

Abstract
Synthetic barbiturate receptors have been utilized for many applications due to their high binding affinities for complementary guests. Although interest in this class of receptors spans from supramolecular to materials chemistry, the effects of receptor steric bulk and pre-organization on guest binding affinity has not been studied systematically. To investigate the roles that steric bulk and pre-organization play in guest binding, we prepared a series of 12 deconstructed Hamilton receptors with varying degrees of steric bulk and pre-organization. Both diethylbarbital and 3-methyl-7-propylxanthine were investigated as guests for the synthetic receptors. The stoichiometry of guest binding was investigated using Job plots for each host-guest pair, and 1H NMR titrations were performed to measure the guest binding affinities. To complement the solution-state studies, DFT calculations at the B3LYP/6-31+G(d,p) level of theory employing the IEF-PCM CHCl3 solvation model were also performed. Calculated guest binding energies correlated well with the experimental findings and provided additional insight into the factors influencing guest binding. Taken together, the results presented highlight the interplay between pre-organization and steric interactions establishing favorable interactions for self-assembled hydrogen-bonded systems.
Intoduction
Hydrogen-bonding interactions are a widely-used structural arrangement found in many synthetic supramolecular structures. Although individual hydrogen bonds are much weaker than covalent bonds, hydrogen-bonding interactions commonly form cooperative networks when multiple donor and acceptor components combine. The fidelity of such networks can be maximized by encoding attractive primary and secondary interactions in the hydrogen-bonding structures1-2 or by increasing the pre-organization of hydrogen-bonding components to reduce the entropic cost for self-assembly.3 Similarly, the reversibility of hydrogen-bond formation allows for errors in the assembly process to be repaired, leading to formation of the thermodynamically-favored product. By engineering complementary hydrogen-bonding arrays into geometrically-controlled molecular components, larger self-assembled structures, including foldamers, homo- and hetero-multimeric structures, and cavity-containing 3D supramolecular host molecules can be accessed.4-15
One such class of self-assembled hydrogen-bonded host-guest complexes are synthetic barbiturate receptors. Also known as Hamilton receptors, this well-studied class of macrocyclic synthetic receptors bind barbituric acid derivatives in complimentary, pre-organized hydrogen-bonding motifs (Figure 1).16-21 Such receptors typically employ two hydrogen-bond donor-acceptor-donor (DAD) units that align with the two acceptor-donor-acceptor (ADA) faces of the barbiturate. The macrocyclic pre-organization found in most prototypical synthetic barbiturate receptors results in high guest binding affinities ranging from 104 − 105 M-1.16,22 In addition to binding barbiturates, this class of receptors accommodates other guests with the appropriate complementary hydrogen-bonding arrays including uracils,23-26 thymines,23-24,26-30 succinimides,24,31 glutarimides,18,24,32 cyanuric acids,23,33-35 and dipyridine-2-ylamines,31-32 demonstrating the versatility of the receptor scaffolds. This diversity has resulted in the use of synthetic barbiturate receptors in different applications including catalysis,36-38 electrooptical materials,34-35,39 and supramolecular dendrimers.33,40-41 Despite the prevalence of this receptor motif in various disciplines, the impacts of ligand pre-organization, such as the importance of the macrocyclic effect or of ligand flexibility, remain unexplored.
Figure 1.

Selected examples of synthetic barbiturate receptors. (a) Prototypical Hamilton receptor; (b) chelating bis(phosphine) barbiturate receptor; and (c) highly-conjugated non-macrocyclic barbiturate receptor.
Although non-macrocyclic barbiturate receptors have been prepared, studies of such scaffolds have primarily focused on interactions in the solid state. For example, though non-macrocyclic barbiturate receptors have been thoroughly exploited in the solid state to bind nanoparticles to surfaces,42-44 the solution state binding behavior of these receptors has not been investigated comprehensively. Investigation of such systems could provide valuable insight into the interplay between steric interactions near the hydrogen-bonding moieties and the requirements of pre-organization required for efficient guest binding.
Toward our goal of understanding the assembly requirements of deconstructed supramolecular systems, and to probe the requirements of ligand rigidity, macrocylization, and pre-organization on barbiturate receptor designs, we have systematically deconstructed barbiturate receptors into simple subunits to determine the effects of ligand bifurcation on barbiturate binding. By measuring the binding affinities and stoichiometries of both barbital and xanthine guests with rigid, flexible, or bifurcated ligands, we directly investigated the pre-organization requirements for self-assembly. To complement the experimental results, we also screened and refined different DFT computational methods to generate a model that correlated well with solution data. Taken together, these results help to establish the requirements for effective barbiturate binding in synthetic host molecules and can be applied to other host-guest systems in which receptor pre-organization is a requirement for self-assembly.
Results and Discussions
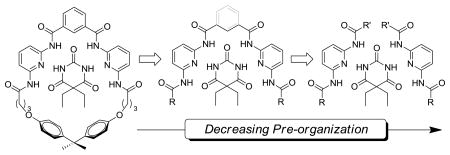
To further understand the effect that pre-organization and steric interactions play in determining the guest binding affinities of barbiturate receptors, we deconstructed prototypical macrocyclic barbiturate receptors into more simple subunits (Figure 2). The impacts of steric constraints on guest binding were investigated by preparing a library of symmetric (1a-c) or unsymmetric (1d-f) bifurcated hosts with methyl, phenyl or tert-butyl groups on the peripheral amides. Similarly, the role of pre-organization on self-assembly was investigated by preparing two receptor sets with either a flexible alkyl spacer between the two 2,6-dicarboxamido pyridine units (3a-c) or a rigid phenyl spacer between the 2,6-diamidopyridine units (4a-c). Both barbital (5) and 3-methyl-7-propylxanthine (6) were used as guests to investigate the structures and stoichiometries of the hydrogen-bonded constructs.
Figure 2.

Deconstruction of Hamilton receptors to determine effects of pre-organization and steric bulk.
Synthesis
Symmetric 2,6-dicarboxamido pyridine hosts 1a-c were prepared from 2,6-diaminopyridine by reaction with the desired acid chloride in the presence of triethylamine. To prepare unsymmetric 2,6-dicarboxamido pyridine receptors 1d-f, 2,6-diaminopyridine was first reacted with one equivalent of the desired acid chloride in the absence of base to afford monoamido pyridines 2a-c, followed by installation of a second amide by treatment with a second acid chloride (Scheme 1). Non-macrocyclic barbiturate receptors were prepared with both flexible and rigid linkers between the two 2,6-dicarboxamido pyridine units. Treatment of glutaric acid with SOCl2 afforded glutaryl chloride, which was treated with mono-amide 2a-c to afford the flexible barbiturate receptors 3a-c. Similarly, treatment of isophthalic acid with SOCl2 generated isophthalolyl chloride, which was treated with monoamines 2a-c to generate rigid backbone ligands 4a-c.
Scheme 1.

Preparation of hydrogen-bonding ligands based on 2,6-dicarboxyamido pyridine scaffolds.
