Figure 1). HN38, the most potent one among the direvatives, exhibits an IC50 value of 0.10±0.05 μmol/L, which is 7-fold potent than XE991 and 40-fold potent than linopirdine. Our work provides a new strategy for the design and development of potent KCNQ2 channel inhibitors.
Experimental section
Representative procedure for the synthesis of compounds HN21, hit 1, HN36-HN10, and HN41- HN410
To a solution of 2-nitro-1,4-phenylenediamine 5 (7.65 g, 0.05 mol) and PTSA (0.25 g) in toluene (200 mL), was added 4-fluorobenzaldehyde (6.8 g, 55.0 mmol). The stirred reaction mixture was heated to reflux under a water sepatator for 3 h. The mixture was filtered quickly through a Büchner funnel packed with silica gel. The filtrate was stirred and allowed to cool to room temperature over 6 h. The orange solid was filtered and washed with petroleum ether (3×15 mL) to give an orange solid (10.0 g, yield: 77.2%). The resulting solid was suspended in a mixture of 1,4-dioxane (40 mL) and methanol (10 mL). To this suspension, was added three separate portions of NaBH4 (1.2 g, 31.6 mmol) at 15-min intervals. The reaction was allowed to stir at room temperature for 2 h, and quenched with water (200 mL). The resulting solid was collected by filtration, washed with water, and dried under vacuum to give compound 6 (4.91 g, yield: 94%). 1H NMR (300 MHz, CDCl3): δ 7.31 (t, J=8.1 Hz, 3H), 7.03 (t, J=8.7 Hz, 2H), 6.84 (dd, J=8.7, 2.7 Hz, 1H), 6.69 (d, J=9.0 Hz, 1H), 5.74 (s, 2H), 4.25 (d, J=5.1 Hz, 2H); LC-MS m/z (%): 284.1 (100) [M+Na]+.
Compound 6 (2.61 g, 10.0 mmol) was hydrogenated under atmospheric pressure of hydrogen for 3 h in MeOH (30 mL) catalysed by Raney-Ni (10%, 261 mg). The catalyst was filtered off and the filtrate was concentrated. The product, compound 7, should be used immediately in the next reaction. To a solution of 7 (1.0 g, 4.32 mmol) in THF: H2O (40 ml, 2:1, v/v), was added Boc2O (2.83 g, 12.96 mmol) and NaHCO3 (1.16 g, 12.96 mmol). The mixture was stirred at room temperature overnight. The mixture was extracted with EtOAc (3×30 mL). The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Purification (5:1 petroleum ether: EtOAc, v/v) by silica gel column chromatography gave compound 8 (1.67 g, yield: 89.5%). 1H NMR (300 MHz, CDCl3): δ 7.28 (t, J=8.7 Hz, 2H), 7.00 (t, J=8.7 Hz, 3H), 6.89 (s, 1H), 6.26 (d, J=8.7 Hz, 1H), 4.25 (s, 2H), 1.48 (s, 18H); LC-MS m/z (%): 454.2 (100) [M+Na]+.
To a solution of 8 (150 mg, 0.348 mmol) in DMF (3 mL), was added allyl bromide (39.0 μL, 0.452 mmol) and DIPEA (99.0 mg, 0.766 mmol). The mixture was stirred at 60 °C for 2 h. The mixture was extracted with EtOAc (3×10 mL). The combined organic phases were washed with water and brine, dried over sodium sulfate and then concentrated in vacuo. Purification (4:1, petroleum ether: EtOAc, v/v) by silica gel column chromatography gave product 9a (143 mg, yield: 87.2%). 1H NMR (300 MHz, CDCl3): δ 7.18 (t, J=6.9 Hz, 2H), 6.90–7.06 (m, 4H), 6.49 (s, 1H), 6.48 (d, J=8.7 Hz, 1H), 5.79–5.88 (m, 1H), 5.18 (s, 1H), 5.13 (s, 1H), 4.44 (s, 2H), 3.92 (d, J=3.6Hz, 2H), 1.49 (s, 18H); 13C NMR (75 MHz, CDCl3): δ 163.5, 160.3, 154.9, 153.5, 147.5, 134.4, 134.3, 133.4, 128.4, 128.3, 127.0, 116.7, 115.5, 115.2, 80.4, 53.4, 53.1, 28.3; LC-MS m/z (%): 494.3 (100) [M+Na]+.
