

Abstract
A hexanucleotide repeat expansion within a non-coding region of the C9ORF72 gene is the most common mutation causative of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). Elucidating how this bidirectionally transcribed G4C2 C4G2 expanded repeat causes “C9FTLD/ALS” has since become an important goal of the field. Likely pathogenic mechanisms include toxicity induced by repeat-containing RNAs, and loss of C9orf72 function due to epigenetic changes resulting in decreased C9ORF72 mRNA expression. With regards to the former, sense and antisense transcripts of the expanded repeat aberrantly interact with various RNA-binding proteins and form discrete nuclear structures, termed RNA foci. These foci have the capacity to sequester select RNA-binding proteins, thereby impairing their function. (G4C2)exp and (C4G2)exp transcripts also succumb to an alternative fate: repeat-associated non-ATG (RAN) translation. This unconventional mode of translation, which occurs in the absence of an initiating codon, results in the abnormal production of poly(GA), poly(GP), poly(GR), poly(PR) and poly(PA) peptides, collectively referred to as C9RAN proteins. C9RAN proteins form neuronal inclusions throughout the central nervous system of C9FTLD/ALS patients and may contribute to disease pathogenesis. This review aims to summarize the important findings from studies examining mechanisms of disease in C9FTLD/ALS, and will also highlight some of the many questions in need of further investigation.
Keywords: amyotrophic lateral sclerosis, bidirectional transcription, C9ORF72, epigenetics, expanded repeat, frontotemporal lobar degeneration, repeat-associated non-ATG translation, RNA foci
Introduction
In 2011, a hexanucleotide repeat expansion in the C9ORF72 gene was identified as the most common genetic cause of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS), two devastating neurological conditions [29,84]. This repeat expansion, located in a non-coding region of C9ORF72, is present in approximately 6% of sporadic ALS and FTLD patients, and in 40% and 25% of familial ALS and FTLD cases, respectively [66]. ALS, the most frequent motor neuron disease, is characterized by the degeneration of upper and lower motor neurons, leading to muscle weakness, spasticity and atrophy. FTLD, a common cause of early-onset dementia [42], results from the degeneration of frontal and temporal lobes, and encompasses a group of disorders distinguished clinically by abnormalities in behavior, language and personality. FTLD-like cognitive and behavioral impairments are present in up to 50% of ALS patients [40,61,80], and approximately 40% of FTLD patients develop motor neuron dysfunction reminiscent of ALS [61].
Despite the fact that only 2 years have passed since the identification of the C9ORF72 repeat expansion in FTLD and ALS, now commonly referred to as C9FTLD/ALS, many advances have been made in gaining a better understanding of the putative pathomechanisms by which it causes disease. The fast pace of research in this field stems in large part from lessons learned from other repeat expansion disorders [10]. Likely mechanisms involved in C9FTLD/ALS include toxicity induced by repeat-containing RNAs, which form nuclear RNA foci and aberrantly interact with various RNA-binding proteins causing transcriptome defects, and loss of C9orf72 function due to decreased C9ORF72 mRNA expression as a result of epigenetic changes (Fig. 1). Adding layers of complexity to the mix are the occurrence of bidirectional transcription of the expanded repeat, and repeat associated non-ATG (RAN) translation of the resulting sense and antisense transcripts, two phenomena that are becoming more widely appreciated in repeat expansion disorders (Fig. 1). This review aims to highlight the potential mechanisms contributing to C9FTLD/ALS and possible therapeutic strategies for treating these devastating diseases.
Figure 1. Potential mechanisms of disease in C9FTLD/ALS.

Expansion of the G4C2 C4G2 repeat within intron 1 of the C9ORF72 gene may cause C9FTLD/ALS through various mechanisms. 1) Abnormal DNA and histone methylation leads to a decrease in C9ORF72 mRNA expression which may consequently result in C9orf2 protein loss of function; 2) (G4C2)exp and (C4G2)exp transcripts are bound by select RNA-binding proteins and this may impair the ability of such proteins to bind their actual RNA targets. In addition, because (G4C2)exp and (C4G2)exp transcripts form nuclear foci, RNA-binding proteins that interact with these transcripts may too be sequestered in foci, also resulting in their loss of function; 3) In the cytosol, (G4C2)exp and (C4G2)exp transcripts are susceptible to repeat-associated non-ATG translation, producing poly(GA), poly(GP), poly(GR), poly(PR) and poly(PA) C9RAN proteins. Each C9RAN protein may have a different toxicity profile, and may contribute to neurodegeneration through the formation of soluble oligomers or their aggregation into insoluble inclusions within neurons.
RNA-mediated toxicity
A role for RNA-mediated toxicity in microsatellite expansion disorders was first described for myotonic dystrophy type 1 (DM1). This most common form of adult muscular dystrophy is caused by a CTG repeat expansion in the 3’UTR of the myotonic dystrophy protein kinase (DMPK) gene [64]. In 1995, RNA transcripts from the CTG repeat expansion were detected, in the form of nuclear foci, in fibroblasts and muscle biopsies from DM1 patients [95]. It was later shown that RNA of long CUG repeats fold into stable structures that bind and sequester select RNA-binding proteins [33,49,70,73,96,101]. For instance, RNA transcripts of expanded CUG repeats [(CUG)exp] are bound by the splicing factor muscleblind-like 1 (MBNL1) [67,73,91,95]. The resulting sequestration of MBNL1 into foci leads to its inactivation and the mis-splicing of a subset of pre-mRNAs, such as muscle-specific chloride ion channel and insulin receptor, which respectively account for the myotonia and insulin insensitivity associated with DM1 [28,68]. In addition to DM1, RNA foci are observed in several neurodegenerative diseases caused by repeat expansions, including the spinocerebellar ataxias SCA8, SCA10 and SCA31, myotonic dystrophy type 2 (DM2), Huntington disease-like 2, fragile X-associated tremor/ataxia syndrome (FXTAS), and C9FTLD/ALS (for review, see[10]).
RNA foci in C9FTLD/ALS
Upon discovery that an expanded G4C2 repeat causes C9FTLD/ALS, the Rademakers group found that RNA foci formed of G4C2-repeat containing RNA are present in the frontal cortex and spinal cord of repeat expansion carriers [29]. These results have since been replicated, and RNA foci composed of sense transcripts from the expanded repeat have also been documented in the motor cortex, temporal lobe, cerebellum and hippocampus, being present mostly in neurons, but also observed in astrocytes, oligodendrocytes, and microglia (Fig. 2) [32,37,57,59,72,110]. Spurred by the fact that, for some microsatellite expansion disorders, such as Huntington’s disease and SCA8, the expanded repeat is bidirectionally transcribed [8,22,23,56,77,102], and the fact that sense and antisense transcripts of the hexanucleotide repeat were detected in cerebellum of C9ORF72 patients [76], we and others investigated whether foci formed of antisense transcripts are present in C9FTLD/ALS. As with sense foci, antisense foci are found in the frontal cortex, motor cortex, hippocampus, cerebellum and spinal cord (Fig. 2) [37,57,72,110]. Sense and antisense foci, while typically found in separate cells, can co-occur within the same cell, and can even colocalize within the nucleus [72,110]. Foci have been detected throughout all layers of the frontal cortex [37], in pyramidal cells of layers III and V of the motor cortex [57], and in cells of the dentate gyrus and CA1 of the hippocampus [57,72]. In the cerebellum, RNA foci are often observed in cerebellar Purkinje cells and in cells in proximity to the Purkinje cell layer, but are also present in cells of the molecular layer, granular layer, and white matter [37,59]. With regards to the spinal cord, foci are observed in motor neurons, interneurons and non-neuronal cells [37,57].
Figure 2. RNA foci and C9RAN protein pathology are present in various regions of the central nervous system in C9FTLD/ALS.