Host-Guest Binding Studies
To obtain binding constants for the deconstructed barbiturate receptors, 1H NMR titrations were performed for each receptor/guest pair. Because guest binding involves hydrogen bonding of the amide N-H groups of both the host and the guest molecules, changes in N-H chemical shift reflect the position of the thermodynamic host-guest equilibrium during the course of the titration. To test the barbiturate binding affinity of each host construct, we used barbital (5) as the guest due to its high solubility and previous use as a guest in similar systems.16,23 Barbital has two hydrogen-bonding ADA faces that can interact with the DAD faces of prototypical barbiturate receptors. For receptors with flexible (3a-c) or rigid (4a-c) backbones, 5 is expected to form a 1:1 host:guest complex with each face of 5 interacting with each DAD face of the ligand. For bifurcated ligands 1a-f, either a 1:1 or a 2:1 host:guest complex could be formed, depending on the relative magnitude of the enthalpic gain upon hydrogen bonding and the entropic penalty for assembly of three components. In addition to using 5 as a guest, we also performed titrations with 3-methyl-7-propylxanthine (6), which has only one ADA face, thus simplifying the possible binding modes (Figure 3). Furthermore, 6 is less likely to self-aggregate in solution, whereas barbital derivatives, such as 5, are known to form self-complementary hydrogen-bonded oligomers.45 Previous studies with similar systems have shown negligible host dimerization.23,32 For each host-guest system, a Job plot was constructed to determine the stoichiometry of guest binding. After establishing the binding stoichiometry, 1H NMR titrations were performed in triplicate and the N-H chemical shifts of the guest were followed during the titrations. The resulting data was fit to the established binding stoichiometry.46 Figure 4 shows the characteristic shift in the N-H 1H NMR resonances used to quantify guest binding.
Figure 3.

Barbital (5) and 3-methyl-7-propylxanthine (6) guests used in the titration studies with ligands 1a-f, 3a-c, and 4a-c.
Figure 4.
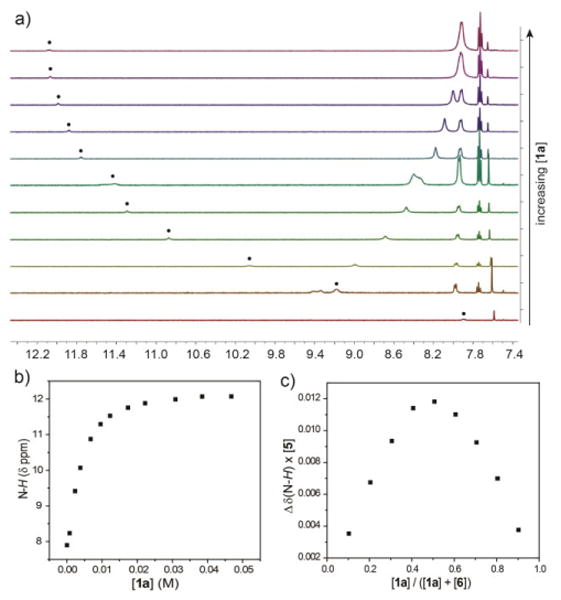
Representative 1H NMR (500 MHz, 25 °C, CDCl3) titration data for 1a with 6. (a) Stacked 1H NMR titration spectra; (b) Plot of the N-H chemical shift data from the 1H NMR titration; and (c) Job plot for 1a binding to 6 confirming a 1:1 binding stoichiometry.
Job plots of bifurcated barbiturate receptors 1a-f with 5 and 6 revealed 1:1 binding stoichiometries, suggesting that the entropic penalty to form the three-component system with 5 was too large for the relatively weak binding of 1a-f with 5 and 6 to overcome. The binding affinities of 2,6-dicarboxamido pyridine hosts 1a-f for guests 5 and 6 depended greatly on the steric bulk at the periphery of the receptor (Table 1). For example, replacing one or both methyl groups of 1a with tert-butyl groups (1d, 1f) reduces the binding affinities of 5 and 6 by almost one order of magnitude per tert-butyl group. This sizeable reduction in binding affinity is likely due to the more twisted guest approach angle required to avoid disfavorable steric interactions between the host and the guest (vide infra). Similar trends are observed for the addition of phenyl groups, although the magnitude of the decrease in binding affinity is attenuated, which is likely due to rotation of the phenyl group away from the guest to minimize disfavored steric interactions. In all cases, binding constants for 6 were larger than those determined for 5, which is consistent with the propensity of 5 to form hydrogen-bonded aggregates. Compared to binding affinities of macrocyclic receptors (Ka ≈ 104 M-1), these results demonstrate that complete bifurcation of Hamilton-derived receptors greatly diminishes guest binding affinity, thus suggesting that greater host pre-organization is required to generate high-fidelity guest binding.
Table 1.
Binding constants for 5 and 6 with deconstructed barbituric acid receptors 1a-f, 3a-c, and 4a-c.a
| Binding Constant (Ka, M-1) | Binding Constant (Ka, M-1) | ||||
|---|---|---|---|---|---|
| 5 | 6 | 5 | 6 | ||
| 1a | 85 ± 25 | 490 ± 70 | 3a | 70 ± 37b | Ka1 = 1230 ± 280b |
| Ka2 = 184 ± 78b | |||||
| 1b | 2 ± 1 | 3 ± 1 | 3b | 40 ± 4 | 74 ± 15 |
| – b,c | 7 ± 1b | ||||
| 1c | 7 ± 2 | 21 ± 3 | 3c | – b,c | 24 ± 7b |
| 1d | 17 ± 2 | 28 ± 2 | 4a | 139 ± 18b | 70 ± 9b |
| 1e | 29 ± 1 | 109 ± 14 | 4b | 174 ± 3 | 40 ± 3 |
| 6 ± 2b | 7 ± 5b | ||||
| 1f | 3 ± 1 | 6 ± 1 | 4c | 4 ± 8b | 22 ± 10b |
Titrations were performed in CDCl3 at 25 °C. All measurements are the average of at least three independent titrations.
Performed in 5% DMSO in CDCl3 due to poor solubility of either the host or the host-guest complex.
Binding constant too low (< 5 M-1) to measure accurately.
To further investigate the degree of pre-organization required for optimal guest binding, receptors 3a-c and 4a-c were prepared. These scaffolds employ either flexible (3a-c) or rigid (4a-c) linkers in the backbone to allow for the degree of pre-organization to be modified. Because both of the 2,6-dicarboxamido pyridine groups in these receptors are tethered together, the entropic penalty for binding 5 should be attenuated. By contrast, hosts 3a-c and 4a-c could potentially form either 1:1 or 1:2 host:guest complexes with 6 because 6 does not have entirely complementary interactions to interface with the two DAD faces of the receptor leaving one side of the receptor face free to interact with a second guest. For these receptors, increased steric bulk of the tethered host will reduce the overall binding affinity and more likely result in 1:1 complex formation due to reduced capacity for multiple guests.