To a solution of 9a (80 mg, 0.17 mmol) in DCM (0.2 mL), was added TFA (1.0 mL), and the mixture was stirred at room temperature for 1 h. The mixture was concentrated in vacuo. The residue was taken up in 1,4-dioxane (10 mL) and to this solution was added DIPEA (66.0 mg, 0.51 mmol). Ethyl chloroformate (18 mg, 0.17 mmol) was added dropwise as a solution in 1,4-dioxane (2.0 mL), and the mixture was stirred at room temperture for 1 h. The mixture was extracted with EtOAc (3×10 mL). The combined organic phases were washed with water and brine, dried over sodium sulfate and concentrated in in vacuo. Purification (4:1 to 2:1, petroleum ether: EtOAc, v/v) by silica gel column chromatography gave HN22 (33.6 mg, yield: 58%). 1H NMR (300 MHz, CDCl3): δ 7.18 (t, J=8.1 Hz, 2H), 6.99 (t, J=8.4 Hz, 2H), 6.91 (d, J=8.7 Hz, 1H), 6.12 (d, J=8.7Hz, 2H), 6.06 (d, J=2.1 Hz, 1H), 5.80-5.86 (m, 1H), 5.19 (s, 1H), 5.14 (d, J=3.0 Hz, 1H), 4.44 (s, 2H), 4.20 (q, J=6.9 Hz, 2H), 3.91 (d, J=4.5 Hz, 2H), 3.59 (brs, 2H), 1.28 (t, J=6.9 Hz, 3H); LC-MS m/z (%): 366.2 (100) [M+Na]+.
Similar methods were used to synthesize compounds hit 1, HN36-HN310, and HN41-HN410
hit 1:
1H NMR (300 MHz, CDCl3): δ 7.18 (dd, J=8.4, 5.4 Hz, 2H), 7.09 (brs, 1H), 6.99 (t, J=8.4 Hz, 2H), 6.60 (m, 2H), 5.96 (m, 2H), 5.67 (m, 1H), 5.47 (m, 1H), 5.35 (d, J=17.4 Hz, 2H) 5.26 (d, J=12.0 Hz, 2H), 4.65 (d, J=5.7 Hz, 4H), 4.48 (s, 2H), 3.95 (d, J=5.7 Hz, 2H), 1.67 (d, J=6.3 Hz, 6H); LC-MS m/z (%): 438.2 (100) [M+Na]+.
HN36:
1H NMR (300 MHz, CDCl3): δ 7.18 (dd, J=7.5, 5.7 Hz, 2H), 7.06 (d, J=8.7 Hz, 1H), 6.99 (t, J=8.7 Hz, 2H), 6.42 (s, 1H), 6.42 (d, J=8.7 Hz, 2H), 5.48–5.62 (m, 2H), 4.45 (s, 2H), 4.20 (m, 4H), 3.88 (d, J=4.5 Hz, 2H), 1.57 (d, J=6.0 Hz, 2H), 1.24–1.32 (m, 6H); LC-MS m/z (%): 452.2 (100) [M+Na]+.
HN37:
1H NMR (300 MHz, CDCl3): δ 7.19 (dd, J=8.4, 5.7 Hz, 2H), 7.07 (d, J=8.7 Hz, 1H), 6.98 (t, J=8.4 Hz, 3H), 6.41 (s, 1H), 6.41 (dd, J=8.7, 2.4 Hz, 2H), 5.23 (t, J=6.3 Hz, 2H), 4.43 (s, 2H), 4.12–4.24 (m, 4H), 3.90 (d, J=6.0 Hz, 2H), 1.71 (s, 3H), 1.62 (s, 3H), 1.28 (t, J=7.2 Hz, 6H); LC-MS m/z (%): 466.2 (100) [M+Na]+.
HN38:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.7, 5.4 Hz, 2H), 7.06 (d, J=9.0 Hz, 1H), 6.99 (t, J=8.7 Hz, 2H), 6.94 (s, 1H), 6.38 (s, 1H), 6.38 (dd, J=8.7, 2.4 Hz, 2H), 4.97 (m, J=6.3 Hz, 2H), 4.88 (s, 1H), 4.81 (s, 1H), 4.50 (s, 2H), 4.11–4.23 (m, 4H), 3.84 (s, 2H), 1.73 (s, 3H), 1.23–1.31 (m, 6H); LC-MS m/z (%): 452.2 (100) [M+Na]+.