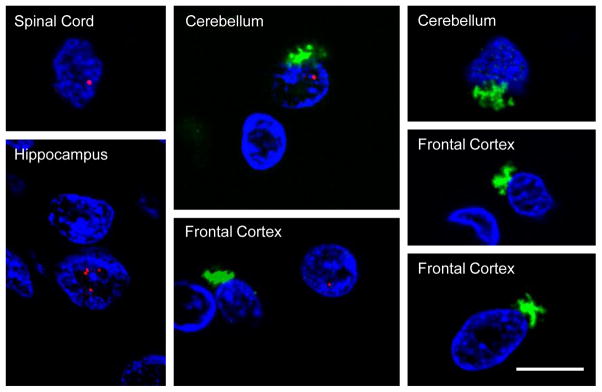
Fluorescence in situ hybridization of C9FTLD/ALS tissue sections using a probe against sense G4C2 transcripts (spinal cord, hippocampus) or antisense C4G2 transcripts (cerebellum, frontal cortex) was followed by immunofluorescence staining to detect poly(GP) inclusions. Note that RNA foci (red) in the nucleus (stained with DAPI, blue), and star-shaped cytoplasmic poly-GP inclusions (green), seldom co-occur in the same cells. Scale bar = 10 μm.
While usually nuclear, foci in brain tissues are occasionally detected in the cytoplasm [72]. Sense and antisense RNA foci are heterogeneous in size, and, while the number of foci per cell can range from few to many, a consistent finding among studies is that the majority of cells have only 1 or 2 foci [29,37,57,72,110]. Nonetheless, the number of foci per cell, and the number of cells bearing foci, may be influenced by brain region and foci type, though there is some inconsistency among studies. For instance, Mizielinska and colleagues report that the highest burden of foci was consistently found in the frontal cortex, the region that suffers the greatest neuronal loss in FTLD. However, a separate study reported a higher frequency of foci in the cerebellum compared to the frontal cortex and temporal lobes [59]. Fewer neurons appear to contain antisense foci than sense foci, but the number of antisense foci per neuron is greater [72]. The latter should be interpreted with caution given that the relative sensitivity of probes used to detect sense and antisense foci is not known. In addition, no difference in the percentage of cells with either sense or antisense foci was observed in two other studies, albeit the number of cases investigated was low, and a comparison of foci type in different brain regions was not carried-out [37,110]. Despite these caveats, a difference in frequency between sense and antisense foci could result from the fact that (G4C2) RNA, but not C-rich antisense (C4G2) RNA, forms stable uni- and multimolecular parallel G-quadruplex structures [35,83], and that such structures may influence foci formation.
Not unexpectedly, marked sense and antisense foci burden was found in a case homozygous for the C9ORF72 repeat expansion. This patient, who developed early-onset behavioral variant frontotemporal dementia and displayed severe clinical and pathological features that still fell within the normal spectrum of disease [36], had a higher proportion of neurons containing foci in the frontal cortex and hippocampus, and also more foci per cell, than heterozygous cases [72]. The severe foci burden and early age of onset of disease in this case is consistent with the observation of Mizielinska et al. that foci burden inversely correlates with age of onset in their cohort of 8 cases. Validating this finding in a larger series, however, remains to be done.
In addition to C9FTLD/ALS brain and spinal cord, nuclear foci, and less frequently cytoplasmic foci, are observed in C9FTLD/ALS skin biopsy-derived fibroblasts [4,32,57], immortalized lymphoblasts [57], peripheral blood leukocytes [110], and human neurons differentiated from induced pluripotent stem cells (iPSCs) [4,32,86]. Of interest, RNA-FISH using probes to sequences either upstream or downstream of the C9ORF72 expanded repeat revealed that these sequences are not primary components of the nuclear RNA foci in C9ORF72 iPSC-derived neurons (iPSNs), suggesting that foci are mainly composed of repeat tracts [32]. In addition to foci, iPSNs recapitulate several of the genomic and pathological abnormalities found in the brain and spinal cord of C9FTLD/ALS patients, including the expression of RAN translated proteins (C9RAN proteins; Table 1). As such, iPSNs provide a valuable model with which to investigate the pathophysiological pathways of C9FTLD/ALS
Table 1.
Comparison of iPSC-derived neuronal models from C9FTLD/ALS patients
| Almeida et al. | Donnelly et al. | Sareen et al. | |
|---|---|---|---|
| Case | FTLD patient and pre-symptomatic carrier from the VSM-20 family with C9FTLD/ALS | ALS | ALS or ALS with FTLD |
| Neuron Type | iPSC-differentiated neurons (~80% MAP2-positive: >30% glutamatergic, <10% GABAergic or dopaminergic) | iPSC-differentiated neurons<br1>Heterogeneous neuronal cell population, of which 30–40% stained positive for the motor neuron marker, HB9 | iPSC-differentiated motor neurons (33–45% SMI32+ motor neurons, 58–75% TuJ1+ pan-neurons, 20–30% GFAP+ and nestin+) |
| C9ORF72 expression |
|
|
|
| Foci |
|
|
|
| RNA-binding protein sequestration |
|
|
|
| C9RAN proteins |
|
|
|
| Other noted features |
|
|
|
Foci formation and protein sequestration in models of C9FTLD/ALS
Lee and colleagues recently provided evidence that cellular toxicity is associated with the nuclear retention of G4C2 transcripts and the appearance of RNA foci. Specifically, they demonstrated that longer G4C2 repeats generate RNA foci that are toxic when expressed in SH-SY5Y cells and zebrafish embryos [59]. Whereas apoptosis was induced upon expression of foci-forming (G4C2)38 or (G4C2)72, no such toxicity was observed upon expression of (G4C2)8, which does not result in foci formation [59]. As with other microsatellite expansion diseases, the presence of sense and antisense transcripts of the expanded C9ORF72 repeat, and the foci resulting thereof, implicates RNA-binding protein sequestration and dysregulation as a pathogenic mechanism of C9FTLD/ALS. In vitro studies have identified several proteins, such as hnRNPA3, hnRNPH, Pur , ASF/SF2 and ADARB2 that bind G4C2 transcripts [4,32,59,75,83,86,106], and these are discussed in more detail below.
hnRNPA3
Using in vitro-transcribed (G4C2)23 RNA incubated with nuclear extracts from HEK 293 cells, Mori et al. identified 20 top candidate RNA-binding proteins that interact with (G4C2)23 RNA. These included heterogeneous ribonucleoproteins (hnRNPs), splicing factors and mRNA-binding proteins having a variety of functions in RNA metabolism, translation and transport. Upon investigating the pathological involvement of select (G4C2)23-RNA-binding proteins for which antibodies were available, hnRNPA3 was found to form neuronal cytoplasmic and intranuclear inclusions in the hippocampus of patients with the C9ORF72 repeat expansion [75]. While the relationship of these inclusions to RNA foci was not examined in this study, hnRNPA3 was found to be a constituent of some TDP-43-negative, p62-positive neuronal inclusions characteristic of C9ORF72 repeat expansion carriers [3,81]. These inclusions are also immunoreactive for ubiquitin and select ubiquitin-binding proteins, most notably ubiquilin-2 [12,14], as well as C9RAN proteins [69,76]. In fact, because of the involvement of hnRNPA3 in mRNA export [62], it was speculated that binding of hnRNPA3 to (G4C2)exp pre-mRNA would result in its export to the cytoplasm where it could be RAN translated [75].
Consistent with binding of hnRNPA3 to (G4C2)exp transcripts causing their export to the cytoplasm rather than being retained in the nucleus, hnRNPA3 was not found to colocalize with nuclear foci in C9FTLD/ALS cerebellar tissues [59]. Likewise, Sareen and colleagues have shown that nuclear foci of (G4C2)exp RNA do not colocalize with hnRNPA3 or hnRNPA2/B1 in C9ALS iPSN, but do colocalize with hnRNPA1. While the colocalization between hnRNPA3 and foci must still be investigated in C9FTLD/ALS brain tissues, this finding suggests that not all proteins capable of binding (G4C2)exp RNA are sequestered within foci. The function of RNA-binding proteins that escape this fate is nonetheless expected to be altered given that, in binding (G4C2)exp RNA, these RNA-binding proteins may fail to bind and regulate their actual RNA targets. The notion of such RNA toxicity is not unique to repeat expansion diseases - defective RNA metabolism is a likely contributor to the pathogenesis of various neurodegenerative disorders. Mutations in hnRNPA1 and hnRNPA2/B1 cause motor neuron disease [50], and mutations in TDP-43 and FUS, which are structurally related to the family of hnRNPs, are causative of ALS and FTLD [41,55,85,100,107]. Some studies [4,75], but not all [106], have shown that TDP-43 and FUS interact with (G4C2)n RNA in vitro. However, neither TDP-43 or FUS show changes in subcellular localization in C9FTLD/ALS iPSCs [4,32], nor colocalize with foci in cultured cell models [59], or in C9ALS patient motor neuron cultures [86]. Similarly, TDP-43-containing aggregates were not found to colocalize with RNA foci in spinal motor neurons from C9ORF72 patients [57]. In C9FTLD frontal cortex, hippocampus and cerebellum, RNA foci are observed in neurons with TDP-43 inclusions, but not at a greater frequency than expected by chance, indicating that the presence of foci does not predict the presence of TDP-43 pathology [72]. Nevertheless, because hnRNPA1 is a binding partner of TDP-43 [15], the sequestration of hnRNPA1 by foci could indirectly influence TDP-43 function, thus providing a potential link between the C9ORF72 repeat expansion and TDP-43 dysfunction in C9FTLD/ALS.