Consistent with the results obtained from bifurcated hosts 1a-f, steric interactions from the receptor periphery greatly impacted guest binding for the scaffolds with flexible backbones 3a-c. For example, replacing the methyl groups in 3a with tert-butyl groups (3b) resulted in a 60-fold decrease in barbital binding. This observation is consistent with the bifurcated 2,6-dicarboxamido pyridine hosts (1a-f) and demonstrates the sensitivity to steric repulsion near the hydrogen-bonding sites. Flexible receptors 3a-c also maintained higher binding affinity for 6 than for 5, although the magnitude of this preference was diminished. Job plots of 3a-c with 5 and 3b,c with 6 confirmed 1:1 host:guest binding. Investigations of 3a with 6, however, revealed two binding events corresponding to the formation of 1:1 and 1:2 host:guest complexes. The first binding event, corresponding to a 1:1 3a:6 complex, had a Ka of 1230 M-1, which was an order of magnitude greater than binding of a second guest with a Ka of 180 M-1. The difference in Ka values between the first and second guest binding events are consistent with the required elongation and corresponding entropic penalty of 3a to accommodate two xanthine guests. Although 2:1 binding was observed with 6, only 1:1 binding was observed with 5. If barbital were to form a 1:2 host:guest complex, then the benefits from the chelate effect would need to be sacrificed in order to accommodate two barbital guests.
Job plots of 5 and 6 with 4a-c confirmed exclusively 1:1 binding and 1H NMR titration data was subsequently fit to a 1:1 model. For 5, the methyl end capped host (4a) had the highest binding affinity, followed by the tert-butyl (4b), and finally the phenyl (4c) analogs. Although phenyl groups are less sterically demanding than tert-butyl groups, the requirement of phenyl group rotation to accommodate a bound guest results in a reduction of the conjugation into the amide and thereby reduces the enthalpic gain upon guest binding. As expected, the rigid backbone hosts had lower affinities for 6 than 5 due to constrained binding pockets present in the host constructs and greater steric bulk of 6 in comparison to 5. Similar to trends observed for other host-guest pairs, steric bulk on the periphery of receptors 4a-c directly affected the binding affinity toward 6.23,32
The interplay between pre-organization and steric interactions between host and guest is also observed across the different types of receptors. For example, by comparing the binding affinities of 5 with tert-butyl substituted receptors 1b, 3b, and 4b, a clear trend is apparent, with the more preorganized structures producing stronger binding. Upon increasing the pre-organization, the binding affinity for 5 increases from 2 M-1 for 1, to 40 M-1 for 3b and finally to 139 M-1 for 4b. This series clearly demonstrates that host pre-organization can offset some of the disfavorable steric interactions present in this series of receptors. A second trend is observed for the same series of host molecules interacting with 6. In this case, because the xanthine guest cannot form favorable interactions with both sides of the symmetric receptors 3b and 4b, the steric influences are more important. The increase of binding affinities from 3 M-1 for 1b to 75 M-1 for 3b is primarily due to the decreased steric bulk from the flexible propyl chain by comparison to a tert-butyl group. Replacement of the flexible backbone of 3b with the rigid phenyl backbone in 4b slightly increases the steric encumbrance on the guest due to the inability of the phenyl group in 4b to completely rotate away from the bound guest. As would be expected from these interactions, the binding constant of 6 with 4b is lower than for 3b, but higher than for 1b. Taken together, these comparisons highlight the delicate balance between pre-organization and minimization of steric interactions for synthetic barbiturate receptors.
To better understand the enthalpic and entropic effects associated with guest binding, we determined ΔH and ΔS using van't Hoff analysis for hosts 1a, 1c, and 1e binding guest 6 (Figure S25). The binding enthalpies and entropies (ΔH, ΔS) were determined to be 1a (4.7 kcal/mol, -4.0 eu), 1c (3.8 kcal/mol, -6.5 eu), and 1e (5.8 kcal/mol, -10.1 eu). Both 1a and 1c have symmetric amide substituents, whereas 1e does not. This desymmetrization results in preferential orientation of the guest to minimize the steric interaction between the propyl tail of 6 and the phenyl substituent of 1e, resulting in a more negative binding entropy than was observed for 1a or 1c. Changes in the binding enthalpies are also observed. For example, 1c has two phenyl substituents that must twist out of conjugation with the amide to allow for guest binding, which results in a lower observed binding enthalpy for 1c by comparison to 1a or 1e.
Computational Studies on Hydrogen-Bonded Adducts
To gain further insight into the factors influencing host-guest binding, we optimized the structure of each host, guest, and hydrogen-bonded adduct using Gaussian 09 at the B3LYP/6-31+G(d,p) level of theory employing the IEF-PCM CHCl3 solvation model. Based on the computed energies for each of the optimized geometries for each ligand, guest, and host-guest component, binding enthalpies were calculated (Table 2).47 Although the DFT calculations over-estimated the absolute magnitude of the binding energies, good correlation between the experimentally-determined binding affinities determined in CDCl3 and the computed binding enthalpies was observed, suggesting that this computation level of theory can be used to reliably estimate trends in binding affinities of future similar barbiturate-binding scaffolds (Figure 6).
Table 2.
Calculated binding energies for 5 and 6 with 1a-f, 3a-c, and 4a-c.a
| –ΔHbinding (kcal/mol) | –ΔHbinding (kcal/mol) | ||||
|---|---|---|---|---|---|
| 5 | 6 | 5 | 6 | ||
| 1a | 8.18 | 8.17 | 3a | 10.88 | 9.40 |
| 1b | 3.69 | 5.19 | 3b | 5.18 | 7.41 |
| 1c | 5.26 | 6.63 | 3c | 3.98 | 8.53 |
| 1d | 6.90 | 7.69 | 4a | 12.69 | 8.13 |
| 1e | 6.44 | 7.91 | 4b | 7.96 | 8.17 |
| 1f | 5.33 | 6.05 | 4c | 10.83 | 6.96 |
Calculated using Gaussian 09, B3LYP/6-31+G(d,p), with IEF-PCM solvation model for CHCl3. Binding energies correspond to the difference in ZPE-corrected energies from the host-guest complexes and the isolated host and guest species.
Figure 6.
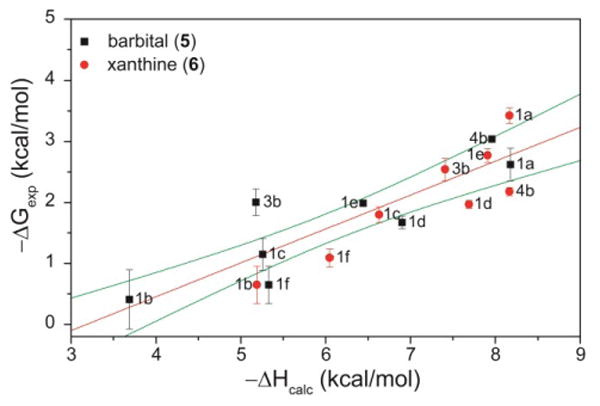
Comparison of experimentally-determined binding affinities (ΔG, kcal/mol) with the calculated binding enthalpies, including linear fit (red) and 95% confidence interval (green).