HN39:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.4, 5.4 Hz, 4H), 7.06 (d, J=9.3 Hz, 2H), 6.99 (t, J=8.7 Hz, 2H), 6.88 (s, 1H), 6.38 (dd, J=9.0, 2.7 Hz, 1H), 6.38 (s, 1H), 4.87 (s, 1H), 4.82 (s, 1H), 4.50 (s, 2H), 4.13–4.22 (m, 4H), 3.86 (s, 2H), 2.00 (t, J=7.2 Hz, 2H), 1.43–1.48 (m, 2H), 1.23–1.35 (m, 10H), 0.89 (t, J=7.5 Hz, 3H); LC-MS m/z (%): 550.3 (100) [M+Na]+.
HN310:
1H NMR (300 MHz, CDCl3): δ 7.17 (t, J=8.7 Hz, 2H), 7.08 (d, J=9.0 Hz, 1H), 6.99 (t, J=8.4 Hz, 2H), 6.91 (s, 1H), 6.73 (s, 1H), 6.32–6.40 (m, 2H), 6.30 (s, 1H), 5.63 (s, 1H), 4.50 (s, 2H), 4.18–4.23 (m, 6H), 3.83 (s, 3H), 1.85–1.91 (m, 6H); LC-MS m/z (%): 496.2 (100) [M+Na]+.
HN41:
1H NMR (300 MHz, CDCl3): δ 7.21 (dd, J=8.7, 5.7 Hz, 2H), 7.09 (d, J=9.0 Hz, 1H), 7.03 (t, J=8.7 Hz, 2H), 6.41 (s, 1H), 6.41 (d, J=8.7 Hz, 2H), 4.92 (s, 1H), 4.85 (s, 1H), 3.88 (s, 2H), 3.78 (s, 3H), 3.77 (s, 3H), 1.77 (s, 3H); LC-MS m/z (%): 424.2 (100) [M+Na]+.
HN42:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.7, 5.4 Hz, 2H), 7.06 (d, J=9.0 Hz, 1H), 6.99 (t, J=8.7 Hz, 2H), 6.95 (s, 1H), 6.36–6.41 (m, 2H), 4.88 (s, 1H), 4.81 (s, 1H), 4.50 (s, 2H), 4.04–4.12 (m, 4H), 3.84 (s, 2H), 1.73 (s, 3H), 1.63–1.69 (m, 4H), 0.91–0.99 (m, 6H); m/z (%): 480.2 (100) [M+Na]+.
HN43:
1H NMR (300 MHz, CDCl3): δ 7.16 (dd, J=8.7, 5.4 Hz, 2H), 7.07 (d, J=9.0 Hz, 1H), 6.99 (t, J=8.7 Hz, 2H), 6.39 (s, 1H), 6.39 (dd, J=8.7, 2.4 Hz, 2H), 5.90–5.97 (m, 2H), 5.34 (d, J=17.1 Hz, 2H), 5.25 (d, J=10.2 Hz, 2H), 4.88 (s, 1H), 4.81 (s, 1H), 4.61–4.68 (m, 4H), 4.51 (s, 2H), 3.84 (s, 2H), 1.73 (s, 3H); LC-MS m/z (%): 476.2 (100) [M+Na]+.
HN44:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.4, 5.7 Hz, 2H), 7.06 (d, J=9.0 Hz, 1H), 6.99 (t, J=8.7 Hz, 2H), 6.86 (s, 1H), 6.38 (s, 1H), 6.38 (dd, J=8.7, 2.4 Hz, 2H), 4.94–4.99 (m, J=6.3 Hz, 2H), 4.88 (s, 1H), 4.81 (s, 1H), 4.51 (s, 2H), 3.88–3.93 (m, 6H), 3.84 (s, 2H), 1.91–1.97 (m, 2H), 0.93 (d, J=6.9 Hz, 6H); LC-MS m/z (%): 508.3 (100) [M+Na]+.
HN45:
1H NMR (300 MHz, CDCl3): δ 7.16 (dd, J=7.5, 5.7 Hz, 2H), 7.06 (d, J=8.7 Hz, 1H), 6.98 (t, J=8.4 Hz, 2H), 6.91 (brs, 1H), 6.74 (s, 1H), 6.37 (s, 1H), 6.37 (d, J=8.7 Hz, 2H), 4.87 (s, 1H), 4.80 (s, 1H), 4.49 (s, 2H), 3.83 (s, 2H), 1.72 (s, 3H), 1.48 (s, 18H); LC-MS m/z (%): 508.3 (100) [M+Na]+.