hnRNPH
To explore which RNA-binding proteins interact with transcripts of G4C2 repeats, Lee and colleagues utilized SH-SY5Y cells transfected with foci-forming (G4C2)72 and carried-out immunofluorescence studies with antibodies to 30 proteins having a known or predicted ability to bind GC-rich sequences. This led to the identification of 3 proteins that colocalized with (G4C2)72 RNA foci in their cell model: hnRNPH, serine-arginine-rich splicing factor 1 (SF2), and serine-arginine-rich splicing factor 2 (SC35) [59]. Among these 3 proteins, hnRNPH immunoreactivity showed the most overlap with foci, and was the only protein to interact with (G4C2)72 RNA, as assessed by RNA immunoprecipitation.
The sequestration of hnRNPH with foci in (G4C2)72-expressing cells was associated with a decrease in exon 7 inclusion in TARBP2, an RNA target of hnRNPH [105]. Given that knockdown of hnRNPH in cells similarly affected TARBP2 RNA splicing, these results suggest that binding of hnRNPH to (G4C2)exp transcripts impairs the splicing function of hnRNPH. Of importance, although the colocalization of SF2 and SC35 with foci was quite rare in C9FTLD/ALS cerebellar tissues (occurring for less than 5% of foci), a striking colocalization of hnRNPH with 70% of RNA foci was observed [59]. While further studies are needed, these findings suggest that the sequestration of hnRNPH by (G4C2)exp foci, and the ensuing dysregulation of RNA processing, could play a pathogenic role in C9FTLD/ALS.
Pur
By incubating (G4C2)10 RNA with whole-cell lysate from mouse spinal cord, Xu and colleagues identified Pur , as well as Pur and Pur , as (G4C2)10 RNA binders [106]. They subsequently found that (G4C2)n RNA was bound by both recombinant mouse and Drosophila Pur in vitro, by exogenous mouse Pur in cultured N2A cells, and by endogenous Pur in brain lysate from mouse and control human frontal cortex. In a similar fashion to hnRNPA3, Pur inclusions were detected in the cerebellum of C9ORF72 repeat expansion carriers, as well as in (G4C2)30-expressing flies. The significance of this finding, however, is unclear since Pur inclusions were also observed in FTLD cases without the expanded repeat [106]. While this study did not investigate whether Pur colocalized with foci, it was later shown that such colocalization was observed in an iPSN motor neuron model of C9FTLD/ALS [86], but no colocalization was observed between (G4C2)exp foci and Pur in C9FTLD/ALS cerebellar tissue [59]. Studies by Xu and colleagues did nonetheless provide compelling data supporting that (G4C2)exp RNA is toxic, and that this toxicity is due, at least in part, to Pur dysregulation. Indeed, expression of (G4C2)30, but not (G4C2)3, in eyes or motor neurons of Drosophila resulted in disruption of eye morphology and decreased locomotor activity, respectively. Of importance, overexpression of Pur suppressed neurodegeneration in the eyes of (G4C2)30-expressing flies. These results suggest that binding of Pur by (G4C2)exp RNA causes a toxic loss of Pur function, a theory that is strengthened by the finding that knockdown of Pur is sufficient to reduce cell viability in N2A cells, which is also rescued by Pur overexpression [106].
ADARB2
Compared to the above-mentioned studies, the Rothstein and Sattler groups employed a different approach, hybridization of Cy5-labeled (G4C2)6.5 RNA to a proteome array containing nearly two-thirds of the annotated human proteome, to identify (G4C2)6.5 RNA-binding proteins [32]. Of the 19 proteins thusly identified, they focused their attention on ADARB2, a known RNA-binding protein and member of the ADAR family of proteins involved in RNA editing. RNA FISH combined with immunofluorescence staining for ADARB2 revealed that ADARB2 colocalized with nuclear (G4C2)exp RNA foci in C9ORF72 iPSNs, as well as in motor cortex of C9ALS patients [32]. In addition, nuclear localization of ADARB2 was increased in C9ORF72 iPSNs and C9ALS postmortem tissue. Notably, siRNA-mediated knockdown of ADARB2 in patient iPSNs decreased the number of cells with RNA foci by half, suggesting that ADARB2 is involved in the formation or maintenance of sense RNA foci, and that the interaction between ADARB2 and (G4C2)exp RNA plays a role in RNA-mediated toxicity. In support of this, it was found that C9ORF72 iPSNs are much more vulnerable to glutamate-induced excitotoxicity compared to control iPSNs, and that ADARB2 knockdown in control iPSNs enhances their susceptibility to glutamate toxicity to levels comparable to that observed in C9ORF72 iPSNs [32].
Transcriptional changes and enhanced vulnerability associated with the C9ORF72 expanded repeat
The above studies highlight that steady progress is being made in identifying proteins bound, and potentially sequestered, by RNA transcripts of expanded G4C2 repeats. No doubt similar studies are underway to identify protein binders of antisense C4G2 repeat transcripts. It should nonetheless be kept in mind that, due to the technical difficulties associated with cloning long GC-rich sequences, the (G4C2)n RNA utilized to identify interacting proteins have had relatively few repeats compared to the hundreds to thousands of repeats in C9FTLD/ALS patients. In addition, using extracts from cultured cells or mouse tissues to identify protein binders of (G4C2)n or (C4G2)n RNA may have limitations given that the pool of proteins present in these models may not accurately reflect the full-complement of proteins expressed in affected regions of C9FTLD/ALS patients. Also to consider when investigating whether (G4C2)n or (C4G2)n RNA-binding proteins are sequestered in foci is that the quantity of expanded repeat RNA used in transfectant cell models is likely to be much higher than is present in C9FTLD/ALS patients and, as such, may lead to the identification of false-positive hits. For example, Almeida and colleagues identified proteins in nuclear mouse brain extracts that bound biotinylated (G4C2)30 RNA and that were sequestered by large foci formed in cultured cells overexpressing G4C2 repeats; however, such sequestration was not observed in iPSC harboring smaller foci. Similar results were observed by the Shaw group: SF2 and SC35 colocalized with foci in cells overexpressing (G4C2)72, but no such colocalization was observed in C9FTLD/ALS cerebellar tissue [59]. Irrespective of whether the binding of proteins to (G4C2)exp or (C4G2)exp RNA causes their sequestration into foci, this abnormal interaction is likely to be deleterious. The transcriptome is affected by the C9ORF72 repeat expansion, and this may be partially due to the aberrant binding of repeat containing-transcripts to a plethora of RNA-binding proteins [32,57,86]. Studies have shown that the transcriptome in fibroblasts [32,57], iPSNs [32,86] and motor cortex [32] of C9ORF72 repeat expansion carriers is different than the transcriptome in respective models derived from control subjects. Of note, the altered gene expression profile in C9ALS iPSN is improved upon treatment of cells with antisense oligonucleotides (ASOs) that decrease C9ORF72 transcript levels and/or foci formation, suggesting that these transcriptome changes are due to a gain of function of the C9ORF72 repeat [32,86]. However, no such reversal of differentially expressed RNAs was observed in C9FTLD/ALS patient fibroblasts treated with ASOs targeting sense strand C9ORF72 transcripts, perhaps because some of the transcriptome abnormalities are due to antisense C4G2 transcripts [57].