Having established the validity of this computation model for estimating the magnitude of the binding interactions with the barbiturate-binding hosts, we used the optimized structures to gain insight into the major factors affecting guest binding affinities. By comparing the optimized geometries 1a-f interacting with 5 and 6, the steric bulk on the periphery of the receptors greatly affected the approach angle of 5 or 6 to the 2,6-dicarboxamido pyridine scaffolds. Comparing the binding affinities of symmetric 1a-c with 5, 1a forms the strongest interaction with 5 due to the limited repulsive steric interactions. The level of deviation from an ideal co-planar guest approach angle can be compared by measuring the angle between the least squares planes of the pyridine ring of 1a and the six-membered ring of 5. Comparing guest approach angles with 5, 1a had the lowest approach angle of 15.1° angle, which increases to 32.3° for symmetric tert-butyl compound 1b, and then decreased for the symmetric phenyl complex 1c (Figure 7). These twist angles correlate strongly with both the experimental and computational binding affinities. Similarly, for hosts tethered with either flexible or rigid backbones, the steric pressure exerted on the guest is greatly dictated by the size of the amide groups (Figure 7b). Although the phenyl groups can rotate to minimize steric interactions with the bound barbital, and potentially generate favorable CH-π interactions, this rotation results in a break of planarity with the amide, thereby reducing the overall conjugation of the system.
Figure 7.

Examples of the optimized geometries for host-guest species 1a-c with 5 (a) and 3a-c with 5 (b). Changing the steric bulk of the periphery of the receptor influences the twist angel of guest approach and the overall hydrogen-bonding fidelity.
Conclusion
Both experimental titration data and DFT calculations show that both pre-organization and steric bulk play a direct role in the binding affinity of the deconstructed Hamilton receptors. For the least preorganized hosts, 1a-f, steric bulk had the largest role in influencing guest binding affinity with the least bulky host 1a maintaining the highest binding affinities toward 5 and 6, whereas the most bulky host, 1b, had the smallest guest binding affinities. For hosts 3a-c with moderate pre-organization, the effects of steric bulk were attenuated. For the most rigid hosts, 4a-c, steric interactions played a direct role in the guest binding affinities with the bulkiest host having the lowest affinity and the least bulky host having the highest binding affinity.
In addition to steric interactions, pre-organization also played a distinct role in guest binding affinities. The more pre-organized hosts, 4a-c, had the highest binding affinity for barbital due to the high complementarity with the two hydrogen-bonding faces of 5. The less pre-organized hosts, 3a-c, had higher binding affinities for 6 than did 4a-c due to the flexibility of the ligand backbone, which allowed for two guests to be accommodated. The completely bifurcated hosts, 1a-f, had much lower binding affinities than 3a-c or 4a-c due to the decreased ligand pre-organization. Importantly, the combined experimental and computational results obtained for the deconstructed barbiturate receptors are also applicable to other hydrogen-bonding systems in which pre-organization must be balanced with disfavorable steric interactions between the host and guest. These results illustrate the important roles that both sterics and pre-organization play in host-guest complexes and how each should be either minimized or maximized in order to obtain the highest affinity for a given host-guest system.
Experimental Section
Materials and Methods
All commercially-available reagents and deuterated solvents were used as received. Anhydrous solvents used for syntheses were collected from a solvent purification system. Reactions were monitored by TLC and the products were purified on an automated flash chromatography instrument. NMR spectra were recorded at the indicated frequencies and chemical shifts are reported in parts per million (δ) and are referenced to residual protic solvent resonances. The following abbreviations are used in describing NMR couplings: (s) singlet, (d) doublet, (t) triplet, (m) multiplet and (b) broad.
General Job plot procedure
Job plots were performed in CDCl3 and monitored by 1H NMR for host molecules 1a-f, 3b, 3b, and 4b. Job plots with the host molecules 3a, 3c, 4a, and 4c were performed in 5% DMSO-d6 in CDCl3 due to poor solubility of the hosts in CDCl3. All Job plots were performed using total (host + guest) concentrations of 10 mM, but compounds 1a-f were also run at 100 mM total concentrations due to weaker guest binding. For a typical Job plot, 3 mL of a host solution and 3 mL of a guest solution were prepared and then divided between 10 NMR tubes in 10 mol% increments. After equilibration, the 1H NMR spectrum for each sample was recorded and the shift in the guest N-H resonance was used to construct the Job plot.
General procedure binding constant determination
Binding studies were performed in CDCl3 for host molecules 1a-f, 3b, and 4b and monitored by 1H NMR spectroscopy. Due to poor solubility of compounds 3a, 3c, 4a, and 4c in CDCl3, these compounds were measured in 5% DMSO-d6 in CDCl3. In order to compare the binding constants obtained in 5% DMSO-d6 in CDCl3 to those obtained in neat CDCl3, titrations with host molecules 3b and 4b were also carried out in 5% DMSO-d6 in CDCl3. In a typical CDCl3 titration, 2 mL of a 1 mM 5 or 6 was prepared. The guest solution was then divided such that 1 mL was placed into an NMR tube and the other 1 mL was used to create a second solution containing 150-300 mM host. An initial spectrum of the guest was recorded, after which aliquots (5-100 µL) of the host solution were added until the N-H resonance of 5 or 6 no longer shifted. In a typical 5% DMSO-d6 in CDCl3 titration, 2 mL of a 3 mM host solution was prepared. The host solution was then divided such that 1 mL was placed into an NMR tube and the other 1 mL was used to create a second stock solution containing 25-100 mM guest. An initial spectrum of the host was recorded, after which aliquots (5-100 µL) of 5 or 6 were be added until the N-H resonance of the 5 or 6 no longer shifted.
General van't Hoff plot procedure
Stock solutions of 1a, 1c, 1e, and 6 were prepared at concentrations of 24 mM, 120 mM, 86 mM, and 2 mM, respectively. These host concentrations were chosen to ensure complete host-guest complexation at the highest concentration. Six NMR samples of varying host:guest ratios were prepared for each host/guest pair and the N-H resonance of 6 was monitored over the temperature range 298 – 328 K. All temperatures were calibrated using a MeOH temperature standard.48
Computational Details
Calculations were performed using the Gaussian 0949 software package with the GaussView50 graphical user interface. Graphical representations were produced using the UCSF Chimera package v1.8.51 Initial conformational searches and optimizations were performed using either the 3-21g or 6-31g basis sets, followed by full geometry optimizations and unscaled frequency calculations at the B3LYP/6-31+G(d,p) level of theory using the IEF-PCM solvation model for chloroform. Frequency calculations were performed on all converged structures confirmed that they corresponded to local minima. Calculated enthalpies are reported as zero-point corrected enthalpies. In all cases, the lowest energy conformer was used to compare the relative energetics of the calculated species.
Syntheses
N,N'-(Pyridine-2,6-diyl)diacetamide (1a)
A round bottom flask was charged with dry THF (50 mL), 2,6-diaminopyridine (3.0 g, 28 mmol), and triethylamine (9.7 ml, 69 mmol). The flask was then lowered into an ice bath and degassed with N2. Acetyl chloride (4.3 mL, 61 mmol) was added to an addition funnel containing dry THF (20 mL), and the resultant solution was then slowly added to the diaminopyridine solution while stirring in the ice bath under N2. Once the addition of the acid chloride was complete, the ice bath was removed, and the reaction was allowed to warm to room temperature overnight while stirring under N2. The reaction mixture was concentrated by rotary evaporation and the crude product was purified by column chromatography (Si2O, EtOAc) to afford 1a as off-white crystals (5.2 g, 96% yield) with spectroscopic properties consistent with literature data.32 Mp = 201-202 °C. 1H NMR (500 MHz, CDCl3) δ: 7.91 (d, J = 7.7, 2H), 7.73 (t, J = 7.9, 1H), 7.59 (s, 2H), 2.22 (s, 6H). 13C{1H} NMR (125 MHz, CDCl3) δ: 168.5, 149.4, 140.9, 109.5, 24.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C9H12N3O2, 194.0930; found 194.0932.