HN46:
1H NMR (300 MHz, CDCl3): δ 8.69 (brs, 2H), 7.58 (s, 1H), 7.46 (d, 1H), 7.40 (d, J=9.0, 8.7 Hz, 1H), 7.18 (dd, J=8.1, 5.4 Hz, 2H), 5.74–5.83 (m, 1H), 5.37 (d, J=5.7 Hz, 1H), 5.33 (s, 1H), 4.58 (s, 2H), 4.15 (d, J=5.7 Hz, 2H), 3.79 (s, 6H); LC-MS m/z (%): 410.2 (100) [M+Na]+.
HN47:
1H NMR (300 MHz, CDCl3): δ 7.19 (dd, J=8.4, 5.4 Hz, 2H), 6.95–7.08 (m, 5H), 6.39–6.43 (m, 2H), 5.79–5.91 (m, 1H), 5.20 (s, 1H), 5.14 (d, J=3.0 Hz, 1H), 4.48 (s, 2H), 4.08–4.13 (m, 4H), 3.96 (d, J=4.5 Hz, 2H), 1.65–1.72 (m, 4H), 0.95 (t, J=7.5 Hz, 6H); LC-MS m/z (%): 466.2 (100) [M+Na]+.
HN48:
1H NMR (300 MHz, CDCl3): δ 7.34 (brs, 2H), 6.80–7.24 (m, 6H), 6.87 (d, J=8.7 Hz, 1H), 5.85–5.98 (m, 3H), 5.10–5.38 (m, 6H), 4.45–4.71 (m, 4H), 4.50 (s, 2H), 4.03 (d, J=4.2 Hz, 2H); LC-MS m/z (%): 462.2 (100) [M+Na]+.
HN49:
1H NMR (300 MHz, CDCl3): δ 7.28 (brs, 2H), 7.18 (dd, J=8.4, 5.4 Hz, 2H), 7.10 (s, 1H), 6.62 (s, 1H), 6.63 (d, J=7.2 Hz, 1H), 5.79–5.83 (m, 1H), 5.26 (s, 1H), 5.22 (d, J=3.0 Hz, 1H), 4.52 (s, 2H), 4.04 (d, J=5.1 Hz, 2H), 3.90–3.96 (m, 4H), 1.93–1.99 (m, 2H), 0.85–0.89 (m, 12H); LC-MS m/z (%): 466.2 (100) [M+Na]+.
HN410:
1H NMR (300 MHz, CDCl3): δ 7.18 (dd, J=8.1, 5.7 Hz, 2H), 7.07 (d, J=8.7 Hz, 1H), 6.98 (t, J=8.7 Hz, 2H), 6.73 (s, 1H), 6.40 (d, J=6.6 Hz, 1H), 6.29 (brs, 1H), 5.18 (s, 1H), 5.13 (d, J=5.4 Hz, 1H), 4.46 (s, 2H), 3.94 (d, J=4.5 Hz, 2H), 1.46 (s, 18H); LC-MS m/z (%): 494.3 (100) [M+Na]+.
Preparation of HN21
To a solution of 7 (1.0 g, 4.32 mmol) in 1,4-dioxane (30 mL) cooled in an ice bath, was added DIPEA (836 mg, 6.48 mmol). Ethyl chloroformate (703 mg, 6.48 mmol) was added dropwise as a solution in 1,4-dioxane (20 mL) and the mixture was stirred for 1 h. The mixture was extracted with EtOAc (3×30 mL). The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Purification (5:1 petroleum ether: EtOAc, v/v) by silica gel column chromatography gave HN21 (1.13 g, yield: 68%). 1H NMR (300 MHz, CDCl3): δ 7.31 (dd, J=8.1, 5.7 Hz, 2H), 7.08 (brs, 2H), 6.95 (t, J=8.7 Hz, 2H), 6.44 (brs, 1H), 6.28 (d, J=6.6 Hz, 1H), 4.24 (s, 2H), 4.06–4.16 (m, 5H), 0.87–0.97 (m, 6H); LC-MS m/z (%): 398.2 (100) [M+Na]+.