While no overt toxicity is observed in iPSCs and iPSNs that harbor the C9ORF72 mutation [4,32,86], defects in their gene expression profile could nonetheless have harmful consequences. Several genes involved in regulating membrane excitability (DPP6 and KCNQ3) and synaptic transmission (CBLN1, CBLN2 and CBLN4) are disrupted in C9ALS iPSC-derived motor neuron cultures compared to control controls, and this may play a role in their decreased electrical excitability [86]. Furthermore, as mentioned above, C9ALS iPSNs are significantly more sensitive to glutamate-induced toxicity than are control iPSNs [32]. The formation of RNA foci, nuclear accumulation of ADARB2, and deregulated gene expression are believed to underlie this heightened susceptibility to excitotoxicity. That the treatment of C9ALS neurons with ASOs targeting sense strand C9ORF72 transcripts was found to mitigate these features and provide partial protection against glutamate-induced cell death supports this notion. In addition to foci and altered gene expression, these C9ORF72 iPSN recapitulate other features of C9FTLD/ALS, namely decreased C9ORF72 transcript levels and the production of poly(GP) C9RAN proteins (Table 1), thus leading to the question as to whether these phenomena also contribute to the increased vulnerability of cells to excitotoxicity. Arguing against the involvement of loss of C9orf72 function, Donnelly and colleagues reported that, in addition to ASOs that target the repeat without altering C9ORF72 RNA levels, ASOs that do decrease C9ORF72 mRNA levels still significantly protected iPSNs against glutamate-induced cell death. They also found that poly(GP) C9RAN proteins persisted following ASO-treatment, suggesting that poly(GP) did not contribute to the observed acute neurotoxicity. Nonetheless, the expression of other C9RAN proteins was not examined, and the possibility remains that greater rescue may be observed at later time-points that allow for C9RAN proteins to be degraded. In fact, it is not yet known whether all C9RAN proteins are expressed in iPSC-derived neurons, and whether they have different expression, stability, solubility and toxicity profiles. While the neurotoxic potential of C9RAN protein will be discussed in more detail below, the findings herein imply that RNA toxicity causes cells affected with the C9ORF72 repeat expansion to be highly sensitive to excitotoxicity. In a similar manner, the Gao group has shown that C9FTLD/ALS iPSN are more sensitive to chloroquine and 3-MA, two inhibitors of autophagy [4]. This heightened sensitivity is not general to all cellular stressors, as no difference in toxicity was observed between control and C9FTLD/ALS iPSN treated with rotenone, an inducer of mitochondrial dysfunction, tunicamycin, an inducer of endoplasmic reticulum stress, and the broad-spectrum kinase inhibitor, staurosporine, which induces apoptosis. These results thus raise the possibility that proper autophagic processing, a biological process known to be disrupted in neurodegenerative diseases [103], is compromised in C9FTLD/ALS iPSNs. This is further supported by the finding that levels of p62, a known substrate of the autophagy pathway and a component in neuronal inclusions in C9ORF72 patients [3,81], were significantly higher in iPSNs from C9ORF72 repeat expansion carriers compared to iPSNs from non-carriers [4]. The cause of the enhanced vulnerability of C9FTLD/ALS iPSNs to autophagy inhibitors is unknown; the Gao group has shown that foci and poly(GP) proteins are present in the C9FTLD/ALS iPSN lines they generated, as is a decrease in mRNA expression of C9ORF72 variant 2 (Table 1). At present, the role of C9orf72 remains unknown, but evidence demonstrates that it is structurally related to DENN domain proteins, GDP-GTP exchange factors for Rab GTPases [60,108], raising the possibility that decreased C9orf72 expression may result in the misregulation of Rab-dependent vesicular trafficking necessary for proper autophagic processing. In addition, the accumulation of C9RAN proteins could indirectly or directly impair the autophagy pathway, perhaps by overwhelming the system. Finally, (G4C2)exp or (C4G2)exp RNA, by binding to and/or sequestering RNA-binding proteins, could influence the expression of various molecular players involved in the autophagy pathway.
RAN translation and C9FTLD/ALS
Translation initiation, a highly regulated process in eukaryotes, involves the formation of a pre-initiation complex that scans along a mRNA from the 5’ end to locate the start codon [1]. The most common start codon is AUG, which is translated into methionine. Recent studies indicate that, although rare, cellular eukaryotic mRNAs can also undergo translation using CUG or ACG as alternative start codons [99]. Furthermore, while investigating the molecular mechanisms of SCA8 and DM1, neurodegenerative disorders caused by CAG and CTG expansion mutations, Ranum and colleages discovered a novel form of translation, namely repeat-associated non-ATG (RAN) translation [109]. RAN translation occurs across expanded repeat tracts despite the absence of an initiating AUG codon. Because RAN translation can occur in all reading frames, various products can be synthesized from a given transcript. For example, RAN translation of transcripts of long expanded CAG CUG repeats leads to the production of homopolymeric polyglutamine (polyQ), polyalanine (polyA), and polyserine (polyS) proteins [109].
Although the mechanisms of RAN translation are unknown, the Ranum group has identified, through a series of elegant experiments, certain biological guidelines for this unconventional mode of translation. First, RAN translation appears to depend on repeat length, with different reading frames having a different length threshold [25,109]. For instance, polyQ proteins RAN translated from the first reading frame are synthesized upon transfection of cells with constructs of 42–107 CAG repeats, but not of only 15–20 repeats. PolyS proteins encoded by the second reading frame are not synthesized from 42 repeats as are polyQ proteins, but are synthesized by longer 58–107 repeats. Finally, polyA proteins encoded by the third reading frame are not expressed with 42 or 58 repeats, only moderately expressed from 73 and 78 repeats, but robustly expressed from 105 and 107 CAG repeats [109]. Next, RAN translation of CAG repeats requires secondary hairpin structure; long tracts of CAG repeats that form hairpins undergo RAN translation but non-hairpin-forming CAA repeats of similar length are not RAN translated [109]. Yet another interesting aspect of RAN translation is that, once started, it can read through the entire expanded repeat without frame-shifting or prematurely stopping [109,110].
In addition to expanded CAG CTG repeats, expanded repeats of CGG [97], G4C2 [5,76] and C4G2 [37,74,110] are now known to be RAN translated, making RAN translation an established occurrence in multiple microsatellite expansion disorders (DM1 [109], SCA8 [109], FXTAS [97], and C9FTLD/ALS [5,37,74,76,110]), with the list likely to grow longer. The discovery of RAN translation, a phenomenon so far considered unique to expanded repeat diseases, implicates that RAN proteins thusly synthesized are involved in pathogenic mechanisms culminating in neurodegeneration. Of importance, polyA and polyG proteins respectively accumulate in disease-relevant tissues of patients with SCA8 and DM1, and their expression in cultured cells is sufficient to cause apoptotic cell death [109]. The quest to determine whether the same is true for C9RAN proteins produced in C9FTLD/ALS is now underway.
RAN translation of sense and antisense transcripts of the expanded C9ORF72 repeat in C9FTLD/ALS
When examining cerebellar C9ORF72 mRNA expression, Mori and colleagues found that, consistent with previous results [29,39], C9ORF72 mRNA levels were decreased in C9FTLD/ALS patients. However, they also observed that both sense and antisense RNA containing intron 1, where the hexanucleotide repeat is located, were strongly increased [76]. This finding raised the possibility that both sense and antisense transcripts of the expanded repeat are available for RAN translation, which was later independently confirmed by several groups, including our own [5,37,69,74,76,110]. RAN translation of sense (G4C2)exp-containing transcripts leads to the synthesis of poly(GA), poly(GP) and poly(GR) proteins, whereas RAN translation of antisense (C4G2)exp transcripts leads to the synthesis of poly(PR), poly(PG) and poly(PA) proteins. The antisense repeat is denoted here as “C4G2” rather than “G2C4”, the reverse complement of the sense G4C2 repeat. The reason for this is that there are Gs downstream of the last sense G4C2 repeat, thus making the first antisense repeat C4G2. Note that C9RAN proteins with the GP repeat motif are predicted in both the sense and antisense directions; however, because of their unique C-terminal region they represent two distinct C9RAN proteins, thus bringing the total to 6 C9RAN proteins in C9FTLD/ALS, excluding putative ATG-initiated antisense poly(GP) and poly(PR) proteins [110].