N,N'-(Pyridine-2,6-diyl)dipivalamide (1b)
The monosubstituted diaminopyridine 2b was prepared according to the general procedure outlined for 1a with the following quantities: 2,6-diaminopyridine (33 mg, 0.30 mmol) in THF (15 mL) and trimethylacetyl chloride (81 µL, 0.66 mmol) in THF (15 mL). The crude product was purified by column chromatography (Si2O, EtOAc) to afford a tan solid (83 mg, 99% yield) with spectroscopic properties consistent with literature data.32 Mp = 112-113 °C. 1H NMR (500 MHz, CDCl3) δ: 7.94 (d, J = 7.8, 2H), 7.76 (s, 2H), 7.71 (t, J = 7.8, 1H), 1.34 (s, 18H) 13C{1H} NMR (125 MHz, CDCl3) δ: 176.8, 149.6, 140.8, 109.3, 39.8, 27.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C15H24N3O2, 278.1869; found 278.1859.
N,N'-(Pyridine-2,6-diyl)dibenzamide (1c)
The monosubstituted diaminopyridine 1c was prepared according to the general procedure outlined for 1a with the following quantities: 2,6-diaminopyridine (31 mg, 0.28 mmol) in THF (15 mL) and benzoyl chloride (72 µL, 0.62 mmol) in THF (15 mL). The crude product was purified by chromatography (Si2O, 1:1 EtOAc:DCM) to afford a tan solid (89 mg, 98% yield) with spectroscopic properties consistent with literature data.52 Mp = 168-170 °C. 1H NMR (500 MHz, CDCl3) δ: 8.53 (s, 2H), 8.14 (d, J = 7.8, 2H), 7.93 (d, J = 7.3, 4H), 7.84 (t, J = 7.7, 1H), 7.60 (t, J = 7.3, 2H), 7.53 (t, J = 7.3, 4H). 13C{1H} NMR (125 MHz, CDCl3) δ: 170.0, 165.5, 149.7, 141.3, 134.1, 133.6, 132.37, 130.0, 128.9, 128.5, 127.2, 110.01. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H16N3O2, 318.1243; found 318.1247.
N-(6-Acetamidopyridin-2-yl)pivalamide (1d)
A round bottom flask was charged with dry THF (75 mL), 2a (2.9 g, 19 mmol), and triethylamine (5.3 mL, 39 mmol). The flask was then lowered into an ice bath and degassed with N2. Trimethylacetyl chloride (3.0 mL, 25 mmol) was added to an addition funnel containing dry THF (25 mL) and the resultant acid chloride solution was then slowly added to the diaminopyridine solution while stirring in the ice bath under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The reaction was concentrated by rotary evaporation and the crude product was purified by column chromatography (SiO2, EtOAc) to afford a white crystalline solid (3.88 g, 66%). Mp = 128-129 °C. 1H NMR (500 MHz, CDCl3) δ: 7.97 (d, J = 7.8, 1H), 7.91 (d, J = 7.5, 1H), 7.74 (m, 1H), 7.71 (s, 1H), 2.23 (s, 3H), 1.35 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 176.9, 168.4, 149.7, 149.3, 140.9, 109.5, 109.3, 39.8, 27.5, 24.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C12H18N3O2, 236.1399; found 236.1402.
N-(6-Acetamidopyridin-2-yl)benzamide (1e)
The disubstituted diaminopyridine 1e was prepared according to the general procedure outlined for 1d with the following quantities: benzoyl chloride (1.9 mL, 16 mmol) in THF (25 mL) was added slowly to 2a (1.9 g, 13 mmol) and triethylamine (3.6 mL, 26 mmol) in THF (50 mL). Purified by column chromatography (Si2O, EtOAc) to afford a white crystalline solid (2.69 g, 81%). Mp = 195-196 °C. 1H NMR (500 MHz, CDCl3) δ: 8.34 (s, 1H), 8.10 (d, J = 7.8, 1H), 7.96 (d, J = 6.4, 1H), 7.92 (d, J = 7.8, 2H), 7.79 (m, 1H), 7.61 (t, J = 7.3, 1H), 7.53 (t, J = 8.3, 2H), 2.24 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ: 165.4, 149.5, 140.0, 134.2, 132.3, 128.9, 127.1, 109.6, 24.8. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C14H14N3O2, 256.1086; found 256.1097.
N-(6-Pivalamidopyridin-2-yl)benzamide (1f)
The disubstituted diaminopyridine 1f was prepared according to the general procedure outlined for 1d with the following quantities: trimethylacetyl chloride (0.24 mL, 2.1 mmol) in THF (25 mL) was added slowly to 2c (0.35 g, 1.6 mmol) and triethylamine (0.34 mL, 2.5 mmol) in THF (50 mL). Purified by column chromatography (Si2O, CH2Cl2) to afford a chalky off-white solid (0.51 g, 82%). Mp = 120-121 °C. 1H NMR (500 MHz, CDCl3) δ: 8.33 (s, 1H), 8.06 (d, J = 8.3, 1H), 7.97 (d, J = 8.3, 1H), 7.94 (d, J = 4.3, 2H), 7.79 (s, 1H), 7.75 (t, J = 8.3, 1H), 7.56 (t, J = 7.3, 1H), 7.49 (t, J = 6.5, 2H), 1.32 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 176.8, 165.4, 149.8, 149.6, 140.9, 134.2, 132.3, 128.9, 127.1, 109.7, 109.6, 39.8, 27.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H20N3O2, 298.1556; found 298.1565.
N-(6-Aminopyridin-2-yl)acetamide (2a)
A round bottom flask was charged with dry THF (10 mL) and 2,6-diaminopyridine (1.0 g, 9.1 mmol). The flask was then lowered into an ice bath and degassed with N2. Acetyl chloride (0.32 mL, 4.6 mmol) was added to an addition funnel containing dry THF (20 mL), and the resultant solution was then added slowly to the diaminopyridine solution over the course of 1 hour while stirring at 0 °C under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The precipitate from the reaction was filtered, and the resultant filtrate was concentrated by rotary evaporation. The crude product was purified by column chromatography (SiO2, EtOAc) to afford a tannish pink solid (0.65 g, 95%), with spectroscopic properties consistent with literature data.32 Mp = 150-152 °C. 1H NMR (300 MHz, CDCl3) δ: 7.70 (s, 2H), 7.54-7.46 (m, 2H), 6.27 (d, J = 7.8, 1H), 4.32 (s, 2H), 2.18 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ: 168.4, 157.0, 149.7, 140.2, 104.3, 103.3, 24.7. HRMS (ESI-TOF) m/z: [M]+ Calcd for C7H9N3O, 151.0746; found 151.0742.