Preparation of HN31-HN35
To a solution of HN21 (75 mg, 0.20 mmol) in DCM (2 mL) was added formaldehyde (75 μL, 40% in water, w/w), one drop of acetic acid and sodium triacetoxyborohydride (43 mg, 0.20 mmol). The mixture was stirred at room temperature for 10 h, then quenched with saturated aqueous K2CO3 (3 mL). The mixture was extracted with EtOAc (3×10 mL). The combined organic phases were washed with water and brine, dried over sodium sulfate and concentrated in vacuo. Purification (4:1 to 2:1 petroleum ether: EtOAc, v/v) by silica gel column chromatography gave HN31 (47.6 mg, yield: 61%). 1H NMR (300 MHz, CDCl3): δ 7.18 (dd, J=8.1, 5.4 Hz, 2H), 7.08 (t, J=8.4 Hz, 1H), 6.99 (d, J=8.7 Hz, 2H), 6.45 (d, J=8.7 Hz, 1H), 6.40 (brs, 1H), 4.47 (s, 2H), 4.17–4.24 (m, 4H), 3.98 (s, 3H), 3.59 (brs, 2H), 1.28 (t, J=6.9 Hz, 6H); LC-MS m/z (%): 412.2 (100) [M+Na]+.
Similar methods were used to synthesize of compounds HN32-HN35
HN32:
1H NMR (300 MHz, CDCl3): δ 7.18 (dd, J=8.7, 5.7 Hz, 2H), 7.11 (s, 1H), 7.02 (d, J=8.4 Hz, 2H), 6.99 (t, J=8.7 Hz, 2H), 6.38 (dd, J=9.0, 2.7 Hz, 1H), 6.37 (brs, 1H), 4.45 (s, 2H), 4.16–4.23 (m, 4H), 3.42 (q, J=7.2 Hz, 2H), 1.28 (t, J=6.9 Hz, 6H), 1.18 (t, J=7.2 Hz, 3H); LC-MS m/z (%): 426.2 (100) [M+Na]+.
HN33:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.1, 5.4 Hz, 2H), 7.02–7.05 (m, 2H), 6.95 (t, J=8.4 Hz, 2H), 6.34–6.38 (m, 2H), 4.48 (s, 2H), 4.15–4.23 (m, 4H), 3.31 (t, J=7.2 Hz, 2H), 1.65 (m, 2H), 1.28 (t, J=7.2 Hz, 6H), 0.92 (t, J=7.2 Hz, 3H); LC-MS m/z (%): 440.2 (100) [M+Na]+.
HN34:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.4, 5.4 Hz, 2H), 6.94–7.06 (m, 5H), 6.35–6.38 (m, 2H), 4.47 (s, 2H), 4.15–4.23 (m, 4H), 3.42 (t, J=7.2Hz, 2H), 1.56–1.66 (m, 2H), 1.33–1.45 (m, 8H), 0.93 (t, J=7.2 Hz, 3H); LC-MS m/z (%): 454.2 (100) [M+Na]+.
HN35:
1H NMR (300 MHz, CDCl3): δ 7.17 (dd, J=8.7, 5.4 Hz, 2H), 6.94–7.06 (m, 5H), 6.35–6.39 (m, 2H), 4.47 (s, 2H), 4.15–4.23 (m, 4H), 3.33 (t, J=7.2 Hz, 2H), 1.57–1.68 (m, 2H), 1.28–1.37 (m, 10H), 0.89 (t, J=7.2 Hz, 3H); LC-MS m/z (%): 468.2 (100) [M+Na]+.
Preparation of HN311 (Scheme 2)
Scheme 2.

Synthesis of HN311.
Reagents and conditions: (i) Methyl vinyl ketone, Et3N, MeOH, reflux; (j) Methyltriphenylphosphonium bromide, n-BuLi, THF, −78 °C;
To a solution of HN21 (150 mg, 0.40 mmol) in MeOH (5 mL), was added methyl vinyl ketone (MVK) (49 μL, 0.6 mmol) and Et3N (115 μL, 0.8 mmol), and the mixture was refluxed for 10 h. The mixture was concentrated in vacuo. Purification (4:1, petroleum ether: EtOAc, v/v) by silica gel column chromatography gave compound 10 (130 mg, yield 73%). 1H NMR (300 MHz, CDCl3): δ 7.15 (dd, J=8.7, J=5.4 Hz, 2H), 7.05 (d, J=8.4 Hz, 1H), 6.97 (t, J=8.4 Hz, 2H), 6.43 (brs, 1H), 6.35 (dd, J=8.4, 2.4 Hz, 2H), 4.49 (s, 2H), 4.18 (m, 4H), 3.65 (t, J=6.9 Hz, 2H), 2.78 (t, J=6.6 Hz, 2H), 2.13 (s, 3H), 1.28 (t, J=6.9 Hz, 6H), LC-MS m/z (%): 468.2 (100) [M+Na]+.