In a similar fashion to what has been observed for expanded CAG CUG repeats [109], RAN translation of expanded G4C2 C4G2 repeats may too be dependent on reading frame and repeat size. Mori and colleagues have reported that, upon transfection of HEK 293 cells with a (G4C2)38 construct, poly(GA) proteins were faintly detected, and these products increased in size and abundance as the repeat lengthened [76]. Unlike the synthesis of poly(GA) proteins, which occurs from the first reading frame, poly(GP) proteins from the second reading frame were only observed when cells were made to overexpress ~145 G4C2 repeats, and no poly(GR) proteins encoded by the third reading frame were detected at any of the repeat lengths tested [76]. With regards to RAN translation of antisense transcripts, we have reported that poly(PR) (first reading frame) and poly(PG) (second reading frame) proteins are synthesized from 66, but not 2, C4G2 repeats. However, poly(PA) protein synthesis from the third reading frame was not observed in cultured cells expression (C4G2)66 [37]. Together, these findings suggest that RAN translation mechanisms become more efficient with increasing repeat length. However, Zu et al. have shown that as few as 30 G4C2 repeats are sufficient to cause RAN translation of poly(GA), poly(GP) and poly(GR) proteins [110]. They have also reported that poly(PR), poly(PG) and poly(PA) proteins are all expressed following transfection of cells with 40 or 50 antisense C4G2 repeats [110]. The disparate results among studies may result from different sensitivities of antibodies used to detect each C9RAN protein, or from the different expression vectors utilized; our group and the Edbauer group included 5’ flanking sequences upstream of the repeat, and these may regulate or influence RAN translation [37,76].
While the mechanisms regulating RAN translation are still unclear, the pathology resulting from RAN translation is no less distinctive. Using multiple antibodies against individual C9RAN proteins, made either to detect dipeptide-repeat regions or unique C-terminal regions, all potential C9RAN proteins have been detected in brain tissues of C9FTLD/ALS patients but not in FTLD/ALS patients without the C9ORF72 repeat expansion [5,37,63,74,76,110] (Table 2). Biochemical analysis has revealed the presence of high-molecular weight, insoluble C9RAN proteins in the cerebellum and frontal cortex of C9FTLD/ALS patients [5,76,110]. Neuropathologically, dot-like and “star-shaped” neuronal cytoplasmic inclusions and intranuclear inclusions of poly(GA), poly(GP) and poly(GR) proteins are found throughout the central nervous system (CNS), being highly abundant in neocortical regions, the hippocampus and cerebellum (Fig. 2) [5,63,69,74,76]. C9RAN pathology, however, is less frequently observed in the spinal cord [5,63,74]. Poly(GA) and poly(GP) inclusions are specifically deposited in neurons and not observed in glia [5,63]. Consistent with this finding, no poly(GP) inclusions are detected in other organs, including peripheral nerves and ganglia, with the exception of testes [5].
Table 2.
Relative regional neuropathology of C9RAN proteins in C9FTLD/ALS
| Poly(GA) | Poly(GP) | Poly(GR) | Poly(PR) | Poly(PA) | |
|---|---|---|---|---|---|
| Cortex | +++ | +++ | +++ | ± | ± |
| Hippocampus | +++ | +++ | +++ | ± | ± |
| Cerebellum | +++ | +++ | +++ | ± | ± |
| Spinal Cord | ± | ± | ± | − | − |
| Amygdala | N.D. | +++ | N.D. | ± | ± |
| Thalamus | N.D. | +++ | N.D. | ± | ± |
| Medulla | + | ++ | N.D. | ± | ± |
| Striatum | ++ | + | N.D. | N.D. | N.D. |
| Substantia Nigra | ± | + | N.D. | N.D. | N.D. |
| XIIth Nerve | − | − | N.D. | N.D. | N.D. |
N.D.: not determined
Similar to inclusions of sense transcript-derived C9RAN proteins, most inclusions of poly(PR) and poly(PA) proteins have a star-shaped morphology [37,74,110]. Somewhat surprisingly, although 80% of poly(GP) inclusions are estimated to derive from the antisense transcript [110], inclusions of poly(PR) and poly(PA), also generated from the antisense transcript, are sparse in the brain of C9FTLD/ALS patients, and not detectable in the spinal cord [37,74,110] (Table 2). This lower frequency of antisense poly(PR) and poly(PA) inclusions could be due to lower RAN translation efficiency in these reading frames, or a decreased capacity of these proteins to form aggregates. It is unlikely to be due to differences in antibody sensitivity given that several groups report the same finding despite using different antibodies [37,69,74,110].
The relationship between C9RAN proteins and other pathological features of C9FTLD/ALS
Prior to the discovery of RAN translation in C9FTLD/ALS, a neuropathological feature highly characteristic of patients with the C9ORF72 repeat expansion was observed in the cerebellum and hippocampus: TDP-43-negative cytoplasmic and intranuclear inclusions immunoreactive for ubiquitin and the ubiquitin-binding proteins, p62 and ubiquilin-2 [3,14,78]. It is now known that all C9RAN proteins, but especially poly(GA), poly(GP) and poly(GR), are among the proteins making up many of these p62-positive inclusions [63,69,74,76].
In addition to the TDP-43-negative, p62-positive inclusions pathognomic to C9FTLD/ALS, TDP-43-positive inclusions typical of classical sporadic ALS and the most common molecular subtype of FTLD (FTLD-TDP) are a consistent feature of C9FTLD/ALS [13,26,29,46,65,78]. Double-label immunofluorescence studies for TDP-43 and poly(GA) revealed that affected neurons in neocortical regions contained either TDP-43 or poly(GA) inclusions [63]. Yet, in cases with abundant TDP-43 pathology, both proteins could be found to co-accumulate within hippocampal dentate granules cells. In such instances, poly(GA) aggregates were present in the center of the neuron and surrounded by TDP-43 [63]. In a similar fashion, Mori and colleagues found neither poly(GA) or poly(GP) proteins co-aggregate with phospho-TDP-43. On the rare occasion, however, small spherical poly(GA) inclusions were found to form a core inside phospho-TDP-43 inclusions, but never vice versa, suggesting that C9RAN protein aggregation may precede TDP-43 pathology [76].
Just as C9RAN protein pathology is rarely observed in the same cells harboring TDP-43 inclusions, we have found that RNA foci and poly(GP) inclusions seldom co-occur within the same cell in the frontal cortex and cerebellum of C9FTLD/ALS patients [37]. Of interest, the brain region sampled (frontal cortex vs. cerebellum) and foci type (antisense vs. sense) both significantly affected the frequency of cells having both foci and inclusions, with sense foci being more likely to co-occur with poly(GP) inclusions than antisense foci. While it remains to be determined whether other C9RAN proteins more commonly co-localize to the same cells as foci, these results suggest there are competing mechanisms in place to determine whether (G4C2)exp and (C4G2)exp transcripts are fated to form foci or instead be RAN translated. Just recently, the Shaw group, using cultured cells transfected with expression vectors in which G4C2 repeats were placed 3’ to EGFP, demonstrated that cells with intranuclear RNA foci expressed little or no EGFP, implicating that foci formed shortly after transcription of the repeat precludes nuclear export of the transcript [59]. Thus, only those transcripts that escape being sequestered in foci are likely to be RAN translated.
Are C9RAN proteins involved in neurodegeneration?
C9RAN proteinaceous inclusions are present in vulnerable areas of the CNS (e.g., neurons of neocortex and hippocampus), but we have shown there is a paucity of inclusions in certain affected areas, such as the spinal cord, making the contribution of RAN translation to disease pathogenesis unclear [5,37]. Mackenzie and colleagues subsequently reported that, unlike TDP-43 pathology, there is no correlation between poly(GA) pathology and the degree of neurodegeneration in C9FTLD/ALS [63]. While one interpretation of this result is that poly(GA) proteins do not directly cause neuronal death, another explanation could be that, as with other aggregation-prone proteins involved in neurodegeneration (e.g. tau [38]), poly(GA) proteins, as well as other C9RAN proteins, may contribute to neurodegeneration through the formation of toxic soluble oligomers. Moreover, given that the different C9RAN proteins are likely to have different expression, stability, and solubility profiles, and that some may be post-translationally modified, their neurotoxic potential may differ from one another. With this in mind, it is interesting to note that Mori and colleagues have observed that poly(GR) inclusions in C9FTLD/ALS contain a mixture of non-methylated, as well as symmetrically and asymmetrically dimethylated, arginine residues [74]. This post-translational modification, which increases protein hydrophobicity, may influence the aggregation and toxicity of poly(GR) proteins.