N-(6-Aminopyridin-2-yl)pivalamide (2b)
The monosubstituted diaminopyridine 2b was prepared according to the general procedure outlined for 2a with the following quantities: 2,6-diaminopyridine (0.51 g, 4.6 mmol) in THF (10 mL) and trimethylacetyl chloride (0.25 mL, 2.2 mmol) in THF (5 mL). Purified by column chromatography (Si2O, EtOAc) to afford a tan solid (0.86 g, 97% yield), with spectroscopic properties consistent with literature data.32 Mp = 131-132 °C. 1H NMR (500 MHz, CDCl3) δ: 7.73 (s, 1H), 7.59 (d, J = 8.3, 1H), 7.46 (t, J = 7.8, 1H), 6.26 (d, J = 7.81 1H), 4.35 (s, 2H), 1.32 (s, 9H). 13C{1H} NMR (125 MHz, CDCl3) δ: 168.3, 157.0, 149.8, 104.3, 103.3, 39.7, 27.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H16N3O, 194.1293; found 194.1295.
N-(6-Aminopyridin-2-yl)benzamide (2c)
The monosubstituted diaminopyridine 2c was prepared according to the general procedure outlined for 2a with the following quantities: 2,6-diaminopyridine (2.0 g, 18 mmol) in THF (50 mL) and benzoyl chloride (1.0 mL, 9.0 mmol) in THF (25 mL). Purified by column chromatography (Si2O, CH2Cl2) to afford a white crystalline solid (3.53 g, 92%). Mp = 184-186 °C. 1H NMR (500 MHz, CDCl3) δ: 8.33 (s, 1H), 7.91 (d, J = 8.0, 2H), 7. 74 (d, J = 8.0, 1H), 7.59-7.50 (m, 4H), 6.32 (d, J = 8.0, 1H), 4.39 (s, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ: 165.4, 157.1,149.9, 104.3, 134.5, 132.1, 128.8, 127.1, 104.6, 103.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C12H12N3O, 214.0980; found 214.0974.
N1,N5-bis(6-Acetamidopyridin-2-yl)glutaramide (3a)
Glutaric acid (0.21g, 1.6 mmol) was stirred in thionyl chloride (3 mL) for 5 hours at room temperature, after which the thionyl chloride was removed under vacuum. A round bottom flask was charged with dry THF (50 mL), 2a (0.40 g, 2.7 mmol), and triethylamine (1.1 mL, 8.1 mmol). The flask was then lowered into an ice bath and degassed with N2. The crude glutaroyl chloride was taken up in THF (10 mL) and added to an addition funnel. The resultant acid chloride solution was then slowly added to the diaminopyridine solution while stirring in the ice bath under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The reaction mixture was concentrated by rotary evaporation, and the residue was then taken up in EtOAc washed with water and then saturated NaHCO3. The organic layer was concentrated and the resultant residue was then taken up in water (20 mL) and heated to 80 °C until all of the solid was dissolved. Upon cooling, the product crystallized as a white crystalline solid, which was collected by filtration and dried under vacuum (0.70 g, 65%). Mp = 221-222 °C. 1H NMR (300 MHz, DMSO) δ: 10.52 (s, 2H), 10.19 (s, 2H), 8.54 (s, 2H), 8.18 (d, J = 7.3, 2H), 7.83 (s, 6H), 7.70 (t, J = 7.6), 3.36 (s, 4H), 2.14 (s, 2H). 13C{1H} NMR (125 MHz, DMSO) δ: 172.2, 169.7, 150.8, 143.3, 109.5 35.8, 24.5, 21.1. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H23N6O4, 399.1781; found 399.1799.
N1,N5-bis(6-Pivalamidopyridin-2-yl)glutaramide (3b)
The alkyl tethered diaminopyridine 3b was prepared according to the general procedure outlined for 3a with the following quantities: glutaroyl dichloride (0.56 g, 3.3 mmol) in THF (20 mL) was added slowly to 2b (1.1 g, 5.6 mmol) and TEA (1.4 mL, 10 mmol) in THF (50 mL). Purified by column chromatography (Si2O, DCM with 5% of a 9:1 MeOH:NH4OH mixture) to afford a white crystalline solid (1.11 g, 41%). Mp = 258 °C (dec). 1H NMR (300 MHz, CDCl3) δ: 7.95 (d, J = 8.1, 2H), 7.90 (s, 2H), 7.86 (d, J = 8.7, 2H), 7.77 (s, 2H), 7.71 (t, J = 8.1, 2H), 2.55 (t, J = 6.9, 4H), 2.16 (m, 2H), 1.33 (s, 18H). 13C{1H} NMR (125 MHz, CDCl3) δ: 176.9, 170.7, 149.8, 149.2, 140.8, 109.6, 109.3, 39.8, 36.1, 27.5, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H35N6O4, 483.2720; found 483.2744.
N1,N5-bis(6-Benzamidopyridin-2-yl)glutaramide (3c)
The alkyl tethered diaminopyridine 3c was prepared according to the general procedure outlined for 3a with the following quantities: glutaroyl dichloride (0.39 g, 2.3 mmol) in THF (20 mL) was added slowly to 2c (0.81 g, 3.8 mmol) and TEA (1.6 mL, 11 mmol) in THF (50 mL). Purified by column chromatography (Si2O, 3:2 hexanes:EtOAc) to afford a white crystalline solid (1.38 g, 67%). Mp = 179 °C (dec). 1H NMR (500 MHz, CDCl3) δ: 8.67 (s, 4H), 8.38 (d, J = 8.5, 2H), 7.84 (m, 6H), 7.50 (d, J = 8.0, 2H), 7.42 (t, J = 7.5, 4H), 6.89 (d, J = 8.0, 2H), 2.76 (t, J = 7.0, 4H), 2.08 (dd, J = 7.5, 8.0, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ: 173.0, 166.0, 152.0, 148.6,140.5, 134.2, 132.2, 128.6, 127.7, 119.7, 114.0, 32.3, 17.0. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C29H27N6O4, 523.2094; found 523.2106.
N1,N5-bis(6-Acetamidopyridin-2-yl)isophthalamide (4a)
Isophthalic acid (0.30 g, 1.8 mmol) was stirred in thionyl chloride (2 mL) at 65 °C with catalytic DMF for 7 hours, after which the excess thionyl chloride was removed under vacuum. A round bottom flask was charged with dry THF (50 mL), 2a (0.44 g, 3.0 mmol), and triethylamine (1.2 mL 8.9 mmol). The flask was then lowered into an ice bath and degassed with N2. The crude isophthaloyl chloride was taken up in dry THF (20 mL) and added to an addition funnel. The acid chloride solution was then slowly added to the diaminopyridine solution while stirring in the ice bath under N2. Once the addition of the acid chloride was complete, the ice bath was removed and the reaction was allowed to warm to room temperature overnight while stirring under N2. The reaction mixture was concentrated by rotary evaporation and the residue was washed with water and then with saturated NaHCO3. The organic layer was concentrated, and the resultant residue was then taken up in water (20 mL) and heated to 80 °C until all of the solid was dissolved. Upon cooling, the product crystallized as a white crystalline solid, which was collected by filtration and dried under vacuum (1.73 g, 45%). Mp = 161-163 °C. 1H NMR (300 MHz, DMSO) δ: 10.51 (s, 2H), 10.18 (s, 2H), 8.53 (s, 1H), 8.16 (d, J = 7.3, 2H), 8.02 (d, J = 7.8, 2H), 7.92 (d, J = 7.3, 2H), 7.83 (m, 1H), 7.70 (t, J = 7.3, 2H), 2.17 (s, 6H). 13C{1H} NMR (125 MHz, DMSO) δ: 169.8 165.8, 151.1, 150.6, 140.5, 134.7, 161.8, 129.3, 127.9, 129.3, 127.9, 110.9, 110.3, 24.4. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C22H21N6O4, 433.1624; found 433.1615.