A solution of methyltriphenylphosphonium bromide (107 mg, 0.3 mmol) in anhydrous THF (5 mL) was cooled to −78 °C in a dry-ice acetone bath. To this solution was added dropwise a solution of n-BuLi (1.6 mol/L in THF, 0.75 mL, 1.2 mmol). The reaction was maintained at −78 °C for 30 min, warmed to room temperature over 30 min. The mixture was cooled to −78 °C, followed by addition of solution of compound 10 (67 mg, 0.15 mmol). The reaction mixture was stirred at −78 °C for 1 h and was allowed to warm to room temperature. The reaction was quenched by addition of saturated aqueous NH4Cl. The mixture was extracted with EtOAc (3×10 mL). The combined organic phases were washed with water and brine, dried over sodium sulfate and concentrated in vacuo. Purification (8:1 to 6:1 petroleum ether: EtOAc, v/v) by silica gel column chromatography gave HN311 (7.3 mg, yield: 11%). 1H NMR(300 MHz, CDCl3): δ 7.18 (dd, J=8.7, 5.4 Hz, 2H), 7.07 (d, J=8.7 Hz, 1H), 6.95–6.99 (m, 2H), 6.72 (brs, 1H), 6.39 (dd, J=8.7, 2.7 Hz, 2H), 4.78 (s, 1H), 4.73 (s, 1H), 4.48 (s, 2H), 4.15–4.23 (m, 4H), 3.47 (dd, J=7.8, 8.1 Hz, 4H), 1.76 (s, 3H), 1.24–1.32 (m, 6H); LC-MS m/z (%): 466.2 (100) [M+Na]+.
Author contribution
Fa-jun NAN, Zhao-bing GAO, Min LI designed the research; Hai-ning HU, Fei CHEN, and Ping-zheng ZHOU performed the research; all authors analyzed data; Hai-ning HU and Ping-zheng ZHOU wrote the paper.
Acknowledgments
The project was supported by National Science and Technology Major Project on “Key New Drug Creation and Manufacturing Program” (2013ZX09103001-016), National Key Basic Research Program of China (2013CB910604), and National Natural Science Foundation of China for Excellent Key Laboratory (81123004).
References
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–95. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–62. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock J, Li M. hERG channel function: beyond long QT. Acta Pharmacol Sin. 2013;34:329–35. doi: 10.1038/aps.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-S, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–3. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008;29:99–107. doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Yang SP, Yue JM. Discovery of structurally diverse and bioactive compounds from plant resources in China. Acta Pharmacol Sin. 2012;33:1147–58. doi: 10.1038/aps.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Pell L, Gofman Y, Haitin Y, Shamgar L, Patrich E, et al. Targeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifier. Proc Natl Acad Sci U S A. 2010;107:15637–42. doi: 10.1073/pnas.0911294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieniaszek HJ, Jr, Fiske WD, Saxton TD, Kim YS, Garner DM, Xilinas M, et al. Single-dose pharmacokinetics, safety, and tolerance of linopirdine (DuP 996) in healthy young adults and elderly volunteers. J Clin Pharmacol. 1995;35:22–30. doi: 10.1002/j.1552-4604.1995.tb04741.x. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, et al. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther. 1998;285:724–30. [PubMed] [Google Scholar]
- Earl RA, Zaczek R, Teleha CA, Fisher BN, Maciag CM, Marynowski ME, et al. 2-Fluoro-4-pyridinylmethyl analogues of linopirdine as orally active acetylcholine release-enhancing agents with good efficacy and duration of action. J Med Chem. 1998;4:4615–22. doi: 10.1021/jm9803424. [DOI] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Tzingounis AV. The specific slow after hyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol Pharmacol. 2010;78:1088–95. doi: 10.1124/mol.110.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung YY, Yu H, Xu K, Zou B, Wu M, McManus OB, et al. Discovery of a series of 2-phenyl-N-(2-(pyrrolidin-1-yl)phenyl) acetamides as novel molecular switches that modulate modes of Kv7.2 (KCNQ2) channel pharmacology: Identification of (S)-2-phenyl-N-(2-(pyrrolidin-1-yl)phenyl)butanamide (ML252) as a potent, brain penetrant Kv7.2 channel inhibitor. J Med Chem. 2012;55:6975–9. doi: 10.1021/jm300700v. [DOI] [PMC free article] [PubMed] [Google Scholar]