While it is difficult to reconcile whether C9RAN proteins are merely benign bystanders or harmful entities in post-mortem tissue, the Ranum group has provided the first evidence that C9RAN proteins, specifically poly(PR) and poly(GP), induce cellular toxicity in cultured cells independently of the accumulation of repeat RNA [110]. The novel antibodies for each C9RAN protein now available provide valuable tools to allow researchers of the field to further investigate the contribution of RAN translation to the development of C9FTLD/ALS. The generation of cell and animal models, especially those that preferentially drive the expression of individual C9RAN proteins, will be critical to decipher whether any of the C9RAN proteins are neurotoxic and, if so, by what means. Whatever the outcome of these studies, C9RAN proteins, if reliably detectable in cerebrospinal fluid, could become a much needed biomarker for C9FTLD/ALS. In fact, determining whether C9RAN proteins accumulate in cerebrospinal fluid prior to or after the first appearance of symptoms will shed light on the role of C9RAN proteins in disease pathogenesis.
C9orf72 loss of function
Through alternative splicing, at least three transcripts are produced from the human C9ORF72 gene, and these are predicted to encode two protein isoforms. The G4C2 repeat is located between two alternatively spliced noncoding first exons (exons 1a and 1b) of C9ORF72. If exon 1a is used, the repeat is transcribed (variant 1, NM_145005.5; variant 3, NM_001256054.1); if exon 1b is used, the repeat is located in the promoter region (variant 2, NM_018325.3). This raises the possibility that the expanded repeat could influence C9ORF72 expression in a transcript-specific manner. Initial studies revealed a decrease in variant 2 expression in lymphoblastoid cell lines and frontal cortical tissue from C9ORF72 repeat expansion carriers [29,84]. Thereafter, decreased expression of multiple C9ORF72 variants were observed in C9FTLD/ALS frontal cortex, motor cortex, cerebellum and cervical spinal cord [9,24,32,36,39,76], lymphoblast cell lines [24], and in some lines of neurons differentiated from patient iPSC [4,32]. Still to be determined is the variability in expression of individual C9ORF72 transcript variants across brain regions in normal subjects and C9FTLD/ALS patients, since regional differences in their expression may account for the susceptibility of certain cell populations to neurodegeneration. Despite the aforementioned decreases in C9ORF72 expression, it was also shown that both sense and antisense transcripts containing intron 1 (where the repeat is located) were strongly increased in C9ORF72 patients, suggesting a selective stabilization of repeat containing pre-mRNA or of the excised intron 1 [76]. In addition, the Baloh group reported that transcription of the repeat was increased in C9ALS iPSC-derived motor neurons due to increased use of exon 1a by the mutant allele [86]. The increase in repeat-containing RNA likely contributes to foci formation and RAN translation, whereas the reduction of other C9ORF72 transcripts could cause C9orf72 depletion. Whether C9orf72 protein expression is decreased in C9FTLD/ALS has not yet been definitively determined, in large part because limitations with currently available C9orf72 antibodies render them unsuitable for quantification analyses. Studies to date have either shown a reduction in C9orf72 protein expression in fibroblast cell lines from ALS patients [84], or have failed to detect marked changes in C9orf72 in brain tissue from expanded repeat carriers [26,29,39,46,89,90,93]. More recently, Sareen et al. have reported that no difference in expression of C9orf72 protein is observed between C9ALS and control iPSC-derived motor neurons; yet, this is not unexpected given that no decrease in C9ORF72 transcripts was observed in their C9ALS iPSN model [86], in contrast to other C9FTLD/ALS iPSN models [4,32] (Table 1).
Because knockdown of C9ORF72 is well tolerated in mice [57] and C9FTLD/ALS iPSN [32,86], and even abrogates toxicity associated with the endogenous C9ORF72 mutation in iPSN [32], it has been put forth that loss of C9orf72 function is not a major cause of C9FTLD/ALS. Prior to drawing this conclusion, however, one must consider that C9orf72 protein expression was not examined in these models, and its degree of knockdown not established. Furthermore, it remains unknown how sustained loss of C9orf72 in the context of the aging brain, especially under circumstances where aberrant foci and proteinaceous inclusions are present, would influence neuronal survival. While it may be telling that a patient homozygous for the C9ORF72 repeat expansion developed severe clinical and pathological features but not sufficiently so to fall outside the usual disease spectrum [36], a loss of function mechanism in C9FTLD/ALS ought not to be discounted based on this one case, especially given the clinical heterogeneity across C9ORF72 mutation carriers. Indeed, that downregulation of the zebrafish C9ORF72 orthologue leads to altered morphology of motor neuron axons and locomotor deficits, and that these features are reversed upon expression of human C9ORF72 [24], support the notion that loss of C9ORF72 is detrimental.
Keeping in mind limitations with current antibodies, C9orf72 is reportedly expressed in swollen dystrophic neurites in the CA1 and molecular layer of the hippocampus of ALS, Alzheimer’s disease, Parkinson’s disease, multiple system atrophy and normal control brains [88]. This observation could suggest that C9orf72 plays a role, protective or otherwise, in the process of neurodegeneration. Of interest, the neuronal populations most sensitive to ALS and FTD transcribe the highest levels of C9ORF72 in mouse, perhaps providing an explanation for this selective vulnerability in individuals harboring the C9ORF72 mutation [94]. Upon availability of reliable C9orf72 antibodies, these questions will be more readily answered as they will provide a means to quantitatively assess C9orf72 protein levels in different regions of the CNS, and to track changes in C9orf72 expression at different stages of disease. Furthermore, a better understanding of C9orf72 function will shed light on whether, and how, its dysregulation contributes to disease. As mentioned, C9orf72 is structurally related to DENN domain proteins [60,108], but nothing about its function in the CNS is currently known. DENN domain proteins are highly conserved Rab-GEFs: GDP/GTP exchange factors that activate Rab-GTPases, the latter serving as master regulators for intracellular membrane trafficking [48]. Several DENN domain proteins have been linked to neurodegeneration [6,30,43], and it has been postulated that depletion of C9orf72 may play a part in C9FTLD/ALS pathogenesis through altered Rab-dependent vesicular trafficking processes, such as autophagy [11,60,108]. Macroautophagy is a bulk degradation process thought critical for the clearance of large protein aggregates and the maintenance of neuronal health [44]. Indeed, increasing evidence links dysregulated autophagy to the pathogenesis of ALS, FTLD and other dementias [19,53,79]. In addition to autophagy, aberrant endocytic trafficking may too contribute to disease. For instance, mutations in GRN, the gene encoding progranulin, cause GRN haploinsufficiency and FTLD-TDP [7,27], highlighting the importance of proper progranulin homeostasis. Sortilin, a neuronal progranulin receptor, mediates progranulin endocytosis and delivery to lysosomes [16,47,58], and we have recently reported that TDP-43 dysfunction, which causes altered splicing of sortilin, may consequently contribute to altered progranulin metabolism [82]. Whether or not C9orf72 influences sortilin-mediated endocytosis is worth investigating, as is determining which Rabs interact with C9orf72, whether C9orf72 does in fact possess GEF activity, and what role in may play in modulating intracellular trafficking. Answers to these questions will provide mechanistic insight into the potential contribution of C9orf72 loss of function to C9FTLD/ALS.
Epigenetic alterations cause decreased C9ORF72 mRNA expression
While many questions remain regarding the consequences of decreased C9ORF72 mRNA expression in C9FTLD/ALS, great progress has been made in elucidating the mechanisms by which this occurs. Epigenetic alterations contribute to the pathogenesis of almost all repeat expansion disorders [45]: chromatin changes are believed to be implicated in DM1 [34,92], SCA8 [18] and SCA31 [87], Freidreich ataxia [2], as well as fragile X syndrome and FXTAS [98]. In addition, levels of proteins involved in methylation processes, such as DNMT1, DNMT3a, and 5-methylcytosine, are reportedly upregulated in motor neurons of ALS patients [20], further supporting a role of epigenetic alterations in disease. Of interest, studies from our group suggest that the decrease in C9ORF72 expression in C9FTLD/ALS is modulated by epigenetic processes. We recently reported that the reduction in C9ORF72 mRNA expression in frontal cortex and cerebellum of expanded repeat carriers is associated with histone trimethylation at residues H3K9, H3K27, H3K79, and H4K20 [9]; these modifications, observed in all C9FTLD/ALS cases tested, are well-known to silence gene expression. In fact, we found that treatment of C9FTLD/ALS-derived fibroblasts with 5-aza-2-deoxycytidine, a DNA and histone demethylating agent, increased C9ORF72 expression, an effect not observed in control fibroblasts. The increase in C9ORF72 expression upon demethylation was accompanied by a decrease in binding of C9ORF72 with trimethylated histones, validating that the reduction in C9ORF72 mRNA expression in C9FTLD/ALS results from aberrant histone methylation [9]. Moreover, because these histone modifications are easily detected in the blood of expanded repeat carriers [9], these epigenetic events may prove useful as biomarkers for C9FTLD/ALS.