N1,N5-bis(6-Pivalamidopyridin-2-yl)isophthalamide (4b)
The alkyl tethered diaminopyridine 3e was prepared according to the general procedure outlined for 3d with the following quantities: isophthaloyl dichloride (1.0 g, 6.2 mmol) in THF (20 mL) was added slowly to 2b (2.0 g, 10 mmol) and triethylamine (3.6 mL, 26 mmol) in THF (100 mL). Purified by column chromatography (Si2O, 3:2 EtOAc:hexanes) to afford a white crystalline solid (2.89 g, 56%). Mp = 135-136 °C. 1H NMR (500 MHz, CDCl3) δ: 9.10 (s, 2H), 8.46 (s, 2H), 8.20-8.17 (m, 3H), 8.05 (d, J = 8.5, 2H), 7.97 (d, J = 8.0, 2H), 7.86 (t, J = 8, 2H), 7.72 (t, J = 8, 1H), 1.13 (s, 18H). 13C{1H} NMR (125 MHz, CDCl3) δ: 177.0, 164.2, 149.9, 149.2, 141.0, 134.8, 130.8, 129.6, 125.8, 110.0, 109.0, 39.8, 27.5. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C28H33N6O4 517.2563; found 517.2574.
N1,N5-bis(6-Benzamidopyridin-2-yl)isophthalamide (4c)
The alkyl tethered diaminopyridine 4c was prepared according to the general procedure outlined for 3d with the following quantities: isophthaloyl dichloride (0.23 g, 1.4 mmol) in THF (20 mL) was added slowly to 2c (0.0.51 g, 2.4 mmol) and triethylamine (0.71 mL, 7.0 mmol) in THF (50 mL). Purified by column chromatography (Si2O, 3:2 hexanes:EtOAc) to afford a white crystalline solid (0.88 g, 66%). Mp = 213-215 °C. 1H NMR (300 MHz, CDCl3) δ: 8.56 (s, 2H), 8.53 (s, 1H), 8.40 (s, 2H), 8.17 (d, J = 8.0, 4H), 8.14 (d, J = 8.0, 2H) 7.94 (d, J = 7.0, 4H), 7.87 (t, J = 8.0, 2H), 7.70 (t, J = 8.0, 1H), 7.61 (t, J = 7.5, 2H), 7.53 (t, J = 7.5, 4H). 13C{1H} NMR (125 MHz, CDCl3) δ: 165.5, 164.2, 149.8, 149.4, 141.2, 134.8, 134.1, 132.4, 130.9, 128.9, 127.2, 110.3, 109.9. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C32H25N6O4, 557.1937; found 557.1940.
Supplementary Material
Figure 5.
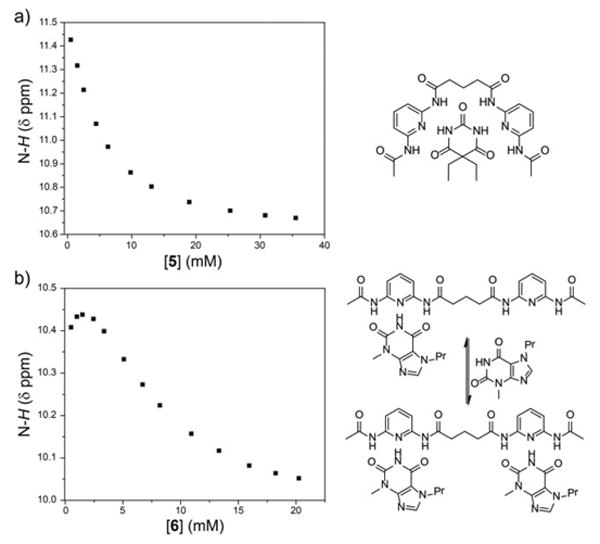
Representative 1H NMR (500 MHz, 25 °C, 5% d6-DMSO in CDCl3) titration data for 1a with 6. Graph of the change in N-H chemical shift with changing concentrations of (a) 5 and (b) 6 in the presence of 3a.
Acknowledgments
We thank Mr. Ryan Hansen for assistance with preliminary titration data and Dr. Jesse Gavette for helpful discussions. This work was supported by funding from the University of Oregon. The NMR facilities at the UO are supported by the NSF/ARRA (CHE-0923589), and the computational infrastructure is supported by the OCI (OCI-096054). The Biomolecular Mass Spectrometry Core of the Environmental Health Sciences Core Center at Oregon State University is supported, in part, by the NIEHS (P30ES000210) and the NIH.