In addition to histone methylation, CpG island hypermethylation can lead to gene expression silencing [51]. Xi et al. report that the CpG island 5’ of the repeat expansion is hypermethylated in blood, frontal cortex, and cervical spinal cord of approximately 40% of ALS patients carrying the expansion, while the CpG island 3’ of the repeat expansion is not hypermethylated [104]. Of note, C9ALS patients with a hypermethylated allele with >50 repeats had reduced expression of C9ORF72 transcripts [104], and the level of methylation 5’ of the G4C2 repeat inversely correlated with disease duration, implicating it may be associated with increased ALS severity. That high methylation of the CpG island near the G4C2 repeat was significantly associated with familial ALS, but more than half of the ALS expansion carriers showed either low or no methylation is intriguing. Xi et al. propose that repeat size may influence CpG methylation and account for the wide range of methylation levels among expansion carriers, akin to the correlation between CpG methylation and repeat size observed in Friedreich ataxia [17]. It is also possible that other CpG sites near the repeat, or the repeat itself, which becomes a new CpG island upon expansion, are aberrantly methylated. Elucidating whether this is the case will be of great interest, as will determining whether global DNA methylation levels at these islands and/or methylation of specific cytosine sites correlate with different disease phenotypes, and consequently act as a disease-modifier. Furthermore, while the studies above convincingly implicate histone and DNA methylation in the C9FTLD/ALS disease cascade, it remains to be determined whether DNA hypermethylation triggers histone trimethylation, whether histone trimethylation leads to DNA hypermethylation, or whether histone trimethylation alone is sufficient to cause disease. Finally, given that both sense and antisense transcripts of the expanded repeat are involved in C9FTLD/ALS, evaluating the methylation state of the antisense strand of DNA, and whether methylation influences sense and antisense repeat transcription, also merits attention.
Conclusion
The findings discussed above highlight the great strides made towards elucidating how the expanded repeat in C9ORF72 causes C9FTLD/ALS. While many questions remain, and the exact mechanisms involved are not yet known, the accomplishments to date provide significant insight into potential C9FTLD/ALS diagnostics and therapeutics. Even as we seek to bridge the gap in our understanding, we can nonetheless pursue the development of therapeutic strategies based on the assumption that repeat-containing RNA is at the crux of C9FTLD/ALS pathogenesis. Whatever the contribution of foci or RAN translation may be, therapeutic strategies aimed at neutralizing or degrading transcripts of the expanded repeat are expected to inhibit downstream neurotoxic cascades. On this front, it is very encouraging that several groups have identified ASOs that target repeat-containing transcripts and thereby decrease RNA foci formation, mitigate disease-specific transcriptional changes, and attenuate susceptibility to excitotoxicity in C9FTLD/ALS patient fibroblasts and iPSN, without significantly affecting the overall level of RNAs encoding for C9orf72 [32,57,86]. Such ASOs could provide a therapeutic approach to suppress the toxic effects of repeat-containing transcripts without decreasing C9orf72 protein expression and contributing to its loss of function. Of importance, intrathecal administration of ASOs have gone to clinical trial for SOD1-related familial ALS [71], with similar trials in the planning for DM1 and Huntington disease [52,71]. In addition to ASOs, small ligands may offer an attractive therapeutic alternative given their permeability and tight binding affinities. Several studies have shown that small molecules can bind RNA and abrogate RNA-toxicity [21,31,54]. Accordingly, small molecules that bind (G4C2)exp or (C4G2)exp RNA could potentially inhibit foci formation or the interaction between these transcripts and RNA-binding proteins, as well as prevent RAN translation by steric blocking mechanisms that impede translation or by altering the RNA structure thought necessary for it to occur. As prospective treatments are investigated, a related need must also be addressed: a means to accurately and reproducibly monitor C9FTLD/ALS disease progression and assess drug efficacy. Biomarkers in the form of molecular indicators of disease pathology could offer sensitive and efficient ways to diagnose and evaluate disease activity. Whether levels of C9RAN proteins in cerebrospinal fluid, or the detection of trimethylated histone residues in blood, can serve this purpose are important questions in need of being answered.
Acknowledgments
This work was supported by Mayo Clinic Foundation; National Institutes of Health/National Institute on Aging [R01 AG026251 (LP)]; National Institutes of Health/National Institute of Neurological Disorders and Stroke [R21 NS074121 (TFG), R21 NS079807 (YZ), R01 NS063964 (LP); R01 NS077402 (LP), R21 NS084528 (LP)]; National Institute of Environmental Health Services [R01 ES20395 (LP)]; Amyotrophic Lateral Sclerosis Association (LP); the Canadian Institutes of Health Research (VVB), and the Siragusa Foundation (VVB).
References
- 1.Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol. 2012;19 (6):568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 2.Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17 (5):735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- 3.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122 (6):691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 4.Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126 (3):385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77 (4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azzedine H, Bolino A, Taieb T, Birouk N, Di Duca M, Bouhouche A, Benamou S, Mrabet A, Hammadouche T, Chkili T, Gouider R, Ravazzolo R, Brice A, Laporte J, LeGuern E. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72 (5):1141–1153. doi: 10.1086/375034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442 (7105):916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 8.Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum Mol Genet. 2010;19 (R1):R77–82. doi: 10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, Pregent L, Daughrity L, Baker MC, Rademakers R, Boylan K, Patel TC, Dickson DW, Petrucelli L. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126 (6):895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belzil VV, Gendron TF, Petrucelli L. RNA-mediated toxicity in neurodegenerative disease. Mol Cell Neurosci. 2012:S1044–7431. doi: 10.1016/j.mcn.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci. 2013;126 (Pt 5):1059–1069. doi: 10.1242/jcs.123075. [DOI] [PubMed] [Google Scholar]
- 12.Bieniek KF, Murray ME, Rutherford NJ, Castanedes-Casey M, DeJesus-Hernandez M, Liesinger AM, Baker MC, Boylan KB, Rademakers R, Dickson DW. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013;125 (2):289–302. doi: 10.1007/s00401-012-1048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Jr, Parisi JE, Lucas JA, Petersen RC, Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135 (Pt 3):765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L, McCluskey L, Irwin DJ, Grossman M, Molina-Porcel L, Lee VM, Trojanowski JQ. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012;123 (6):825–839. doi: 10.1007/s00401-012-0970-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280 (45):37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 16.Carrasquillo MM, Nicholson AM, Finch N, Gibbs JR, Baker M, Rutherford NJ, Hunter TA, DeJesus-Hernandez M, Bisceglio GD, Mackenzie IR, Singleton A, Cookson MR, Crook JE, Dillman A, Hernandez D, Petersen RC, Graff-Radford NR, Younkin SG, Rademakers R. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87 (6):890–897. doi: 10.1016/j.ajhg.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castaldo I, Pinelli M, Monticelli A, Acquaviva F, Giacchetti M, Filla A, Sacchetti S, Keller S, Avvedimento VE, Chiariotti L, Cocozza S. DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J Med Genet. 2008;45 (12):808–812. doi: 10.1136/jmg.2008.058594. [DOI] [PubMed] [Google Scholar]
- 18.Chen IC, Lin HY, Lee GC, Kao SH, Chen CM, Wu YR, Hsieh-Li HM, Su MT, Lee-Chen GJ. Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Mol Biol. 2009;10:9. doi: 10.1186/1471-2199-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, Zhang X, Song L, Le W. Autophagy dysregulation in amyotrophic lateral sclerosis. Brain Pathol. 2012;22 (1):110–116. doi: 10.1111/j.1750-3639.2011.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31 (46):16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. 2012;7 (5):856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, Tapscott SJ. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol Cell. 2005;20 (3):483–489. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Chung DW, Rudnicki DD, Yu L, Margolis RL. A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum Mol Genet. 2011;20 (17):3467–3477. doi: 10.1093/hmg/ddr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of Amyotrophic Lateral Sclerosis. Ann Neurol. 2013 doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 25.Cleary JD, Ranum LP. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet. 2013;22 (R1):R45–51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K, McDermott CJ, Traynor BJ, Kirby J, Wharton SB, Ince PG, Hardy J, Shaw PJ. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135 (Pt 3):751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442 (7105):920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 28.Dansithong W, Paul S, Comai L, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem. 2005;280 (7):5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 29.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72 (2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Villar K, Miller CA. Down-regulation of DENN/MADD, a TNF receptor binding protein, correlates with neuronal cell death in Alzheimer's disease brain and hippocampal neurons. Proc Natl Acad Sci U S A. 2004;101 (12):4210–4215. doi: 10.1073/pnas.0307349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Disney MD, Liu B, Yang WY, Sellier C, Tran T, Charlet-Berguerand N, Childs-Disney JL. A small molecule that targets r(CGG)(exp) and improves defects in fragile X-associated tremor ataxia syndrome. ACS Chem Biol. 2012;7 (10):1711–1718. doi: 10.1021/cb300135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron. 2013;80 (2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, Brook JD. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11 (7):805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 34.Filippova GN, Thienes CP, Penn BH, Cho DH, Hu YJ, Moore JM, Klesert TR, Lobanenkov VV, Tapscott SJ. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet. 2001;28 (4):335–343. doi: 10.1038/ng570. [DOI] [PubMed] [Google Scholar]
- 35.Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, Lai MC, Gendron TF, Petrucelli L, Fisher EM, Revesz T, Warren JD, Collinge J, Isaacs AM, Mead S. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126 (3):401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126 (6):829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Baumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11 (1):54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 40.Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurol Sci. 2011;32 (1):9–16. doi: 10.1007/s10072-010-0439-6. [DOI] [PubMed] [Google Scholar]
- 41.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63 (4):535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27 (1):48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- 43.Hadano S, Otomo A, Kunita R, Suzuki-Utsunomiya K, Akatsuka A, Koike M, Aoki M, Uchiyama Y, Itoyama Y, Ikeda JE. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One. 2010;5 (3):e9805. doi: 10.1371/journal.pone.0009805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol. 2012;8 (2):108–117. doi: 10.1038/nrneurol.2011.200. [DOI] [PubMed] [Google Scholar]
- 45.He F, Todd PK. Epigenetics in nucleotide repeat expansion disorders. Semin Neurol. 2011;31 (5):470–483. doi: 10.1055/s-0031-1299786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsiung GY, Dejesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ, Baker M, Rademakers R, Mackenzie IR. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135 (Pt 3):709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68 (4):654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91 (1):119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Human Molecular Genetics. 2004;13 (24):3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 50.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495 (7442):467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31 (2):89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74 (6):1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kragh CL, Ubhi K, Wyss-Coray T, Masliah E. Autophagy in dementias. Brain Pathol. 2012;22 (1):99–109. doi: 10.1111/j.1750-3639.2011.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar A, Parkesh R, Sznajder LJ, Childs-Disney JL, Sobczak K, Disney MD. Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem Biol. 2012;7 (3):496–505. doi: 10.1021/cb200413a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323 (5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 56.Ladd PD, Smith LE, Rabaia NA, Moore JM, Georges SA, Hansen RS, Hagerman RJ, Tassone F, Tapscott SJ, Filippova GN. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16 (24):3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- 57.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110 (47):E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee WC, Almeida S, Prudencio M, Caulfield TR, Zhang YJ, Tay WM, Bauer PO, Chew J, Sasaguri H, Jansen-West KR, Gendron TF, Stetler CT, Finch N, Mackenzie IR, Rademakers R, Gao FB, Petrucelli L. Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide Repeats in ALS/FTD Form Length-Dependent RNA Foci, Sequester RNA Binding Proteins, and Are Neurotoxic. Cell Rep. 2013 doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29 (4):499–503. doi: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60 (7):1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 62.Ma AS, Moran-Jones K, Shan J, Munro TP, Snee MJ, Hoek KS, Smith R. Heterogeneous nuclear ribonucleoprotein A3, a novel RNA trafficking response element-binding protein. J Biol Chem. 2002;277 (20):18010–18020. doi: 10.1074/jbc.M200050200. [DOI] [PubMed] [Google Scholar]
- 63.Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, Neumann M. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126 (6):859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 64.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science. 1992;255 (5049):1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 65.Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, Collinge J, Revesz T, Mead S, Warren JD. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135 (Pt 3):736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11 (4):323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289 (5485):1769–1773. doi: 10.1126/science.289.5485.1769. 8803 [pii] [DOI] [PubMed] [Google Scholar]
- 68.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10 (1):35–44. doi: 10.1016/s1097-2765(02)00563-4. S1097276502005634 [pii] [DOI] [PubMed] [Google Scholar]
- 69.Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson Y, Pickering-Brown S. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1 (1):68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19 (17):4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12 (5):435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126 (6):845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mooers BH, Logue JS, Berglund JA. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proceedings of the National Academy of Sciences of the United States of America. 2005;102 (46):16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126 (6):881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 75.Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125 (3):413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 76.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339 (6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 77.Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38 (7):758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 78.Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122 (6):673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pasquali L, Ruffoli R, Fulceri F, Pietracupa S, Siciliano G, Paparelli A, Fornai F. The role of autophagy: what can be learned from the genetic forms of amyotrophic lateral sclerosis. CNS Neurol Disord Drug Targets. 2010;9 (3):268–278. doi: 10.2174/187152710791292594. BSP/CDTCNSND/E-Pub/00031 [pii] [DOI] [PubMed] [Google Scholar]
- 80.Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6 (11):994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- 81.Pikkarainen M, Hartikainen P, Alafuzoff I. Ubiquitinated p62-positive, TDP-43-negative inclusions in cerebellum in frontotemporal lobar degeneration with TAR DNA binding protein 43. Neuropathology. 2010;30 (2):197–199. doi: 10.1111/j.1440-1789.2009.01043.x. [DOI] [PubMed] [Google Scholar]
- 82.Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, Dickson DW, Buratti E, Petrucelli L. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A. 2012;109 (52):21510–21515. doi: 10.1073/pnas.1211577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reddy K, Zamiri B, Stanley SY, Macgregor RB, Jr, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288 (14):9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72 (2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4 (9):e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci Transl Med. 2013;5 (208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85 (5):544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Satoh JI, Tabunoki H, Ishida T, Saito Y, Arima K. Dystrophic neurites express C9orf72 in Alzheimer's disease brains. Alzheimers Res Ther. 2012;4 (4):33. doi: 10.1186/alzrt136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Willemsen R, Pijnenburg YA, Heutink P, van Swieten JC. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135 (Pt 3):723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- 90.Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM, Pickering-Brown SM. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135 (Pt 3):693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31 (19):5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steinbach P, Glaser D, Vogel W, Wolf M, Schwemmle S. The DMPK gene of severely affected myotonic dystrophy patients is hypermethylated proximal to the largely expanded CTG repeat. Am J Hum Genet. 1998;62 (2):278–285. doi: 10.1086/301711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, Dejesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IR. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012;123 (3):409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suzuki N, Maroof AM, Merkle FT, Koszka K, Intoh A, Armstrong I, Moccia R, Davis-Dusenbery BN, Eggan K. The mouse C9ORF72 ortholog is enriched in neurons known to degenerate in ALS and FTD. Nat Neurosci. 2013;16 (12):1725–1727. doi: 10.1038/nn.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. The Journal of cell biology. 1995;128 (6):995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic acids research. 1996;24 (22):4407–4414. doi: 10.1093/nar/24.22.4407. 6e0437 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. CGG Repeat-Associated Translation Mediates Neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron. 2013;78 (3):440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Todd PK, Oh SY, Krans A, Pandey UB, Di Prospero NA, Min KT, Taylor JP, Paulson HL. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010;6 (12):e1001240. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol Cell. 2003;95 (3–4):169–178. doi: 10.1016/s0248-4900(03)00033-9. S0248490003000339 [pii] [DOI] [PubMed] [Google Scholar]
- 100.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323 (5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117 (10):2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70 (3):427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13 (7):805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E. Hypermethylation of the CpG Island Near the G4C2 Repeat in ALS with a C9orf72 Expansion. Am J Hum Genet. 2013;92 (6):981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xiao X, Wang Z, Jang M, Nutiu R, Wang ET, Burge CB. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol. 2009;16 (10):1094–1100. doi: 10.1038/nsmb.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110 (19):7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63 (4):538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 108.Zhang D, Iyer LM, He F, Aravind L. Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet. 2012;3:283. doi: 10.3389/fgene.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108 (1):260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]