Footnotes
Supporting Information. NMR spectra of new compounds, optimized geometries from DFT calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sijbesma RP, Meijer EW. Chem Commun. 2003:5–16. doi: 10.1039/b205873c. [DOI] [PubMed] [Google Scholar]
- 2.Sontjens SHM, Meijer JT, Kooijman H, Spek AL, van Genderen MHP, Sijbesma RP, Meijer EW. Org Lett. 2001;3:3887–3889. doi: 10.1021/ol016750a. [DOI] [PubMed] [Google Scholar]
- 3.Mardis KL. J Phys Chem B. 2006;110:971–975. doi: 10.1021/jp054964u. [DOI] [PubMed] [Google Scholar]
- 4.Beijer FH, Kooijman H, Spek AL, Sijbesma RP, Meijer EW. Angew Chem Int Ed. 1998;37:75–78. [Google Scholar]
- 5.Prins LJ, Reinhoudt DN, Timmerman P. Angew Chem Int Ed. 2001;40:2382–2426. doi: 10.1002/1521-3773(20010702)40:13<2382::aid-anie2382>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 6.Whitesides GM, Simanek EE, Mathias JP, Seto CT, Chin DN, Mammen M, Gordon DM. Acc Chem Res. 1995;28:37–44. [Google Scholar]
- 7.ten Cate AT, Sijbesma RP. Macromol Rapid Commun. 2002;23:1094–1112. [Google Scholar]
- 8.Schmuck C, Wienand W. Angew Chem Int Ed. 2001;40:4363–4364. doi: 10.1002/1521-3773(20011203)40:23<4363::aid-anie4363>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 9.Desiraju GR. Acc Chem Res. 1996;29:441–449. doi: 10.1021/ar950135n. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong G, Buggy M. J Mater Sci. 2005;40:547–559. [Google Scholar]
- 11.Adriaenssens L, Ballester P. Chem Soc Rev. 2013;42:3261–3277. doi: 10.1039/c2cs35461f. [DOI] [PubMed] [Google Scholar]
- 12.Rebek J. Chem Soc Rev. 1996;25:255–264. [Google Scholar]
- 13.Sijbesma RP, Meijer EW. Curr Opin Colloid Interface Sci. 1999;4:24–32. [Google Scholar]
- 14.Jasat A, Sherman JC. Chem Rev. 1999;99:931–967. doi: 10.1021/cr960048o. [DOI] [PubMed] [Google Scholar]
- 15.Price SL. Crystengcomm. 2004;6:344–353. [Google Scholar]
- 16.Chang SK, Hamilton AD. J Am Chem Soc. 1988;110:1318–1319. [Google Scholar]
- 17.Berl V, Huc I, Lehn JM, DeCian A, Fischer J. Eur J Org Chem. 1999:3089–3094. [Google Scholar]
- 18.Chang SK, Vanengen D, Fan E, Hamilton AD. J Am Chem Soc. 1991;113:7640–7645. [Google Scholar]
- 19.Collinson SR, Gelbrich T, Hursthouse MB, Tucker JHR. Chem Commun. 2001:555–556. [Google Scholar]
- 20.Shivanyuk AN, Rudkevich DM, Reinhoudt DN. Tetrahedron Lett. 1996;37:9341–9344. [Google Scholar]
- 21.Tecilla P, Jubian V, Hamilton AD. Tetrahedron. 1995;51:435–448. [Google Scholar]
- 22.Schmidt J, Schmidt R, Wurthner F. J Org Chem. 2008;73:6355–6362. doi: 10.1021/jo801083b. [DOI] [PubMed] [Google Scholar]
- 23.Beijer FH, Sijbesma RP, Vekemans J, Meijer EW, Kooijman H, Spek AL. J Org Chem. 1996;61:6371–6380. doi: 10.1021/jo960612v. [DOI] [PubMed] [Google Scholar]
- 24.Feibush B, Figueroa A, Charles R, Onan KD, Feibush P, Karger BL. J Am Chem Soc. 1986;108:3310–3318. [Google Scholar]
- 25.Kotera M, Lehn JM, Vigneron JP. J Chem Soc Chem Comm. 1994:197–199. [Google Scholar]
- 26.Brienne MJ, Gabard J, Lehn JM, Stibor I. J Chem Soc Chem Comm. 1989:1868–1870. [Google Scholar]
- 27.Yu LH, Schneider HJ. Eur J Org Chem. 1999:1619–1625. [Google Scholar]
- 28.Muehldorf AV, Vanengen D, Warner JC, Hamilton AD. J Am Chem Soc. 1988;110:6561–6562. [Google Scholar]
- 29.Hamilton AD, Little D. J Chem Soc Chem Comm. 1990:297–300. [Google Scholar]
- 30.Goodman MS, Rose SD. J Am Chem Soc. 1991;113:9380–9382. [Google Scholar]
- 31.Osmialowski B, Kolehmainen E, Gawinecki R, Kauppinen R, Koivukorpi J, Valkonen A. Struct Chem. 2010;21:1061–1067. [Google Scholar]
- 32.Osmialowski B, Kolehmainen E, Gawinecki R, Dobosz R, Kauppinen R. J Phys Chem A. 2010;114:12881–12887. doi: 10.1021/jp1084857. [DOI] [PubMed] [Google Scholar]
- 33.Eckelmann J, Dethlefs C, Brammer S, Dogan A, Uphoff A, Luning U. Chem -- Eur J. 2012;18:8498–8507. doi: 10.1002/chem.201200181. [DOI] [PubMed] [Google Scholar]
- 34.Gnichwitz JF, Wielopolski M, Hartnagel K, Hartnagel U, Guldi DM, Hirsch A. J Am Chem Soc. 2008;130:8491–8501. doi: 10.1021/ja8018065. [DOI] [PubMed] [Google Scholar]
- 35.Wessendorf F, Grimm B, Guldi DM, Hirsch A. J Am Chem Soc. 2010;132:10786–10795. doi: 10.1021/ja101937w. [DOI] [PubMed] [Google Scholar]
- 36.Larsen J, Rasmussen BS, Hazell RG, Skrydstrup T. Chem Commun. 2004:202–203. doi: 10.1039/b309863j. [DOI] [PubMed] [Google Scholar]
- 37.Sorensen HS, Larsen J, Rasmussen BS, Laursen B, Hansen SG, Skrydstrup T, Amatore C, Jutand A. Organometallics. 2002;21:5243–5253. [Google Scholar]
- 38.Li Y, He YM, Li ZW, Zhang F, Fan QH. Org Biomol Chem. 2009;7:1890–1895. doi: 10.1039/b823047a. [DOI] [PubMed] [Google Scholar]
- 39.Wurthner F, Schmidt J, Stolte M, Wortmann R. Angew Chem Int Ed. 2006;45:3842–3846. doi: 10.1002/anie.200504581. [DOI] [PubMed] [Google Scholar]
- 40.Grimm F, Hartnagel K, Wessendorf F, Hirsch A. Chem Commun. 2009:1331–1333. doi: 10.1039/b822043c. [DOI] [PubMed] [Google Scholar]
- 41.Dirksen A, Hahn U, Schwanke F, Nieger M, Reek JNH, Vogtle F, De Cola L. Chem -- Eur J. 2004;10:2036–2047. doi: 10.1002/chem.200305461. [DOI] [PubMed] [Google Scholar]
- 42.Binder WH, Kluger C, Straif CJ, Friedbacher G. Macromolecules. 2005;38:9405–9410. [Google Scholar]
- 43.Binder WH, Kluger C, Josipovic M, Straif CJ, Friedbacher G. Macromolecules. 2006;39:8092–8101. [Google Scholar]
- 44.Binder WH, Lomoschitz M, Sachsenhofer R, Friedbacher G. J Nanomater. 2009:613813. [Google Scholar]
- 45.Bolton W. Acta Cryst. 1962;16:166–173. [Google Scholar]
- 46.Thordarson P. Chem Soc Rev. 2011;40:1305–1323. doi: 10.1039/c0cs00062k. [DOI] [PubMed] [Google Scholar]
- 47.We also optimized and calculated binding enthalpies for the 2:1 barbital:2,6-biscarboxamido pyridine scaffolds even though we did not observe formation of these in solution, with the exception of 1a. The binding affinity of the second 2,6-biscarboxamido pyridine ligand was slightly lower than the first, and the entropic penalty required for forming the 2:1 adducts may explain the lack of observation in solution. See the Supporting Information for tabulated 2:1 binding enthalpies.
- 48.Findeisen M, Brand T, Berger S. Magn Reson Chem. 2007;45:175–178. doi: 10.1002/mrc.1941. [DOI] [PubMed] [Google Scholar]
- 49.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C.01. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 50.Dennington R, Keith T, Millam J. GaussView, Version 5. Semichem Inc.; Shawnee Mission KS: 2009. [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Langer P, Amiri S, Bodtke A, Saleh NNR, Weisz K, Gorls H, Schreiner PR. J Org Chem. 2008;73:5048–5063. doi: 10.1021/jo8005123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.